

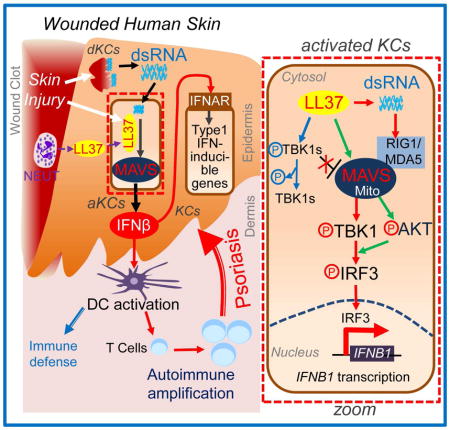
SUMMARY
Type 1 interferons (IFN) promote inflammation in the skin but the mechanisms responsible for inducing these cytokines are not well understood. We found that IFNβ was abundantly produced by epidermal keratinocytes (KCs) in psoriasis and during wound repair. KC IFNβ production depended on stimulation of mitochondrial antiviral-signaling protein (MAVS) by the antimicrobial peptide LL37 and double stranded-RNA released from necrotic cells. MAVS activated downstream TBK1 (TANK-Binding Kinase 1)-AKT (AKT serine/threonine kinase 1)-IRF3 (interferon regulatory factor 3) signaling cascade leading to IFNβ production, and then promoted maturation of dendritic cells. In mice, the production of epidermal IFNβ by LL37 required MAVS, and human wounded and/or psoriatic skin showed activation of MAVS-associated IRF3 and induction of MAVS and IFNβ gene signatures. These findings show that KCs are an important source of IFNβ and MAVS is critical to this function, and demonstrates how the epidermis triggers unwanted skin inflammation under disease conditions.
Graphical Abstract
INTRODUCTION
Psoriasis is a chronic inflammatory skin disease that affects approximately 3% of the US population, 2.5% of the European population and 0.1~0.5% of Asians (Yin et al., 2015). Progress in understanding the etiology of this disease has revealed it to be a complicated immune system disorder involving the dynamic interplay of innate and adaptive immunity, with participation of several cell types including epidermal keratinocytes (KC), T lymphocytes, dendritic cells (DCs) and neutrophils (Lowes et al., 2007; Sweeney et al., 2011). Although successful treatment regimens for psoriasis have been established by inhibiting TNFα (tumor necrosis factor alpha) or IL-17 (interleukin 17) cytokine signaling processes, the primary pathogenic mechanism responsible for the onset of the disease remains poorly understood. Approximately half of patients with psoriasis experience a phenomenon known as the Koebner reaction in which a new psoriatic plaque is formed where the skin is injured (Weiss et al., 2002). The cell types and mechanisms through which injury stimulates psoriasis are unclear but have been hypothesized to involve damage associated molecular patterns (DAMPs) such as release of DNA and RNA from necrotic cells (Bijlmakers et al., 2011; Ganguly et al., 2009).
Type1 interferons (IFNα and IFNβ) are key cytokines induced during viral infection that influence both innate and adaptive antiviral host immune responses (Gonzalez-Navajas et al., 2012). Associations between chronic viral infections and the development of psoriasis have suggested that IFNα or IFNβ may play a role in initiating this disease (Erkek et al., 2000; Yamamoto et al., 1995). Furthermore, treatment of patients with type1 IFNs often directly induces or exacerbates psoriasis or psoriatic arthritis (Erkek et al., 2000; Funk et al., 1991; Toussirot et al., 2014). Further evidence for a role of IFNα or IFNβ in psoriasis has also been seen in mice lacking the type1 IFN receptor, or mice treated with IFNα or IFNβ neutralizing antibodies, as both systems fail to develop T cell and T helper 17 (Th17)-mediated skin inflammation (Gregorio et al., 2010; Nestle et al., 2005). Additionally, mice lacking IRF2 (interferon regulatory factor 2), a negative regulator of type I IFN signaling, develop spontaneous psoriasis-like inflammatory skin disease (Hida et al., 2000). In line with these observations, transcriptome analyses have identified type1 IFN-inducible gene signatures as being upregulated in psoriatic skin, outscoring the upregulation of TNFα and TNFα-inducible gene signatures (Baldwin et al., 2013; Ruano et al., 2016; Schmid et al., 1994; van der Fits et al., 2004; Yao et al., 2008).
Classical immunocytes such as DCs and T cells have been shown to play a major role in psoriasis. Type I IFNs can be induced in these cell types either through toll-like receptor (TLR)-dependent pathways or TLR-independent pathways such as the RIG1 (retinoic acid-inducible gene 1)-MDA5 (melanoma differentiation-associated protein 5)-MAVS pathway (Baccala et al., 2007). However, it is becoming increasingly recognized that the epidermal KCs also participate in the initiation of the cutaneous inflammatory response. KCs comprise the outermost layer of the skin, and are a well-described source of inflammatory mediators (Lowes et al., 2007). During early stages of wound healing, KCs are activated and release proinflammatory mediators such as IL-6, IL-8 and TNFα, and antimicrobial peptides (AMPs) including cathelicidin and β-defensins (Dorschner et al., 2001; Gallo et al., 1994; Heilborn et al., 2003). The mature form of cathelicidin in humans is known as LL37 and is a potent immunomodulator (Lai and Gallo, 2009; Zhang and Gallo, 2016) that is highly expressed in psoriasis and can facilitate plasmacytoid DC (pDC) recognition of self-DNA and production of IFNα in this cell type (Lande et al., 2007). Although the production of IL-6, IL-8 and TNFα from KCs has been clearly established, the source of type 1 IFNs in the skin has been primarily attributed to DCs.
As KCs are the most abundant cell type in the epidermis, and occupy a position in the skin as the outermost barrier exposed to injury, we hypothesized that KCs could also serve as a source of type 1 IFN, thus rapidly and enhancing DC functions and later T cell activation events. We describe how the MAVS signaling pathway is activated by LL37 and dsRNA in KC, and how this leads to IFNβ expression and the subsequent activation and maturation of DCs. These observations provide a more comprehensive understanding of how the epidermis can respond to skin injury, thus linking innate and adaptive immune responses in skin inflammation.
RESULTS
IFNβ1 is produced by the epidermis in response to LL37
To test our hypothesis that KCs serve as a source of type1 IFN, we examined the expression of IFNβ in human skin under both normal and inflammatory conditions. Consistent with previous reports (Nestle et al., 2005), the expression of IFNα was seen primarily in dermal DCs (Figures 1A and S1A). In contrast, IFNβ1 was abundantly expressed in the epidermis, and localized in KCs of psoriatic skin and at the edge of wounds (Figures 1A–C and S1A). Co-immunostaining revealed that IFNβ was detected only in KCs that also expressed LL37 (Figures 1B–C). Such co-localization of IFNβ and LL37 was not seen in the dermis where immunocytes such as pDCs and T cells were located (Figure 1C). The comparable response of IFNβ and LL37 in psoriasis and wounds was also observed for expression of keratin 6 (KRT6), a marker for wound-activated KCs (Wong and Coulombe, 2003) (Figure S1B). These similar cell responses in psoriasis and normal wound repair were consistent with prior observations that psoriasis has immunological similarities with the persistent wounding response (Morhenn, 1988; Nickoloff et al., 2006).
Fig. 1. Epidermal IFNβ1 expression in wounded and psoriatic skin is associated with LL37.

(A). Skin sections of wounded (day3) or psoriatic skin were immunostained with IFNβ or IFNα antibodies as indicated. White dashed line indicates the epidermal and dermal junction. (B–C) Co-immunostaining analyses of normal human unwounded (ctrl) and wounded skin sections (B) or psoriasis non-lesional (PNL) and psoriasis lesional (PSO) (C) skin sections with IFNβ (red) and LL37 (green) antibodies as indicated. Zoom-in polyICtures highlighted in red box are shown on the right panel. (D–E) 3D human skin constructs made with NHEKs (neonatal human epidermal KCs) transfected with shCtrl or shCAMP. (D) CAMP expression in the skin constructs (n=3) was confirmed by RTqPCR analysis (normalized to GAPDH). (E), Skin sections of wounded skin constructs made with NHEKs transfected with shCtrl or shCAMP were immunostained with IFNβ antibody. All images are representative of 3~5 independent experiments. All scale bars = 100 μm. Error bars indicate mean ± s.e.m. *** P<0.001 (one way Anova).
We next investigated whether an increase in LL37 and IFNβ also occurred in an ex vivo model of human skin wounds. In this model, immune cell infiltration into the skin from the periphery (such as neutrophils) is absent (Roupe et al., 2010), thus permitting better assessment of the intrinsic innate response of the tissue. In ex vivo human skin wounds, KRT6 (Figure S1C) as well as LL37 and IFNβ (Figure S1D) were expressed by KCs at the wound edge. These results suggest that induction of LL37 and IFNβ is at least in part an intrinsic response to injury by KCs.
Next, to evaluate the dependence of IFNβ production on LL37, we generated three-dimentional (3D) human skin constructs using normal human epidermal KCs in which the gene for LL37 (CAMP) was silenced (Figure 1D and S1E). IFNβ was induced by injury in control epidermal constructs but silencing of CAMP suppressed this increase (Figures 1E and S1F). These results show that KCs could intrinsically express IFNβ, and that LL37 facilitated epidermal IFNβ expression in response to injury.
dsRNA acts with LL37 to enhance IFNβ production by KCs
To define the mechanisms by which LL37 induces IFNβ expression in keratinocytes, we evaluated the effect of LL37 on IFNβ1 expression by primary monolayer cultures of normal human KCs (hKCs). The double-stranded (ds)RNA polyIC was a potent inducer of IFNB1 mRNA in hKCs (Figures 2A and S2A–E). LL37 alone did not induce IFNB1 but LL37 pretreatment strongly increased polyIC-mediated IFNB1 expression (Figure 2A). LL37 also enhanced IFNβ1 production in response to a crude extract of damaged KCs, and this response was decreased when the damaged KCs extract was first treated with RNAseIII to partially remove dsRNA (Figure 2B). Furthermore, noncoding dsRNAs (U1, U2, U4, U6 and U12) that are released upon KC injury (Bernard et al., 2012; Lai et al., 2009; Borkowski et al., 2015) also induced IFNβ (Figures 2C and S2F). The response of human 3D skin constructs to these endogenous dsRNAs was dependent on LL37 since shRNA-mediated silencing of CAMP expression decreased IFNβ production (Figure 2D) and the induction of IFNB1 and IFN-inducible gene ISG15 (interferon-stimulated gene 15) was completely inhibited (Figures S2G–H). These results indicated that physiologically relevant dsRNAs drove IFNβ production by KCs in the presence of LL37.
Fig. 2. LL37 enhances IFNβ production by dsRNA in keratinocytes.

(A) NHEKs pretreated with LL37 or vehicle control were exposed to various PRR ligands as indicated for 4 hrs (The corresponding TLR# is indicated between parentheses; M=MAVS). IFNB1 mRNA fold induction was analyzed by RTqPCR analyses (n=3). (B) RTqPCR analyses of IFNB1 mRNA induction in NHEKs after stimulation with cell extracts from damaged NHEK (dNHEK) with or without LL37 pretreatment. In some experiments, RNA was depleted in the dNHEK by pretreatment with RnaseIII (a RNase specific for dsRNA) as indicated (n=3). (C). IFNβ protein secretion from NHEKs stimulated with various self-noncoding RNAs or pIC (polyIC) with or without LL37 pretreatment as indicated. (n=3). (D). Control or CAMP silenced 3D skin constructs were treated with vehicle control or pIC (polyIC) for 6 hrs prior to being subjected to immunostaining with IFNβ antibodies (E). RTqPCR analyses of various type1 IFN mRNA levels in NHEKs treated with different combination of polyIC and LL37 as indicated (n=3). (F). Rador plot comparing the expression of type1 IFNs in LL37 and polyIC activated NHEKs and LL37 and CpGA activated pDCs (average of 3 donors). All error bars indicate mean ± s.e.m. * P<0.05, ** P<0.01, *** P<0.001 (one way Anova).
Analyses of the expression of various type1 IFNs revealed that IFNB1 is the major type 1 IFN expressed by KCs exposed to LL37 and dsRNA (Figure 2E). In contrast, human pDCs were not responsive to LL37 and polyIC but rather were able to produce several family members of IFNα when activated by the DNA CpGA combined with LL37 (Figures 2F and S2I). KCs treated with DNA and LL37 did not show a substantial induction of IFNB1 mRNA (Figure S2A). Of note, activated KC expressed relatively higher levels of IFNB1 mRNA when treated with LL37 and polyIC than pDCs activated by LL37 and CpGA (Figures 2E–F and S2J). These results were consistent with the observation in skin that IFNβ was detected abundantly in the epidermis of wounded and psoriatic skin while IFNα was primarily detected in dermal pDCs (Figure 1A). Together, we have demonstrated that LL37 enable KC’s responsiveness to dsRNA to produce type_1 IFNs, mainly IFNβ.
dsRNA and LL37 activates the MAVS-RIG1-MDA5 pathway in KCs
To determine the capacity of different cell types to respond to nucleic acids or IFN, we next compared human KCs, pDC and monocyte-derived classical DCs (cDC) for the relative expression of mRNA for relevant PRRs and the IFNAR1 (IFN-I receptor 1). KCs expressed the highest amounts of MAVS mRNA and moderate mRNA of TLR3, while other TLRs including TLR4, TLR7, TLR8 and TLR9 were barely detectable (Figure 3A). In contrast, pDCs expressed lower mRNA level of MAVS but similar mRNA level of TLR3, and cDC expressed higher mRNA levels of TLR4 and TLR8 (Figure 3A). pDC expressed the highest mRNA levels of TLR7, TLR9 and IRF7 (Figure 3A and S3A). IFNAR1 was highly expressed in all cell types (Figure 3A), suggesting that both DCs and KCs are potential targets of IFNβ. In whole skin, MAVS mRNA was found predominantly in the epidermis whereas TLR4, 7, 8 and 9 were found only in the dermis where DCs are located (Figures 3B and S3B). Analysis of K10 (Keratin 10) and PDGFRA (platelet-derived growth factor receptor alpha) confirmed the adequate isolation of the epidermal and dermal layers from the normal skin sample (Fig. S3B). Similar results were found in epidermis and dermis separated from mouse skin (Figures S3C–D). These results provide a potential explanation for the lack of IFNβ response to TLR4, , 7, 8, or 9 ligands in KCs and strong IFNα response to TLR9 ligand in pDCs (Figure 2F).
Fig. 3. IFNβ production from epidermal keratinocyte in response to LL37 and dsRNA is mediated by MAVS.

(A–B). RTqPCR of mRNA expression of MAVS, various TLRs and IFNAR1 in KC (NHEK), pDC analyses or cDC (A) (n=3; ratio to GAPDH mRNA is shown), or separated epidermis and dermis from normal human skin (B) (fold changes are shown relative to TLR3) (n=3). (C). RTqPCR analysis of IFNB1 mRNA expression in NHEKs transfected with control siRNA, TLR3 siRNA or MAVS siRNA then stimulated with polyIC with or without LL37 pretreatment. (n=3). (D). RTqPCR analysis of Ifnb1 mRNA expression in LL37/polyIC stimulated mouse KCs isolated from Mavs−/+ HET or Mavs−/− KO littermate mice (n=3). (E). RTqPCR analysis of IFNB1 mRNA expression in NHEKs transfected with control siRNA, TLR3 siRNA or MAVS siRNA then stimulated with LL37 and U1-RNA as indicated (n=3). (F). NHEKs treated with various combination of polyIC and LL37 were co-immunostained with anti-MAVS antibody and mitochondrial tracker (MitoV) or phosphor-tyrosine (pTyr) as indicated. (G). Time course analyses of ISG15 mRNA expression in NHEKs treated with LL37 and polyIC as indicated (n=3). (H–I). ISG15 expression in siRNA mediated silencing of TLR3 or MAVS (H) or IFNB1 or IFNAR1. (I) NHEKs treated with LL37 and polyIC (n=3). (J) Proposed scheme for induction of proinflammatory cytokines and type-1 IFN responsive genes mediated by LL37 and dsRNA mediated through MAVS or TLR3. All error bars indicate mean ± s.e.m. * P<0.05, ** P<0.01, *** P<0.001 (one way Anova). L/P, LL37 and polyIC.
The results presented thus far suggested that MAVS and TLR3 were ideally positioned in KCs to detect dsRNA within the epidermis. To test if TLR3 or MAVS could stimulate IFNβ production in response to LL37 and dsRNA, TLR3 or MAVS was silenced in KCs (Figure S3E). Without the addition of LL37, IFNβ production in response to dsRNA was dependent on TLR3 but not MAVS (Figure 3C). In contrast, when LL37 was present, IFNβ expression was strongly inhibited in both cells in which MAVS or TLR3 expression was reduced using short interfering RNAs (siRNAs) (Figure 3C), suggesting LL37 enabled dsRNA recognition through the MAVS pathway. In primary mouse KCs, LL37 strongly boosted dsRNA-mediated induction of Ifnb1 in KCs derived from wild-type mice while KCs from Mavs−/− mice had minimal Ifnb1 gene expression (Figure 3D). In contrast, CRAMP, the mouse homolog of LL37, failed to enhance polyIC-mediated Ifnb1 response (Figure S3F), suggesting that this effect was specific for the human cathelicidin peptide LL37. MAVS was also required for LL37 and U1-RNA mediated induction of IFNB1 (Figure 3E).
MAVS has been shown to cluster to the perinuclear region upon activation (Chen et al., 2007). After exposure to LL37 and polyIC, MAVS localized to this perinuclear region (Figure 3F). A similar perinuclear clustering of RIG1 was also observed in LL37 and polyIC treated cells (Figure S3G). MAVS also migrated slower on SDS-PAGE gels when cells were activated by LL37 and polyIC (Figure S3H), an observation consistent with prior observations that post-translational modifications are associated with activation of MAVS (Wen et al., 2012). Supporting this, co-immunostaining of MAVS with an antibody specific for phospho-tyrosine (pTyr) showed that MAVS colocalized with pTyr (Figure 3F). These results demonstrated that MAVS was abundantly expressed in epidermal KCs and depended on LL37 to be activated by dsRNA.
Multiple cytokines and IFN-inducible genes are activated by LL37 and dsRNA
mRNA expression of several other inflammatory cytokines and type 1 IFN-inducible genes were also induced in KCs exposed to LL37 and dsRNA. Time course studies revealed two distinct induction patterns for these genes. Similar to IFNB1, inflammatory cytokines including IL6, IL8 (CXCL8) and TNFα were induced early and transiently, peaking at 4 hrs after treatment (Figures S4A–C) whereas the expression of CCL5, RIG1 (DDX58), MDA5 (IFIH1) and IFN-inducible genes including ISG15, IRF7 (interferon regulatory factor 7) and STAT1 (signal transducer and activator of transcription 1) were induced later but continuously within the 20 hr time course (Figures 3G and S4D–H). siRNA-mediated silencing of either TLR3 or MAVS demonstrated that LL37 and dsRNA-mediated induction of IL-6 was dependent on TLR3 but not MAVS (Figure S4I), whereas the induction of other genes induced by LL37 and dsRNA was partially dependent on both TLR3 and MAVS (Figures 3H and S4J–O).
We next investigated whether late response genes were secondarily induced by the activation of the IFNβ-IFNAR pathway. siRNA-mediated silencing of either IFNB1 or IFNAR1 (Figure S4P–Q) led to reduction of ISG15, IRF7, STAT1 and RIG1 expression in LL37 and dsRNA treated cells (Figures 3I and S4R–T). However, LL37 and dsRNA-mediated induction of CCL5 (chemokine (C-C motif) ligand 5), IL6, TNF and IL8 was not altered when IFNB1 or IFNAR was silenced by siRNA(Figures S4U–X). Together, these findings establish a selective role for MAVS and IFNβ in regulating the expression of IFN-inducible genes in response to LL37 and dsRNA in KCs (proposed model in Fig. 3J).
TBK1-AKT-IRF3 pathways are activated by LL37 and dsRNA
To investigate the pathways involved in IFNβ production by LL37 and dsRNA, we next assessed key signaling molecules upstream of IFNβ transcriptional events. Activation of TBK1, the kinase downstream of both TLR3 and MAVS, is critically involved in the subsequent phosphorylation and nuclear translocation of transcription factor IRF3, which in turn binds to IFNB1 gene enhancer resulting in IFNB1 mRNA production (Goutagny et al., 2006). Kinase AKT also plays an important role in the activation of IRF3 by interacting with TBK1 (Joung et al., 2011; Sarkar et al., 2004). In hKCs, dsRNA-mediated TBK1 phosphorylation was enhanced by LL37 (Figure 4A). Interestingly, while LL37 itself had no effect on phosphorylation of full length TBK1, it robustly dephosphorylated a smaller spliced variant of TBK1 that is heavily phosphorylated in KCs (Figure 4A). It has been reported that this alternative spliced isoform of TBK1 (IKKe), termed TBK1s, negatively regulates IRF3 phosphorylation by inhibiting RIG1 and MAVS function (Deng et al., 2008; Koop et al., 2011). Thus, LL37 may enable RIG1/MAVS function by deactivating their negative regulator TBK1s. Furthermore, LL37 alone activated AKT (Figure 4A) while polyIC did not significantly change AKT phosphorylation. Time course studies of LL37 revealed that phosphorylation of AKT and dephosphorylation of TBK1 by LL37 peaked at 1 hr (Figure 4B). Phosphorylation of ERK1 and 2 (extracellular signal-regulated kinase 1 and 2) was not affected by LL37 or polyIC (Figures 4A–B), thus ERK1 and 2 was used as a negative control in our studies. IRF3 was phosphorylated by LL37 and polyIC (Figure 4A) and translocated to the nucleus (Figure 4C). Co-immunostaining of TBK1 with the mitochondrial marker Aconitase showed that TBK1 was recruited to mitochondria only in cells treated with LL37 and polyIC (Figure 4D), supporting the involvement of mitochondria in the dsRNA response in the presence of LL37.
Fig. 4. AKT-TBK1-IRF3 pathways are activated by LL37 and dsRNA.

(A). NHEKs were pretreated with 2 μM LL37 then stimulated with 0.4 μg/ml polyIC for 2 hrs. Cell extracts were subjected to immunoblotting analyses using indicated antibodies. Abbreviations: SV: spliced variant; FL: full length; arrow indicates specific band and asterisk indicates non-specific band. In IRF3 blot, form I is non-phosphorylated IRF3 and the form II is phosphorylated IRF3. (B). NHEKs were stimulated with LL37 for indicated time and cell extracts were subjected to immunoblotting analyses using indicated antibodies. (C). IRF3 immunostaining in NHEKs stimulated with indicated treatment for 2 hrs. Nuclei were counterstained with DAPI. (D). Stimulated NHEKs were stained with pTBK1 (red) and mitochondrial marker Aconitase (blue) as indicated.
IFNβ production in response to dsRNA and LL37 is dependent on TBK1-AKT and MAVS
Having shown that TBK1 and AKT were activated by LL37 and polyIC, we next sought to determine the role of TBK1 and AKT in IRF3 phosphorylation and IFNβ gene induction. The TBK1 inhibitor Cay10567 had no effect on AKT or ERK phosphorylation, but it blocked IRF3 phosphorylation in response to LL37 and polyIC (Figure 5A). Consistent with a previous report (Reilly et al., 2013), inhibition of TBK1 activity by Cay10567 increased TBK1 phosphorylation (Figure 5A), presumably due to blockage of feedback inhibition. Inhibition of AKT phosphorylation by Wortmannin did not alter TBK1 phosphorylation but it blocked IRF3 phosphorylation in response to LL37 and polyIC (Figure 5A). In addition, blockage of endosomal acidification using bafilomycin-A1 (BafA1) led to decreased phosphorylation of both TBK1 and AKT, resulting in loss of the subsequent IRF3 phosphorylation (Figure 5A). Consistent with the decrease in TBK1-AKT-IRF3 phosphorylation, IFNB1 mRNA induction in response to LL37 and polyIC was blocked when TBK1, AKT, or endosomal acidification was inhibited (Figure 5B and S5A). Together these results show that both TBK1 and AKT are required for IRF3 activation and the subsequent induction of IFNβ in response to LL37 and dsRNA (see proposed model in Figure S5B).
Fig. 5. IFNβ production in response to dsRNA and LL37 is dependent on TBK1-AKT and MAVS.

(A). NHEKs were pretreated with BafilomycinA (to block endosomal acidification) or inhibitors specific for TBK1 (1μM Cay10567), AKT (100 nM Wortmannin) or ERK (1μM U0126) for 30 mins before treating with LL37 then polyIC for 2 hrs as indicated. Cell extracts were subjected to immunoblotting analyses using indicated antibodies. (B). NHEKs were pretreated with indicated inhibitors prior to LL37 and polyIC treatment for 4 hrs. Cells were subjected to RTqPCR analyses for relative mRNA expression of IFNβ (n=3). −/+ indicates presence or absence of LL37 and pIC or pIC alone. (C). NHEKs were transfected with control siRNA, TLR3 siRNA or MAVS siRNA for 48 hrs before treating with vehicle control, polyIC or LL37 then polyIC for 2 hrs. Cell extracts were subjected to immunoblotting analyses using indicated antibodies. (D–E). Quantification of the ratio of pTBK1 to tTBK1 (D) and the ratio of pAKT to tAKT (E) from immunoblots (n=3). All error bars indicate mean ± s.e.m. * P<0.05, ** P<0.01, *** P<0.001 (one way Anova).
We also observed that siRNA-mediated silencing of either TLR3 or MAVS led to ~50% reduction of TBK1 phosphorylation (Figure 5C–D), suggesting that both TLR3 and MAVS are involved in TBK1 activation in response to LL37 and polyIC. In contrast, AKT phosphorylation was largely dependent on MAVS, but not TLR3 (Figure 5C and 5E). Together, these results show that LL37 and dsRNA activate TBK1, AKT and IRF3 through a MAVS dependent pathway (see proposed model in Figure S5C).
MAVS-mediated secretion of IFNβ from LP-activated keratinocytes promotes activation and maturation of dendritic cells
To evaluate the physiological consequences of IFNβ expression by KCs, we determined if KCs could promote DC activation by stimulating KCs with LL37 and polyIC and then adding this conditioned culture media (CM) to DCs (Figure S6A). KCs derived from Mavs−/− mice were not able to produce IFNβ protein following stimulation with LL37 and polyIC (Figure 6A). CM from WT or Mavs−/+ (HET) mouse KCs (mKCs) potently promoted activation and maturation of mouse bone marrow derived cDC as defined by an increase in the surface expression of CD80 (cluster of differentiation 80) and CD86 (Figures S6B–G and 6B) as well as expression of Mx1, Irf7 and Cd83 (Figure 6C). In contrast, CM from Mavs−/− (KO) mKCs failed to stimulate DC activation and maturation (Figures 6A–C and S6B–G). CM from human KCs treated with LL37 and polyIC also promoted activation and maturation of human monocyte derived cDCs (Figures 6D–E). IFNβ was the major cytokine from the KC-CM that was driving this effect because IFNβ neutralizing antibody, but not control IgG or IFNα neutralizing antibodies inhibited the capacity of KC-CM to promote DC maturation (Figures 6D–E). In addition, DCs that were matured with the CM of KC activated with LL37 and polyIC was able to stimulate T cell proliferation whereas treatment of this conditioned medium with IFNβ neutralizing Ab could not stimulate T cells (Figure 6F). Together, these results demonstrate that IFNβ from KCs promotes functional activation and maturation of DCs.
Fig. 6. MAVS-mediated secretion of IFNβ from keratinocytes promotes activation and maturation of dendritic cells.

(A). IFNβ concentration in CM from control or LL37 and polyIC activated mouse KCs isolated from WT (Mavs+/+), HET (Mavs−/+) or KO (Mavs−/−) mouse skin (n=3). (B). FACS analyses of the geometric MFI of CD80 surface expression in mouse BMDC stimulated KC-CM (n=3). (C) RTqPCR analyses of Mx1 (myxovirus resistance 1), Irf7 and Cd83 mRNA expression in mouse BMDC treated with KC-CM as indicated (n=3). (D–E). RTqPCR analyses of MX1, IRF7 and CD83 mRNA expression (D) or FACS analyses of the geometric MFI of CD86 surface expression (E) in human monocyte-derived DCs stimulated with CM from hKC stimulated with LL37 and polyIC together with control IgG or neutralizing antibodies against for IFNα or IFNβ as indicated (n=3). (F) CFSE (carboxyfluorescein succinimidyl ester)-labeled CD3+ T cells were cultured alone or with control DCs or DCs treated with KCLP-CM with control IgG or IFNβ neutralizing antibody. Proliferation of T cells was analyzed 5 days later by FACS. Representative data from 3 independent experiments are shown. All error bars indicate mean ± s.e.m. * P<0.05, ** P<0.01, *** P<0.001 (one way Anova).
MAVS-IRF3-IFNβ signaling cascade is activated in the epidermis of wounds and psoriatic skin
Similar to the IFNβ expression pattern seen in Figure 1, MAVS was expressed predominantly in basal KCs and greatly increased at the wound edge or in psoriatic epidermis (Figures 7A and S7A–D). MAVS mRNA was also elevated in psoriasis lesional skin compared to non-lesional skin (Figure 7B). Furthermore, Mavs expression was elevated in the epidermis of a K5-IL-17C mice, in which IL17C is overexpressed in KCs and a mouse model of psoriasis (Figure 7C), (Johnston et al., 2013).
Fig. 7. Activation of MAVS-IRF3-IFNβ signaling in psoriatic and wounded skin epidermis.

(A) MAVS immunostaining of skin sections from normal control skin (NC), wounded (W), psoriasis non-lesional (PNL) or psoriasis lesional (PSO) skin as indicated. (B–C) Measurement of relative MAVS mRNA levels in (B) non-lesional (NL) and psoriasis lesional (PSO) skin (n=7~8/group), or (C) in non-lesional (NL) and lesional (L) of K5-IL-17C mouse skin compared with littermate control skin (n=4~5/group). (D–F) Human psoriatic or wounded skin sections were stained with RNA dye (green) and DAPI (D), or RNA, MAVS antibody and DAPI (E), or anti-MAVS and anti-pIRF3 antibodies (F), or ISG15 (G) as indicated. Zoom-in polyICtures highlighted in red box are shown on the right panel. White dashed line indicates the junction of the epidermis and dermis. (H) Representative image of IFNβ immunostaining in Mavs−/+ or Mavs−/− mouse skin injected intradermally with LL37. All scale bars = 100 μm.
Co-staining of skin with an RNA selective dye and DAPI (4′,6-diamidino-2-phenylindole) demonstrated that in normal control or in non-lesional skin the RNA signal predominantly co-localized in the nucleus with DAPI (Figures 7D–E). In contrast, in the suprabasal layers of psoriatic or wounded epidermis, a RNA signal was detected in the cytosol (Figures 7D–E) and colocalized in a perinuclear pattern with MAVS (Figure 3F) Perinuclear clustering of MAVS was also associated with strong phosphorylated IRF3 signal in the suprabasal layers of psoriatic lesional skin and in the wounded edge of normal skin (Figures 7G and S7E,F). A similar increase of phosphorylated IRF3 (pIRF3) and MAVS was also observed in the epithelium of ex vivo skin wounds (Figure S7G). These observations were consistent with data previously shown for cultured KCs in Figures 4–5 that demonstrated clustering of MAVS resulted in IRF3 phosphorylation and induction of IFNβ. Furthermore, the expression of ISG15, IRF7 and RIG1 were elevated in the epidermis of wounded and psoriatic skin (Figures 7H and S7H, I). All IFNβ-positive or LL37-positive cells also co-expressed MAVS (Figures S7J,K).
Mice have a cathelicidin gene (Camp) that is similar to the human CAMP gene encoding LL37, but the mature peptide encoded by exon 4 of the Camp gene is less than 50% identical to human LL37 and lacks the capacity to facilitate dsRNA recognition (Figure S3F). Therefore, further examine the role of MAVS in an animal model, we applied LL37 peptide to wild-type mice or Mavs−/+ mice. Administration of LL37 induced IFNβ expression in control Mavs−/+ mouse epidermis, but this was not seen in Mavs−/− skin (Figures 7I and S7L), suggesting that LL37 triggers MAVS activation by endogenous dsRNA and thus further demonstrating that IFNβ production by the epidermis is dependent on MAVS. Taken together, these results support the conclusion that LL37 enables IFNβ production through a MAVS-dsRNA pathway and that this response is likely to be biologically relevant.
DISCUSSION
In this report, we showed that IFNβ was produced by KCs that were stimulated by molecules present in damaged skin. This observation contrasts with the widely held assumption that type 1 IFNs are primarily produced by DCs. We further demonstrated that this response of KCs acts through the MAVS signaling pathway, a dsRNA recognition system not previously known to be relevant in psoriasis or wound repair. Based on these results and prior reports of activation of skin inflammation by dsRNA (Lai and Gallo, 2009), we propose the following model for IFNβ production in the skin following normal injury: (1) Upon acute injury dsRNA and other DAMPs are released from damaged tissue, (2) LL37 enables dsRNA recognition by MAVS, and this triggers downstream activation of TBK1 and AKT resulting in IRF3 activation and the subsequent production of IFNβ. (3). IFNβ secreted by activated KCs then further promotes activation and maturation of DCs and T cell stimulation. We therefore propose keratinocyte MAVS is a critical pathway for recognition of skin injury and the resulting production of IFNβ may represent a critical link between skin injury and the immune cascade that leads to inflammatory disorders such as psoriasis.
Psoriasis is a T-cell mediated inflammatory disease. However, recent studies using keratinocyte-specific transgenic mouse models have demonstrated that the inflammatory phenotype of psoriasis can be initiated by abnormal cytokine production by KCs in response to external stimuli. Examples of this include epidermal specific deletion of c-JUN (jun proto-oncogene) (Zenz et al., 2005) or IKK2 (inhibitor of nuclear factor kappa-B) (Pasparakis et al., 2002), and epidermal specific overexpression of Tie2 (endothelial-specific receptor tyrosine kinase) (Wolfram et al., 2009), IL17C (Johnston et al., 2013) or the active form of Stat3 (Ivashkiv and Donlin, 2014) leads to keratinocyte activation followed by induction of inflammatory cytokines and the subsequent development of psoriasis-like skin inflammation. These studies demonstrate that KCs can be a potent trigger to initiate the autoimmune cascade and drive psoriasis pathogenesis. However, direct evidence showing that KCs can activate immunocytes has been lacking. Using an in vitro approach, we demonstrated that IFNβ secreted from LL37 and dsRNA activated KCs directly promoted activation and maturation of cDCs, which in turn stimulated the proliferation of autologous naïve CD3 T cells (Figure 6). This directly demonstrates that IFNβ from activated KCs was sufficient to drive the DC-T cell response. In addition to cDCs, pDCs also expressed high level of IFNAR1 and can be activated by activated KCs (Figure 3A and S6G), suggesting that KC may also contribute to pDC activation in skin wounds or psoriasis. These observations support the conclusion that keratinocytes in the epidermis are an active source of IFNβ and can participate with DCs in shaping the immunological environment of the skin.
The endogenous ligands that trigger KC activation during psoriasis pathogenesis are not known. Our study shows that endogenous dsRNAs, such as U1 RNA, were potent activators of INFβ and may be one of the endogenous ligands that trigger this inflammatory response. An important role for dsRNA in influencing both psoriasis and wound repair is also supported by several other lines of evidence. Psoriasis is often triggered following viral infection and skin trauma, both sources of dsRNA release. Extracellular RNA complexes have been previously shown in psoriatic skin (Ganguly et al., 2009) and functional analysis of a psoriasis susceptibility genes have implicated innate immune responses to dsRNA as a pathogenic pathway of psoriasis (Bijlmakers et al., 2011). We have previously reported that dsRNA-mediated activation of TLR3 is required for normal inflammation after wounding (Lai et al., 2009), and in a setting of ultraviolet radiation injury (Bernard et al., 2012). Furthermore, a recent study found that dsRNA released by tissue damage drives skin regeneration (Nelson et al., 2015). In our experiment shown in Figure 2B, RNaseIII did not completely remove the IFN response to the extracts. This suggests that dsRNA in the extract was only partially digested by RNaseIII. Alternatively, the partial inhibition of activity could also be due to self-dsRNA present in the cytoplasm of healthy cells where it was protected from the extracellular RNaseIII but activated by LL37. Together, our study and these previous reports support the conclusion that endogenous dsRNA is an important physiological trigger of inflammation in both psoriasis and in normal wound repair.
MAVS mediates the activation of IRF3 and the induction of type1 IFNs in response to cytosolic dsRNA (Ivashkiv et al., 2014). However, a contribution of MAVS in skin immunity was largely unknown. We found that LL37 shifted dsRNA recognition from a solely TLR3-dependent mechanism to a pathway that was largely MAVS-dependent, allowing abundant expression of IFNβ by KCs. In pDCs, LL37 complexes with self-DNA to induce IFNα through TLR9 (Lande et al., 2007) and LL37 also interacts with extracellular self-RNA to activate pDC to produce IFNα through TLR7 and TLR8 (Ganguly et al., 2009). In contrast to IFNα, which is predominantly produced by dermal pDCs, we show here that IFNβ expression was highly abundant in KCs, a cell type that greatly outnumbers DCs in the skin and are poised directly at the interface with the external environment. KCs are likely to add to the total expression of type-1 interferons by expressing IFNβ through an alternative pathway. Increased expression and perinuclear clustering of MAVS in the suprabasal layers of wounded and psoriatic epidermis allows direct contact with dsRNA released from damaged cells and can promote inflammatory response necessary for an appropriate response to wounding.
In general, type1 IFNs are transiently expressed and are therefore difficult to detect by methods such as transcriptome analysis, whereas the expression of type1 IFN-inducible genes are relative stable. In line with this, we found that KCs exposed to LL37 and dsRNA had the capacity to transiently produce high levels of IFNβ followed by stable induction of type1 IFN-inducible genes including ISG15 and IRF7. This finding is consistent with previous protein and transcriptome analyses of human psoriasis samples (Schmid et al., 1994; van der Fits et al., 2004; Yao et al., 2008).
We have demonstrated that LL37 enabled activation of TBK1-AKT-IRF3 signal events downstream of MAVS in response to dsRNA in keratinocytes, and that LL37 modulated MAVS signaling through multiple mechanisms. First, LL37 triggered AKT phosphorylation through a MAVS-dependent pathway. Second, LL37 and dsRNA induced robust phosphorylation of TBK1, which was partially dependent on both TLR3 and MAVS, suggesting that there is a cross-talk between TLR3 and MAVS. Third, LL37 dephosphorylated the small spliced variant of TBK1 (TBK1s), which was heavily phosphorylated in basal KCs and a known negative regulator of RIG1 and MAVS function (Deng et al., 2008; Koop et al., 2011). This suggests that LL37 promotes the MAVS signaling pathway by removing TBK1s as a negative regulator of this pathway. Further studies will be required to identify the phosphatase(s) activated by LL37 leading to dephosphorylated TBKs.
In summary, our findings provide new insight into how skin injury activates the type1 IFN pathway and illustrates how epidermal keratinocytes participate in this process. We have shown that the production of IFNβ in the skin involves LL37-mediated activation of MAVS by dsRNA. These results demonstrate how injury can trigger innate and adaptive immune responses and how the epidermis may contribute to unwanted inflammation under disease conditions.
EXPERIMENTAL PROCEDURES
Human skin sample collection
All sample acquisition, including skin biopsies of wounded skin, was approved and regulated by the University of California San Diego Institutional Review Board (reference number 140205). The informed consent was obtained from all subjects prior to skin wounding and biopsies. Non-wounded human skin was obtained by taking 3mm punch biopsies from healthy donors (NC) or the lesional skin (PSO) and the control non-lesional (PNL) skin from psoriasis patients. At day1 or day3 after wounding, skin wound samples were retrieved by making new punch biopsies from the edges of the initial biopsies. These samples were freshly embedded in OCT for immunohistochemistry.
shRNA-mediated silencing of CAMP expressionin 3D skin constructs
CAMP expression was reduced using the GIPZ lentiviral system. CAMP shRNAs (GIPZ) were purchased from GE Dharmacon (RHS4531-EG820). Control and CAMP GIPZ lentiviral vectors along with viral packaging vectors were transfected into 293T cells. Viral supernatants were collected 48 hours after transfection and used to infect primary human keratinocytes as previously described (Mistry et al., 2012). Cells were incubated in the viral supernatants, polybrene added (5ug/ml), and centrifuged for 1 hour at 1000rpm. Cells were selected using puromycin (1ug/ml) 24 hours post-transduction for cells stably expressing the lentiviral construct. 3D skin constructs were generated by placing 1 million control or CAMP silenced cells on devitalized human dermis as previously described (Mistry et al., 2014). 3D skin epidermis was wounded by 3mm punch biopsy and skin was collected at day3 for analysis.
Primary human and mouse keratinocytes culture
Primary neonatal human epidermal keratinocytes (NHEKs) was purchased from Invitrogen. NHEKs were grown in serum free EpiLife medium supplemented with 0.06mM CaCl2, EpiLife Defined growth supplements (EDGS) (Invitrogen) and antibiotics, and passage 4~7 cells were used for experiment. Cells at 60~80% confluence were starved overnight without EDGS prior to treatment. Primary neonatal mouse epidermal keratinocytes (mKCs) were isolated from newborn Mavs+/+, Mavs−/+ or Mavs−/− littermate pups as described previously (Zhang et al., 2012) and mKCs were cultured in EpiLife supplemented with 0.04mM CaCl2 and EDGS until cells reached 80% confluency before starvation overnight followed by indicated treatments. Control siRNA, TLR3 siRNA and MAVS siRNA were purchased from Dharmacon (Chicago, IL). 5 nM of each siRNA was electroporated into NHEKs as described previously (Borkowski et al., 2013). To prepare cell extract from damaged cells, post-confluent NHEKs were harvested and briefly sonicated in PBS to release cellular content. The resulting cell extract (40 μg total protein) was treated with or without RNaseIII (10 Unit/ml) according to manufacturer’s manual before adding to 2 × 105 subconfluent NHEKs in culture dishes. To generate KC-CM, NHEKs were treated with LL37 then pIC for 2 hrs then cells were switched to culture medium without LL37 then pIC for additional 18 hrs. Cleared culture supernatant was then collected and supplemented with 10% FBS and 0.42 μM CaCl2 before adding it to DC culture.
Dendritic cell culture
Human pDCs were purified from fresh PBMCs (purchased from ixcells Biotechnology, San Diego, CA) using pDC MACS isolation kit from Miltenyl Biotec (San Diego, CA). Human cDCs were differentiated from PBMC by human GM-CSF and IL4, and mouse cDCs were differentiated from mouse BM cells using mouse GM-CSF. Purity of each cell type is greater than 80% as confirmed by FACS analyses. To stimulate DCs, LPS (0.5 μg/ml) or CpGA (1 μM), or LL37 (2μM), poly IC (0.4 μg/ml), or 1:1 v/v of KC-CM (supplemented with 10% FBS and 0.42 μM CaCl2) was added to DCs. Control IgG or IFNα or IFNβ neutralizing antibodies were added to KC-CM at 10 μg/ml to neutralize IFNα or. RNA was collected at 6 hrs after stimulation and FACS analyses for surface expression of CD11c, CD80, CD86 and CD83 were performed by flow cytometry 24~48 hrs after stimulation and analyzed by FlowJo V10 software.
Statistics
Experiments were repeated at least 3 times with similar results. Statistical significance was determined using Student’s unpaired two-tailed t-test, or one way ANOVA multiple comparison test as indicated in the legend (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001).
Supplementary Material
Highlights.
Keratinocytes co-express IFNβ, LL37 and MAVS in skin wounds and in psoriasis
LL37 enables keratinocytes to produce IFNβ in response to dsRNA from dying cells
LL37 acts through MAVS-dependent activation of TBK1-AKT-IRF3 signaling pathway
IFNβ secreted by activated keratinocytes promotes maturation of dendritic cells
Acknowledgments
This work was supported by NIH grants RO1 AI052453, AR064781, AI116576 and T32 AR062496 to RLG. NLW is supported by P30 AR39750, RO1 AR063437, RO1 AR062546; R21 AR063852 and the National Psoriasis Foundation, NLW. AJ is supported by NIH grant K01 AR064765.
Footnotes
AUTHOR CONTRIBUTIONS
L-J.Z. performed experiments, interpreted data and wrote the manuscript. G.S and Y.C generated CAMP-silenced 3D skin constructs. N.L.W. and Y.F. performed the qPCR analysis using K5;IL17C skin cDNA samples. A.J. and J.B. performed the qPCR analysis using psoriasis skin cDNA samples. T.H. and K.C. wrote the human IRB protocol and provided human tissues. C.A. performed RNA staining. J.A.S. performed IFNα and IFNβ ELISA. M.C. assisted with RTqPCRs. N.G. and E.S. were involved in DC culture. M.W. synthesized various snRNAs. R.L.G. directed the studies and wrote the paper with L-J.Z.
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med. 2007;13:543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- Baldwin HM, Pallas K, King V, Jamieson T, McKimmie CS, Nibbs RJ, Carballido JM, Jaritz M, Rot A, Graham GJ. Microarray analyses demonstrate the involvement of type I interferons in psoriasiform pathology development in D6-deficient mice. J Biol Chem. 2013;288:36473–36483. doi: 10.1074/jbc.M113.491563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard JJ, Cowing-Zitron C, Nakatsuji T, Muehleisen B, Muto J, Borkowski AW, Martinez L, Greidinger EL, Yu BD, Gallo RL. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat Med. 2012;18:1286. doi: 10.1038/nm.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijlmakers MJ, Kanneganti SK, Barker JN, Trembath RC, Capon F. Functional analysis of the RNF114 psoriasis susceptibility gene implicates innate immune responses to double-stranded RNA in disease pathogenesis. Human molecular genetics. 2011;20:3129–3137. doi: 10.1093/hmg/ddr215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkowski AW, Kuo IH, Bernard JJ, Yoshida T, Williams MR, Hung NJ, Yu BD, Beck LA, Gallo RL. Toll-Like Receptor 3 Activation Is Required for Normal Skin Barrier Repair Following UV Damage. J Invest Dermatol. 2015;135:569–578. doi: 10.1038/jid.2014.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkowski AW, Park K, Uchida Y, Gallo RL. Activation of TLR3 in keratinocytes increases expression of genes involved in formation of the epidermis, lipid accumulation, and epidermal organelles. J Invest Dermatol. 2013;133:2031–2040. doi: 10.1038/jid.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZH, Benureau Y, Rijnbrand R, Yi JZ, Wang T, Warter L, Lanford RE, Weinman SA, Lemon SM, Martin A, Li K. GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J Virol. 2007;81:964–976. doi: 10.1128/JVI.02076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng WW, Shi MD, Han MF, Zhong J, Li ZH, Li WN, Hu Y, Yan LC, Wang J, He Y, et al. Negative Regulation of Virus-triggered IFN-beta Signaling Pathway by Alternative Splicing of TBK1. J Biol Chem. 2008;283:35590–35597. doi: 10.1074/jbc.M805775200. [DOI] [PubMed] [Google Scholar]
- Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, Agerberth B, Gudmundsson GH, Gallo RL. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol. 2001;117:91–97. doi: 10.1046/j.1523-1747.2001.01340.x. [DOI] [PubMed] [Google Scholar]
- Erkek E, Karaduman A, Akcan Y, Sokmensuer C, Bukulmez G. Psoriasis associated with HCV and exacerbated by interferon alpha: complete clearance with acitretin during interferon alpha treatment for chronic active hepatitis. Dermatology. 2000;201:179–181. doi: 10.1159/000018447. [DOI] [PubMed] [Google Scholar]
- Funk J, Langeland T, Schrumpf E, Hanssen LE. Psoriasis induced by interferon-alpha. The British journal of dermatology. 1991;125:463–465. doi: 10.1111/j.1365-2133.1991.tb14774.x. [DOI] [PubMed] [Google Scholar]
- Gallo RL, Ono M, Povsic T, Page C, Eriksson E, Klagsbrun M, Bernfield M. Syndecans, Cell-Surface Heparan-Sulfate Proteoglycans, Are Induced by a Proline-Rich Antimicrobial Peptide from Wounds. P Natl Acad Sci USA. 1994;91:11035–11039. doi: 10.1073/pnas.91.23.11035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, Homey B, Barrat FJ, Zal T, Gilliet M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. Journal of Experimental Medicine. 2009;206:1983–1994. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutagny N, Severa M, Fitzgerald KA. Pin-ning down immune responses to RNA viruses. Nat Immunol. 2006;7:555–557. doi: 10.1038/ni0606-555. [DOI] [PubMed] [Google Scholar]
- Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, Arai N, Gallo RL, DiGiovanni J, Gilliet M. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. Journal of Experimental Medicine. 2010;207:2921–2930. doi: 10.1084/jem.20101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilborn JD, Nilsson MF, Kratz G, Weber G, Sorensen O, Borregaard N, Stahle-Backdahl M. The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J Invest Dermatol. 2003;120:379–389. doi: 10.1046/j.1523-1747.2003.12069.x. [DOI] [PubMed] [Google Scholar]
- Hida S, Ogasawara K, Sato K, Abe M, Takayanagi H, Yokochi T, Sato T, Hirose S, Shirai T, Taki S, Taniguchi T. CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity. 2000;13:643–655. doi: 10.1016/s1074-7613(00)00064-9. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, Chen CS, Fu W, Gudjonsson JE, McCormick TS, Ward NL. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol. 2013;190:2252–2262. doi: 10.4049/jimmunol.1201505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung SM, Park ZY, Rani S, Takeuchi O, Akira S, Lee JY. Akt Contributes to Activation of the TRIF-Dependent Signaling Pathways of TLRs by Interacting with TANK-Binding Kinase 1. J Immunol. 2011;186:499–507. doi: 10.4049/jimmunol.0903534. [DOI] [PubMed] [Google Scholar]
- Koop A, Lepenies I, Braum O, Davarnia P, Scherer G, Fickenscher H, Kabelitz D, Adam-Klages S. Novel splice variants of human IKK epsilon negatively regulate IKK epsilon-induced IRF3 and NF-kB activation. Eur J Immunol. 2011;41:224–234. doi: 10.1002/eji.201040814. [DOI] [PubMed] [Google Scholar]
- Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu ZR, Hooper LV, Schmidt RR, von Aulock S, et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med. 2009;15:1377–1382. doi: 10.1038/nm.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YP, Gallo RL. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009;30:131–141. doi: 10.1016/j.it.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Wang YH, Su B, Nestle FO, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–U566. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- Li AG, Wang D, Feng XH, Wang XJ. Latent TGF beta 1 overexpression in keratinocytes results in a severe psoriasis-like skin disorder. Embo J. 2004;23:1770–1781. doi: 10.1038/sj.emboj.7600183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–873. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- Mistry DS, Chen YF, Sen GL. Progenitor Function in Self-Renewing Human Epidermis Is Maintained by the Exosome. Cell stem cell. 2012;11:127–135. doi: 10.1016/j.stem.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry DS, Chen YF, Wang Y, Zhang K, Sen GL. SNAI2 Controls the Undifferentiated State of Human Epidermal Progenitor Cells. Stem Cells. 2014;32:3209–3218. doi: 10.1002/stem.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morhenn VB. Keratinocyte proliferation in wound healing and skin diseases. Immunology today. 1988;9:104–107. doi: 10.1016/0167-5699(88)91278-9. [DOI] [PubMed] [Google Scholar]
- Nelson AM, Reddy SK, Ratliff TS, Hossain MZ, Katseff AS, Zhu AS, Chang E, Resnik SR, Page C, Kim D, et al. dsRNA Released by Tissue Damage Activates TLR3 to Drive Skin Regeneration. Cell stem cell. 2015;17:139–151. doi: 10.1016/j.stem.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, Burg G, Liu YJ, Gilliet M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. Journal of Experimental Medicine. 2005;202:135–143. doi: 10.1084/jem.20050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickoloff BJ, Bonish BK, Marble DJ, Schriedel KA, DiPietro LA, Gordon KB, Lingen MW. Lessons learned from psoriatic plaques concerning mechanisms of tissue repair, remodeling, and inflammation. The journal of investigative dermatology. Symposium proceedings/the Society for Investigative Dermatology, Inc. [and] European Society for Dermatological Research. 2006;11:16–29. doi: 10.1038/sj.jidsymp.5650010. [DOI] [PubMed] [Google Scholar]
- Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, Krampert M, Goebeler M, Gillitzer R, Israel A, et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417:861–866. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, Rubin JR, Mowers J, White NM, Hochberg I, et al. An inhibitor of the protein kinases TBK1 and IKK-epsilon improves obesity-related metabolic dysfunctions in mice. Nat Med. 2013;19:313–321. doi: 10.1038/nm.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roupe KM, Nybo M, Sjobring U, Alberius P, Schmidtchen A, Sorensen OE. Injury is a major inducer of epidermal innate immune responses during wound healing. J Invest Dermatol. 2010;130:1167–1177. doi: 10.1038/jid.2009.284. [DOI] [PubMed] [Google Scholar]
- Ruano J, Suárez-Fariñas M, Shemer A, Oliva M, Guttman-Yassky E, Krueger JG. Molecular and Cellular Profiling of Scalp Psoriasis Reveals Differences and Similarities Compared to Skin Psoriasis. PLoS One. 2016;11:e0148450. doi: 10.1371/journal.pone.0148450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SN, Peters KL, Elco CP, Sakamoto S, Pal S, Sen GC. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat Struct Mol Biol. 2004;11:1060–1067. doi: 10.1038/nsmb847. [DOI] [PubMed] [Google Scholar]
- Schmid P, Itin P, Cox D, Mcmaster GK, Horisberger MA. The Type-I Interferon System Is Locally Activated in Psoriatic Lesions. J Interferon Res. 1994;14:229–234. doi: 10.1089/jir.1994.14.229. [DOI] [PubMed] [Google Scholar]
- Sweeney CM, Tobin AM, Kirby B. Innate immunity in the pathogenesis of psoriasis. Archives of dermatological research. 2011;303:691–705. doi: 10.1007/s00403-011-1169-1. [DOI] [PubMed] [Google Scholar]
- Toussirot E, Bereau M, Bossert M, Malkoun I, Lohse A. Occurrence of Psoriatic Arthritis during Interferon Beta 1a Treatment for Multiple Sclerosis. Case reports in rheumatology. 2014;2014:949317. doi: 10.1155/2014/949317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Fits L, van der Wel LI, Laman JD, Prens EP, Verschuren MCM. In psoriasis lesional skin the type I interferon signaling pathway is activated, whereas interferon-alpha sensitivity is unaltered (vol 122, pg 51, 2004) J Invest Dermatol. 2004;123:415–415. doi: 10.1046/j.0022-202X.2003.22113.x. [DOI] [PubMed] [Google Scholar]
- Weiss G, Shemer A, Trau H. The Koebner phenomenon: review of the literature. Journal of the European Academy of Dermatology and Venereology: JEADV. 2002;16:241–248. doi: 10.1046/j.1473-2165.2002.00406.x. [DOI] [PubMed] [Google Scholar]
- Wen CY, Yan ZF, Yang XL, Guan K, Xu CZ, Song T, Zheng ZR, Wang WJ, Wang Y, Zhao M, et al. Identification of Tyrosine-9 of MAVS as Critical Target for Inducible Phosphorylation That Determines Activation. PloS one. 2012;7 doi: 10.1371/journal.pone.0041687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfram J, Diaconu D, Hatala D, Rastegar J, Knutsen D, Lowther A, Askew D, Gilliam A, McCormick T, Ward N. Keratinocyte but not endothelial cell-specific overexpression of Tie2 leads to the development of psoriasis. 2009:1443–1458. doi: 10.2353/ajpath.2009.080858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P, Coulombe PA. Loss of keratin 6 (K6) proteins reveals a function for intermediate filaments during wound repair. The Journal of cell biology. 2003;163:327–337. doi: 10.1083/jcb.200305032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Katayama I, Nishioka K. Psoriasis and hepatitis C virus. Acta dermato-venereologica. 1995;75:482–483. doi: 10.2340/0001555575482483. [DOI] [PubMed] [Google Scholar]
- Yamasaki K, Di Nardo A, Bardan A, Murakami M, Ohtake T, Coda A, Dorschner RA, Bonnart C, Descargues P, Hovnanian A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975–980. doi: 10.1038/nm1616. [DOI] [PubMed] [Google Scholar]
- Yao Y, Richman L, Morehouse C, de los Reyes M, Higgs BW, Boutrin A, White B, Coyle A, Krueger J, Kiener PA, Jallal B. Type I interferon: potential therapeutic target for psoriasis? PloS one. 2008;3:e2737. doi: 10.1371/journal.pone.0002737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Low HQ, Wang L, Li Y, Ellinghaus E, Han J, Estivill X, Sun L, Zuo X, Shen C, et al. Genome-wide meta-analysis identifies multiple novel associations and ethnic heterogeneity of psoriasis susceptibility. Nature communications. 2015;6:6916. doi: 10.1038/ncomms7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenz R, Eferl R, Kenner L, Florin L, Hummerich L, Mehic D, Scheuch H, Angel P, Tschachler E, Wagner EF. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–375. doi: 10.1038/nature03963. [DOI] [PubMed] [Google Scholar]
- Zhang LJ, Bhattacharya S, Leid M, Ganguli-Indra G, Indra AK. Ctip2 is a dynamic regulator of epidermal proliferation and differentiation by integrating EGFR and Notch signaling. Journal of cell science. 2012;125:5733–5744. doi: 10.1242/jcs.108969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LJ, Gallo RL. Antimicrobial peptides. Current biology: CB. 2016;26:R14–19. doi: 10.1016/j.cub.2015.11.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.