

Abstract
While HIV‐1 continues to spread, the use of antivirals in preexposure prophylaxis (PrEP) has recently been suggested. Here we present a modular systems pharmacology modeling pipeline, predicting PrEP efficacy of nucleotide reverse transcriptase inhibitors (NRTIs) at the scale of reverse transcription, target‐cell, and systemic infection and after repeated viral exposures, akin to clinical trials. We use this pipeline to benchmark the prophylactic efficacy of all currently approved NRTIs in wildtype and mutant viruses. By integrating pharmacokinetic models, we find that intracellular tenofovir‐diphosphate builds up too slowly to halt infection when taken “on demand” and that lamivudine may substitute emtricitabine in PrEP combinations. Lastly, we delineate factors confounding clinical PrEP efficacy estimates and provide a method to overcome these. The presented framework is useful to screen and optimize PrEP candidates and strategies and to understand their clinical efficacy by integrating the diverse scales which determine PrEP efficacy.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Preexposure prophylaxis using tenofovir, with or without emtricitabine, may reduce HIV infection.
• WHAT QUESTION DID THIS STUDY ADDRESS?
☑ How do molecular parameters, pharmacokinetics, virus dynamics, mode of transmission, transmitter virus loads, and risk behavior influence PrEP‐efficacy endpoints against wildtype and resistant viruses? Are other NRTIs suitable?
• WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ We present a modular systems pharmacology modeling pipeline for NRTIs, predicting their effect at the scale of reverse transcription ε, target‐cell infection η, and PrEP efficacy after a single ψ and repeated viral exposure ωT. Novel aspects include the mechanistic multiscale integration of these efficacy endpoints, novel infection, and exposure models (modules III–IV) and the ability to simulate clinical trials (module V).
• HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS
☑ PK‐PD studies for PrEP are unethical, leaving a knowledge gap when designing phase III studies. Our framework provides guidance by identifying pharmacological requirements for PrEP candidates and strategies and may help planning and evaluating clinical trials.
Despite intensive research, HIV cannot be cured to date,1 necessitating life‐long treatment of infected individuals to contain the virus and prevent immunodeficiency. At the same time the HIV epidemic continues to spread, with ∼2.1 million new infections in 2014.2 While an effective vaccine remains to be developed3 a current way forward lies in the repurposing of existing antiviral drugs to prevent transmission, or to develop novel compounds for that purpose.4 Two strategies have been proposed in this context:
Treatment‐as‐prevention (TasP) involves therapy initiation shortly after infection.5 As a consequence, the treated individuals' virus load decreases, which also decreases the contagiousness originating from this individual.6, 7 A recent study,8 however, indicates that onwards transmission may occur very soon after infection, when individuals are unaware of their serologic status and consequently have not yet initiated TasP, which potentially limits its epidemiologic impact.
Preexposure prophylaxis (PrEP) involves antiviral drug administration to uninfected individuals at risk of acquiring HIV infection.9 Early studies have investigated chronic administration of the nucleoside reverse transcriptase inhibitors (NRTI) tenofovir disoproxil fumarate (TDF) alone or in combination with emtricitabine (FTC), with variable outcomes.10 Recent studies also investigated PrEP “on demand,” i.e., PrEP administered shortly before or around viral exposure.23
The goal of this work is to develop an integrated mechanistic modeling pipeline to determine PrEP efficacy of NRTIs, integrating pharmacokinetics (PK) and pharmacodynamics (PD), as well as parameters related to the mode and timing of viral challenge. The pipeline has a building block structure and different parts can be used to assess the PrEP efficacy of other drug classes as well.
NRTIs are administered as prodrugs, which are taken up by target cells and successively phosphorylated by cellular kinases. Their tri‐phosphorylated moieties compete with endogenous nucleotides for incorporation into nascent proviral DNA during reverse transcription,12 effectively halting the process and thus preventing target cell infection. The uptake and intracellular activation of these compounds causes an asynchrony between plasma prodrug concentrations and the concentrations of the active (triphosphorylated) moiety at the target‐site, so that prodrug plasma pharmacokinetics poorly predicts their efficacy.13 Moreover, due to the competitive mode of inhibition, NRTI efficacy can be target‐cell‐dependent.14, 15 While only some of these issues are addressed by most modeling efforts,11, 16 we have recently developed and validated a molecular mechanism of action (MMOA) model17 for this inhibitor class, allowing to determine the compounds' effect on reverse transcription ε and target cell infection η. Moreover, we developed pharmacokinetic models linking prodrug administration with effect‐site concentrations for the NRTIs TDF,18 FTC and 3TC.17 In this work, we link the MMOA model with pharmacokinetic models, which allows exploring the impact of pharmacokinetic attributes, as well as pharmacodynamic parameters, including drug resistance, on drug efficacy. We will then take this approach one step further, by extending the framework to assess the inhibitors' potential for repurposing as PrEP compounds, estimating the compounds' effect on preventing systemic infection ψ after a single exposure with n viruses. The latter allows assessing different PrEP schemes (e.g., chronic administration vs. “on demand”). In a last step, to assess the epidemiologic impact of these compounds, we derive a statistical model linking transmitter virology with virus exposure in the individual at risk for different modes of transmission. We then estimate the long‐term efficacy of PrEP ωT after repeated viral challenges, akin to a clinical study. The final framework is readily integrable into epidemiologic models aiming to assess PrEP or TasP or both. All intermediate steps of this pipeline have been validated with available data.
METHODS
Pharmacokinetics
We will use previously developed models for TDF, FTC, and 3TC, which link oral prodrug application with intracellular tri‐phosphate pharmacokinetics.17, 18 In brief, the plasma pharmacokinetics of their dominant circulating forms (tenofovir (TFV), FTC, and 3TC) are best described by a two‐compartment model with first‐order absorption. Intracellular uptake and phosphorylation was described by Michaelis‐Menten‐type saturable kinetics and elimination was modeled by first‐order kinetics. Details and parameterizations can be found in Supplementary Note 1.
We chose to predict average patients' pharmacokinetic profiles, but extensions to virtual patient populations from Pop‐PK models are straightforward. For the modeled NRTI combinations, we assume no pharmacokinetic interaction at the level of intracellularly active NRTI‐triphosphates (NRTI‐TP), but extensions are possible.19
Molecular mechanism of action
We will utilize a previously developed15 and validated17 MMOA model for NRTIs, which explicitly considers reverse transcriptase (RT)‐mediated polymerization of nascent viral DNA. NRTI‐TPs interfere with polymerization by competing with endogenous nucleotides for incorporation into viral DNA. For as long as they are integrated in the primer, they halt the RT process, which allows the cell to eliminate crucial viral components intracellularly, reducing the virus' chance to infect the cell by integrating its proviral DNA. The MMOA model takes in vitro measurable microkinetic parameters as input (binding affinity, maximum catalytic rate, excision efficacy) and computes the inhibition of reverse transcription ε. This measure is subsequently converted into inhibition of target‐cell infection η following a challenge by a single virus, with corresponding . The MMOA model, including its parametrization is exemplified in Supplementary Note 2.
For NRTI combinations, we assume that the presence of one NRTIs does not affect the microkinetic parameters of the respective other NRTI. The MMOA model readily allows assessing combinatorial effects and this is outlined in Supplementary Note 2.
Probability of infection after challenge with n viruses
After virus exposure during, e.g., intercourse, viruses need to overcome several physiological barriers to reach a target‐cell environment. Assuming n viruses reach an immediate target‐cell environment, the probability of infection is given by:
| (1) |
where and are the probabilities of establishing infection if 1 or virus(es) reach a target‐cell environment, respectively. Thus, 1) the number of viruses reaching a target‐cell environment n (next section) and 2) the infection probability given a single virus (this section) need to be appropriately modeled.
Typically, HIV produces ≈1,000 daughter viruses for each virus completing its replication cycle, making its subsequent extinction unlikely. Consequently, for all cases considered here the probability of the virus completing its first replication cycle provides a good approximation for the probability of establishing infection (see Discussion for limitation). To compute the infection probability, we used two different mathematical approaches, based on the chemical master equation (CME), and a branching process, which delivers an analytical solution of the CME for .
The CME can be directly derived from an established viral dynamics model25 and is detailed in Supplementary Note 3. The probability of target‐cell infection in the presence of NRTIs η is an integral part of this CME, providing a link to the MMOA model.
We used the CME, whenever the effect of NRTIs change on the time‐scale of interest, i.e., to simulate the effect of NRTIs shortly after initiation of prophylaxis (“PrEP on demand”). When the concentrations of NRTI‐TPs are almost constant over time (e.g., “chronic administration”) the branching process is sufficient.
PrEP efficacy
The efficacy of PrEP , defined as the reduction of infection per challenge with viruses (e.g., after coitus with an infected individual) is then readily computed by:
| (2) |
Here, and denote the infection probabilities after exposure to viruses when a PrEP strategy S was applied vs. PrEP was not applied . The PrEP efficacy per typical exposure ψ is then defined by:
| (3) |
which is , where denote the infection probabilities for a typical exposure during coitus. In the equation above, is the conditional probability that viruses reach a target‐site compartment after exposure (e.g., coitus) among all cases where there was an actual exposure that could have led to infection ( viruses reach a replication‐relevant compartment). The exposure probabilities are detailed next and in Supplementary Note 4.
Viral exposure module
The infection probability after coitus is strongly correlated with the donor viral load.6 This correlation is likely attributed to an increased number of transmitted viruses in high viral load donors. While there is strong evidence that only very few founder viruses establish infection,21 the distribution of the number of transmitted viruses and its dependence on the donor viral load is unclear. Here, we propose a model to bridge the donor viral load with the distribution of transmitted viruses in the recipient.
We assume that the number of viruses transmitted and reaching a target‐cell environment n is a binomially distributed random variable, parameterized by the donor viral load. The probability of transmitting exactly n viruses to the recipient when the viral load in the donor is k is then given by:
| (4) |
where m is an exponent of the viral load is the next integer function, and is the success probability. The parametrization and model derivations are outlined in Supplementary Note 4. From here, , can be computed (it is needed in Eq. (3)), i.e., , as shown in Figure 2.
Figure 2.
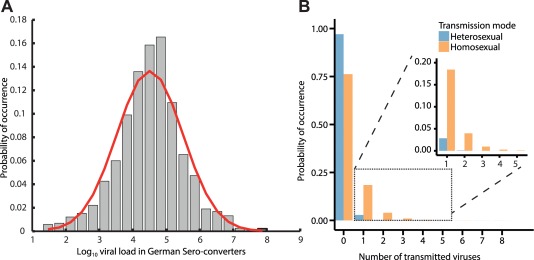
Virus exposure model (module IV). a: Virus load distribution ( scale) in a representative transmitter population (German Sero‐converter study8, 27). b: Estimated distribution of virus exposure in a target cell environment n following unprotected hetero‐ and homosexual intercourse (blue and orange bars) with an infected individual. Inset: Probability that virus enters a replication‐relevant compartment. Derivations are provided in Supplementary Note 4.
Efficacy against repeated viral challenges
Up to now, we assessed PrEP efficacy per exposure ψ. However, clinical trials report the ratio of incidence rates in the treated and placebo arms as a measure of efficacy5, 22, 23, 24 (see also Supplementary Note 5 for derivations). The latter may be a consequence of an individual being repeatedly exposed, and subject to, e.g., risk behavior and trial follow‐up duration making this estimate poorly comparable between trials. The relation between average PrEP efficacy (per challenge) ψ and clinical trial efficacy ωT is given by:
| (5) |
where and denote the number of unprotected sex acts with an infected individual in the PrEP arm S and the placebo arm per person per month, respectively, and T denotes the trial duration in months. denotes the probability (frequency) of infection in the placebo arm per challenge and ωT denotes the estimated PrEP efficacy from the incidence rates in a clinical trial of duration T.
Software
We used MatLab R2015a (MathWorks, Natick, MA; v. 8.5, including optimization and bioinformatics toolbox) and R (v. 3.1.2, Vienna, Austria) for modeling and simulation. Sample codes are provided as Online Supplementary Materials.
RESULTS
Modular modeling framework
We have constructed a modular pipeline to assess the efficacy of different NRTIs in prophylactic regimen. The pipeline (Figure 1) consists of five modules that can be combined, depending on the scientific question. Pharmacokinetic models (module I) for the NRTIs TDF, FTC, and 3TC, linking oral drug administration with the pharmacokinetics of the active intracellular moiety (TFV‐DP, FTC‐TP, and 3TC‐TP), have been developed previously,17, 18 allowing to assess different dosing schedules, adherence, etc. The intracellular concentrations can be coupled to an MMOA model15 (module II), which enables quantifying the effect of NRTIs, alone and in combination, on target cell infection η, for wildtype and mutant viruses, after exposure to a single virion as exemplified in Supplementary Note 2. A previously developed viral replication model25 (module III), that has also been shown to be predictive in17, 18, 26 can then be used to predict the infection probability, given an exposure to n viruses during, e.g., coitus with an infected person. The latter can be used to assess PrEP efficacy ψ per coitus (see Eq. (3)). Module IV simulates viral exposure, depending on the transmitter viral load. The derivation of module IV and its parametrization are elaborated in Supplementary Note 4 and the next section. Module V assesses the long‐term efficacy of PrEP. That is, after repeated viral challenges, akin to a clinical trial.
Figure 1.
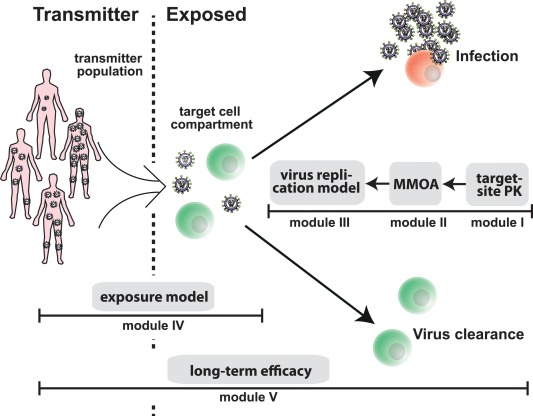
Modular modeling framework. The virus replication model (module III) can be used to compute the probability of infection of an exposed person after viral challenge, given a particular drug inhibition (input from module II) and viral exposure (input from module IV). Model details are elaborated in Supplementary Note 3. Module IV represents a statistical model of the relation between the viral load in a transmitter, the mode of transmission (e.g., homosexual contact) and the number of viruses entering a target cell compartment in the exposed person. It is derived in Supplementary Note 4, where the parametrization is also given. The mechanisms of action model (MMOA) provides the link between intracellular NRTI‐TP concentrations, target process inhibition ε (reverse transcriptase‐mediated polymerization), and inhibition of target cell infection η. It can be used to quantify effects of all currently approved NRTIs and NRTI combinations, including inhibition of mutant viruses; see Supplementary Note 2 for details and model parameters. Pharmacokinetic models (module I), which establish the link between prodrug administration and intracellularly active NRTI‐TPs have been developed for TDF, FTC, and 3TC and allow to evaluate different PrEP strategies (e.g., dosing regimen), related to these compounds (summarized in Supplementary Note 1). Finally, module V can be used to assess the efficacy of PrEP strategies in preventing infection after multiple viral challenges ωT, akin to clinical trials (see Supplementary Note 5 for derivations).
Mode of transmission and viral exposure
We analyzed virus load data from the German Sero‐converter cohort, which is a nationwide, multicenter, open, prospective long‐term observational cohort initiated in 1997.8, 27 We restricted the analysis to untreated individuals with a known seroconversion‐date and risk group (N = 1,213). We found this data source particularly relevant, since viral load data from both early as well as chronic infection is included (median duration of infection: 18 weeks, IQR: 3–42 weeks), acknowledging that HIV‐1 onwards transmission may preferentially occur rather shortly after infection.8 The viral load in this cohort was log‐normal distributed, with mean and (see Figure 2 a), in agreement with other studies.28
Figure 2 b shows the probability distribution of viral exposure following hetero‐ and homosexual intercourse, which was computed by combining data depicted in Figure 2 a with Eq. (4). Note that in the majority of cases no virus enters a replication compartment and subsequently infection will not occur, whereas a few viruses (one to five) may be transmitted occasionally and may subsequently establish infection. This result is in line with Keele et al.,21 who report that only very few founder viruses establish infection. Overall, our results indicate that viral exposure is stronger during homosexual than during heterosexual intercourse (compare blue and orange bars in Figure 2 b).
Concentration vs. risk reduction in wildtype and mutant viruses
The MMOA model allows assessing the inhibition of target cell infection η by different NRTIs. We used this information as part of our modeling pipeline (see Methods and Supplementary Note 3) to assess the concentration–response (percentage of systemic infections prevented ψ, Eq. (3)) for zidovudine (AZT), TDF, 3TC, FTC, stavudine (D4T), and abacavir (ABC). The profiles are shown in Figure 3 a and allow for the first assessment of the suitability of these drugs for repurposing as PrEP compounds. For further assessment, toxicity needs to be included. Note that the solid lines and the background shading in Figure 3 a show the expected efficacy and interquartile range at clinically relevant concentrations (see Supplementary Note 3). We predict that AZT can prevent 14–53% of infections at clinically relevant concentrations, followed by TDF (24–89%), D4T (55–95%), ABC (73–84%), 3TC (64–96%), and FTC (92–99%), with corresponding values of 0.046, 0.1, 0.025, 0.15, 1.72, and 0.82 [μM] (NRTI‐TP concentrations with respect to preventing target‐cell infection following exposure with a single virion (η)). We further assessed the efficacy of TDF, FTC, and 3TC in preventing infection due to transmitted drug resistance.29 Resistance to FTC and 3TC is associated with the M184V mutation, whereas resistance to TDF is associated with the K65R mutation.30, 31 Note that inhibition of the mutant viruses can readily be assessed in the MMOA model (see Supplementary Note 2). Furthermore, the MMOA model allows to assess the fitness costs associated with these mutations. In line with ex vivo experiments,32, 33 both the M184V and K65R mutant conferred a fitness disadvantage predicted by the MMOA model. ( 63, 55 and 46% of the wildtype fitness for M184V, K65R, and the M184V/K65R double mutant). The predicted PrEP efficacy of TDF, FTC, and 3TC against wildtype (WT) and mutant viruses (M184V, K65R, and M184V/K65R) is shown in Figure 3 b–d. We assessed the percentage of infections prevented by prophylaxis after exposure to the mutant virus relative to the wildtype virus in the absence of drugs, i.e.:
| (6) |
where “ ” denotes the mutant virus (M184V, K65R or M184V/K65R) and “ ” denotes the wildtype virus. Thus, both the effect of the drugs, as well as inherent fitness costs, are simultaneously evaluated, allowing to assess whether PrEP fosters the transmission of resistant viruses (this is the case whenever mutant transmission is more effective; i.e., whenever the dashed line is below the solid line in Figure 3 b–d). The simulations show that the K65R mutation may decrease the PrEP efficacy of TDF, while the M184V‐containing virus is hyper‐susceptible to TDF. The M184V/K65R double mutant is almost as susceptible as the wildtype, but has a profound fitness deficit. In the case of FTC, both the M184V and K65R mutation, as well as the double mutant, diminish its PrEP efficacy from 92–99% (wildtype) to 72–92% (K65R) and 47–71% (M184V). In the case of 3TC, mutations K65R, M184V, and the double mutant gradually diminish its efficacy down to complete resistance (in case of the double mutant). At low drug concentrations, the fitness defect of the resistant viruses leads their reduced transmissibility (≈37–54% less likely to be transmitted than the wildtype in the absence of drugs).
Figure 3.
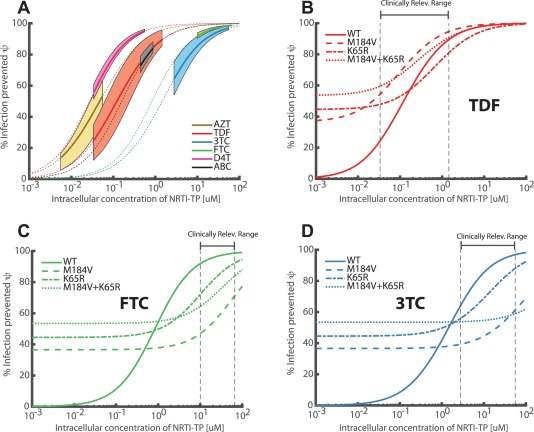
Target‐cell NRTI‐TP concentration vs. risk reduction in wildtype and mutant viruses (modules II‐IV) ψ. a: Mean efficacies (% infections prevented) following viral exposure during a single unprotected homosexual intercourse (Eq. 3) are illustrated by the dotted lines. Solid thick lines mark the risk reduction profile at clinically relevant ranges for the respective drugs (indicated ranges only provide a rough guidance as outlined in Supplementary Note 3). Shaded areas indicate the corresponding IQR of the efficacy estimate, taking variability in microscopic parameters (module II) and virus exposure (module IV, Figure 2b) into account. b–d: Mean efficacies ψ of TDF, FTC, and 3TC against the wildtype virus are highlighted by solid lines. Efficacies against mutant viruses combine both drug effects and inherent fitness defects of the mutants. The relative reduction in infection with the mutant virus in the presence of drug vs. the wildtype virus in the absence of drugs is evaluated (dashed line: M184V, dash‐dotted line: K65R, dotted line M184V/K65R double mutant), see section “Concentration vs. risk reduction in wildtype and mutant viruses” for details. Vertical black dashed lines indicate the clinically relevant drug concentrations range after chronic therapy.
Efficacy shortly after PrEP initiation
Next, we assessed the prophylactic efficacy of TDF, FTC and 3TC alone, or in combination, when initiated shortly before exposure (“on demand”), akin to the IPERGAY protocol.23 In this protocol, individuals initiate PrEP up to 24 hours before viral exposure with a double‐dose and then take two more pills on days 1 and 2. Evaluated pill sizes were 200 mg (FTC) or 300 mg (3TC or TDF). Based on previously developed PK models,17, 18 we simulated the (population‐average) plasma and intracellular pharmacokinetics for TFV, FTC, and 3TC, respectively TFV‐DP, FTC‐TP, and 3TC‐TP. As can be seen in Figure 4 a–c, intracellular concentrations (dashed lines) quickly increase to almost steady state levels for FTC‐TP and 3TC‐TP after ≈6–12 hours, but not for TFV‐DP, arguing that TFV‐DP may not reach protective levels when applied “on demand.” For an exposure occurring either 1, 3, 6, 12, 18, or 24 hours after PrEP initiation, Figure 4 d–f shows the prophylactic efficacy ψ of the different drugs used in isolation. All tested drugs are more efficiently preventing infection, if the viral challenge occurs late with respect to PrEP initiation. Emtricitabine seems to be most efficacious, preventing 73–90% of potential infections, followed by 3TC (55–71%). Tenofovir seems to poorly prevent infection when taken “on demand,” only preventing 15–40% of potential infections after virus exposure. The latter corroborates the hypothesis that protective TFV‐DP levels may build up too slowly in the intracellular compartment to provide sufficient protection.34 The efficacy of the combination 3TC+TDF was 59–77%, whereas the efficacy of the combination FTC+TDF mirrored the efficacy profile of FTC alone (74–92%, see Figure 4 g). The observed clinical trial efficacy estimate for FTC+TDF in IPERGAY was 86%.23 Technical details of the drug combination model are elaborated in Supplementary Note 2.
Figure 4.
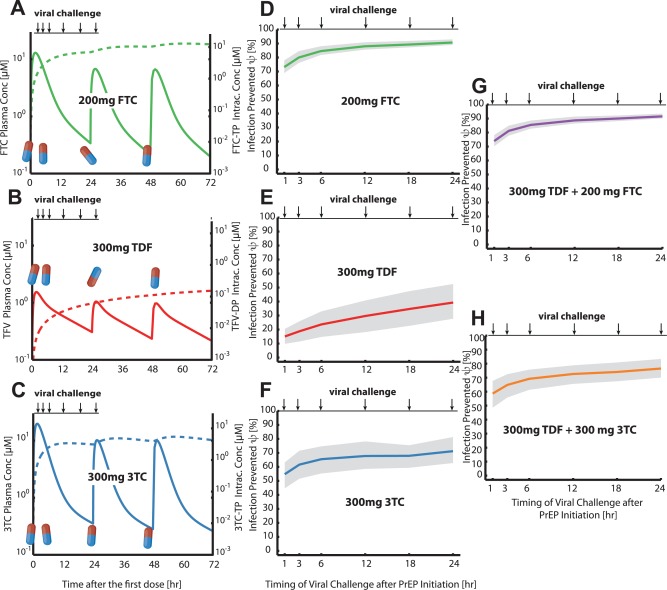
Efficacy ψ of PrEP “on demand” against infection following unprotected homosexual intercourse within 24hours after PrEP initiation (modules I‐IV). a–c: Pharmacokinetic profiles during PrEP “on demand” for the circulating NRTI prodrug (solid lines) and the intracellular, active NRTI‐TP moiety (dashed lines). FTC oral dose was 400 mg at 0 hours, followed by 200 mg at 24 and 48 hours (a), while TDF or 3TC dosage was 600 mg at 0 hours, followed by 300 mg at 24 and 48 hours, respectively. d–e: Infections averted for PrEP “on demand” when viral challenge occurred either 1, 3, 6, 12, 18, or 24 hours after PrEP initiation with either FTC (d), TDF (e), or 3TC (f). Solid lines indicate the mean % infections averted (see Eq. 3), while shaded areas indicate interquartile ranges of this estimate, taking variability in microscopic parameters (module II) and virus exposure during homosexual intercourse (module IV, Figure 2b) into account. g,h: Infections averted for combinations of TDF+FTC (g) and TDF+3TC (h), taken “on demand” (double doses at day 0, followed by single doses at days 1, 2). Combination predictions assumed that no significant pharmacokinetic interactions occur, pharmacodynamic interactions were modeled as outlined in Supplementary Note 2.
Efficacy after PrEP discontinuation
The prophylactic efficacy of TDF, FTC, and 3TC, alone or in combination, during chronic PrEP and after its discontinuation is assessed in Figure 5 a–e, based on (population‐average) pharmacokinetics after oral administration of 200 mg FTC or 300 mg TDF or 300 mg 3TC daily, or combinations thereof. Daily administration of FTC, TDF, and 3TC for 30 days prior to viral exposure lead to a prophylactic efficacy ψ of ≈95, 74, and 75%, respectively. After discontinuation, FTC, TDF, and 3TC remain effective for about 7, 10, and 2 days, respectively, with the PrEP efficacy of 3TC declining most rapidly. The combination FTC+TDF and 3TC+TDF prevent ≈96% and 87% of infections, respectively, after 30 days of daily administration. Corresponding observed clinical trial estimates for high‐level adherence FTC+TDF PrEP are 35 and .24 We predict that both combinations remain effective for about 10 days after discontinuation. Figure 5 f,g shows the efficacy of the combination, with the efficacy of the single drugs superimposed (note that the combinatorial effects are not independent, Supplementary Note 2). The graphic indicates that tenofovir preserves the prophylactic efficacy after discontinuation of the combination and thus makes the regimen robust to poor adherence.
Figure 5.
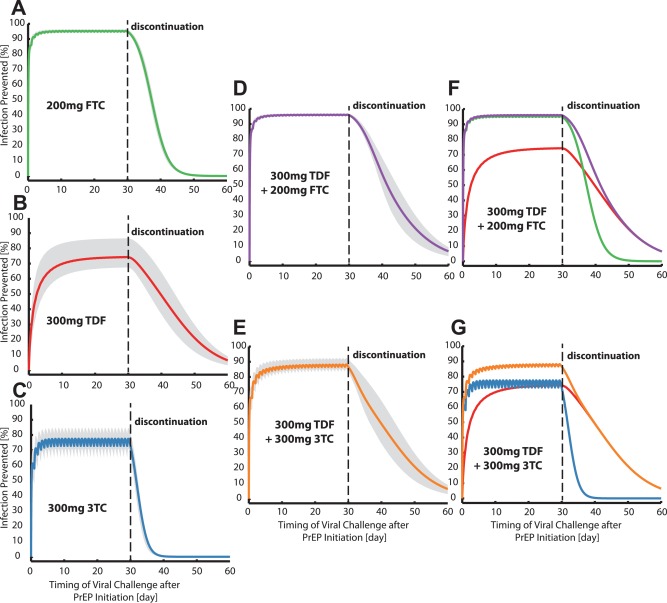
Risk reduction profile ψ for an unprotected homosexual intercourse occurring within 30 days of PrEP or after its discontinuation (modules I‐IV). a–e: Mean risk reduction profiles (see Eq. 3) when either oral doses of 200 mg FTC (a), 300 mg TDF (b), 300 mg 3TC (c), 300 mg TDF+200 mg FTC (d), 300 mg TDF+300 mg 3TC (e) were administered daily for 30 days and discontinued thereafter are illustrated by solid lines. Shaded areas indicate interquartile ranges of this estimate, taking variability in microscopic parameters (module II) and virus exposure during homosexual intercourse (module IV, Figure 2b) into account. f: The mean risk reduction profile for the combination 300 mg TDF + 200 mg FTC (violet solid line) is shown together with the mean risk reduction profiles for the single drugs FTC (green) and TDF (red). g: The mean risk reduction profile for the combination 300 mg TDF + 300 mg 3TC (orange solid line) is shown together with the mean risk reduction profiles for the single drugs 3TC (blue) and TDF (red). Combination predictions assumed that no significant pharmacokinetic interactions occur, pharmacodynamic interactions were modeled as outlined in Supplementary Note 2.
Long‐term efficacy (against repeated virus challenges)
Table 1 depicts the results of a simulated clinical trial with untreated/placebo and PrEP‐treated arms in men who have sex with men (MSM) for different levels of risk compensation and follow‐up durations (T = 6, 12, 18, 24, and 36 months). The infection probability per unprotected sex act with an uninfected individual was fixed to 3%.36, 37 We considered the PrEP strategy to prevent infections per challenge with probability 70, 80, and 90%, respectively. For each efficacy, 0, 10, and 20% risk compensation (additional percentage of risky sex acts in the treated arm compared to the untreated/placebo arm) were assessed. The number of risky sex act per month and individual was set to 1.19, based on a reported value of seven risky acts38 and assuming a prevalence of ≈17% in MSM.39 For all cases, the clinical trial estimated efficacy ωT is lower than the PrEP efficacy per challenge ψ and it decreases with increasing follow‐up time. The decrease is more pronounced when the PrEP efficacy per challenge ψ is low. At 36 months of follow‐up, without risk compensation, the clinical trial efficacy estimate ωT underestimated the actual PrEP efficacy per challenge ψ by 14, 11, and 7%, respectively, for ψ = 70, 80, and 90%. This underestimation becomes even more pronounced when risk compensation occurs.
Table 1.
Bias of clinical‐trial efficacy estimates through risk compensation and follow‐up duration
| Follow‐up durations in months T | Trial‐based PrEP efficacy estimates | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ψ = 70% | ψ = 80% | ψ = 90% | |||||||
| Risk compensation | Risk compensation | Risk compensation | |||||||
| 0% | 10% | 20% | 0% | 10% | 20% | 0% | 10% | 20% | |
| 6 | 68.01 | 64.93 | 61.86 | 78.48 | 76.38 | 74.28 | 89.14 | 88.06 | 86.99 |
| 12 | 65.66 | 62.46 | 59.30 | 76.65 | 74.42 | 72.21 | 88.09 | 86.93 | 85.77 |
| 18 | 63.26 | 59.96 | 56.73 | 74.76 | 72.41 | 70.09 | 86.99 | 85.73 | 84.49 |
| 24 | 60.83 | 57.44 | 54.14 | 72.82 | 70.35 | 67.92 | 85.84 | 84.49 | 83.15 |
| 36 | 55.93 | 52.39 | 48.99 | 68.81 | 66.11 | 63.48 | 83.42 | 81.88 | 80.36 |
Trial‐based PrEP efficacy estimates (after repeated viral challenges) for different levels of risk compensation (reported as and trial durations T were estimated using Eq. (5) (with verifications provided in Supplementary Note 5). The number of unprotected sex acts per month with an infected individual in the untreated arm was set to 1.19. The infection risk per coitus was set to 3% and the prophylactic efficacy per coitus ψ was set to 70, 80, and 90%, respectively.
Taken together, our simulations point to a profound limitation in estimating and comparing PrEP efficacy from incidence rates in clinical trials (as currently done): On the one hand, a clinical trial has to be long enough to provide a statistically reasonable estimate of the incidence rate (a considerable number of individuals have to become infected). On the other hand, the longer the trial, the more confounded will the efficacy estimate ωT be in relation to the actual PrEP efficacy ψ (see Table 1). For this reason we provide the following formula, allowing to convert clinical efficacy estimates ωT into unbiased PrEP efficacies per challenge ψ, which can be compared between different studies (see Supplementary Note 5).
| (7) |
where the subscript S, denote the PrEP and untreated/placebo arms, respectively.
DISCUSSION
In this work, we presented a modular multiscale systems pharmacology modeling pipeline that can be assembled in a building block manner to assess the PrEP efficacy of NRTIs at various levels, ranging from target process inhibition ε, inhibition of target‐cell infection η, and systemic infection ψ, and finally long‐term efficacy after multiple viral challenges ωT. The model allows a flexible integration of processes occurring on different scales: We integrated the microscale interaction between intracellularly active NRTI‐TPs with RT‐mediated viral DNA polymerization, with meso‐, macro‐, and population‐scale processes, such as the pharmacokinetics, replication dynamics, viral transfer, up to the long‐term infection probability after repeated virus exposure, akin to a clinical trial.
Module I (pharmacokinetics) is obviously drug‐specific. We utilized previously developed models17, 18 for 3TC, FTC, and TDF. The module was used to assess the efficacy ψ of PrEP “on demand” and after its discontinuation (see Figures 4 and 5). We observed that TFV‐DP accumulation may be too slow for PrEP “on demand,” in agreement with Ref. 40, who put forward similar concerns. Our analysis also showed, in contrast to dominating views, that FTC is more effective than TDF for PrEP, owing to the fact that higher concentrations may be achieved in target cells and that effective concentrations build up faster than for TDF. On the other hand, TDF seems to be less susceptible to imperfect adherence owing to its long terminal half‐life.18 Moreover, FTC's efficacy is more profoundly reduced by drug‐resistant strains.29 The latter highlights the complementary roles of the two drugs. The Partner PrEP study41 compared the efficacy of TDF alone vs. TDF+FTC, which is partly motivated by cost‐effectiveness considerations. As previously mentioned, our analysis discourages the use of TDF alone for PrEP. In addition, we showed that the drug combination 3TC+TDF may be a cost‐effective alternative to TDF+FTC.
In a previous study42, 43 PrEP efficacy was analyzed in a TDF+FTC combination and an fmol/106cells (≈ 0.09 μM) for TFV‐DP was estimated ( M). This estimate, however, discarded the role of FTC‐TP in the analyzed PrEP combination. In the light of FTC's efficacy (see Figures 3, 4, 5) the previous estimate may vastly underpredict TFV‐DP's actual . We predicted actual single‐drug potencies ( s) of 0.1 and 0.82 μM for TFV‐DP and FTC‐TP, respectively.
Module II (molecular mechanism of effect) allows to translate in vitro measurable microparameters into measures of ex vivo efficacy (prevention of target cell infection η). The model is applicable to all currently approved NRTIs and furthermore allows studying drug efficacy against mutant viruses and mutation‐associated fitness deficits. The latter is particularly useful, since it is unethical to test PrEP in individuals exposed to drug‐resistant viruses. Furthermore, coupled to modules III–V and embedded into epidemiologic models, this allows studying the effect of PrEP on drug resistance spread. We used module II in conjunction with modules III–IV to benchmark the PrEP suitability of various NRTIs. Similar approaches may be used to benchmark PrEP compounds currently under development,4, 44 i.e., nonnucleoside reverse transcriptase inhibitors (NNRTI) or integrase inhibitors, or approved drugs for repurposing, with NNRTIs possibly being cost‐effective alternatives. Obviously, toxicity endpoints have to be included. Note that the MMOA model can in principle predict inhibition of mitochondrial polymerase‐γ, which is frequently associated with toxicity after NRTI administration.45 However, since uptake and anabolism of NRTI is cell‐type‐specific, NRTI‐TP concentrations need to be determined in toxicity‐relevant compartments.46
Motivated by the fact that NRTIs are competitive inhibitors,14 a recent work11 aimed to predict PrEP efficacy solely from the relation of intracellular active TDF and FTC moieties vs. endogenous nucleotide concentrations dNTP in different tissue homogenates. Because they found higher TFV‐DP:dATP ratios in rectal vs. female genital tissue homogenates, they concluded that TDF is more effective in males. The inverse relation was observed for FTC, which was taken as evidence for higher efficacy in females. However, serious drawbacks of this work are the use of tissue homogenates, which may not represent HIV‐1 target cells (more below) and the application of an incomplete and incorrect translational model: The assumption therein11 is that the ratio of NRTI‐TP vs. dNTP determines its effect and may thus explain different PrEP efficacy observed in males and females in clinical trials. This translation of a molecular marker to clinical efficacy lacks substantiation, given that a mechanistic model that assesses the potency at each step from its molecular effect to its clinical efficacy is missing. Moreover, it is evidently wrong at the molecular level: Cottrell et al.'s interpretation would permit that two different active agent concentrations [NRTI‐TP]1 and [NRTI‐TP]2 exert the same effect as long as the ratio NRTI‐TP:dNTP remains fixed. This assumption is incorrect and misleading, since the interactions of NRTIs and dNTPs with the RT‐mediated polymerization process are inherently nonlinear and saturable. We strongly recommend the use of an MMOA model instead (i.e., module II) to capture all involved processes and to translate the considerations mechanistically into clinical effects. In a previous work15 (the basis of module II), we derived a simple formula, which allows to roughly assess how the efficacy of NRTI‐TPs against reverse transcription changes with dNTP concentrations
| (8) |
where denotes the (potentially cell‐specific) rate of NRTI excision, and denote the dissociation constants of the inhibitor and the competing endogenous substrate to their target, respectively, and denotes the incorporation/polymerization constant for the considered NRTI‐TP. When substituting realistic dNTP concentrations from HIV‐1 target cells47 and values from Table SN2.1 (Supplementary Note 2), one can easily see that the ratio for deoxycytosine‐triphosphate (dCTP). Thus, FTC efficacy is not increased by decreasing dCTP concentrations, in contrast to Cottrell et al.'s interpretations.11 For TDF, decreasing deoxyadenosine‐triphosphate (dATP) concentrations may increase its potency up to two‐fold. However, differences may also arise through cell‐dependent rates of excision (different amounts of excision substrates: ATP, PPi), or cell‐dependent differences in NRTI‐TP concentrations, all of which warrant further investigation once the cellular compartments responsible for the early steps of infection for the various routes of transmission are identified. Besides these molecular factors, different clinical PrEP outcomes in the group of women and men can arise through the magnitude of virus exposure after contact (inoculum size; compare Figure 2 b) or through differences in adherence, trial duration, and risk compensation (compare Eq. (7)). Note that the interplay between these putative factors can be assessed within the presented framework once the corresponding data are available.
Module III allows computing infection probabilities and drug efficacies ψ following viral exposure and is based on a validated model of the viral replication cycle.25 The module assumes a “boom or bust” process (see Supplementary Note 3), where successful completion of the first viral replication cycle approximates the probability of infection. The latter assumption is violated whenever subsequent replication cycles cannot be neglected. Examples include prophylaxis with protease inhibitors, which reduce the number of viral progeny after one replication cycle. Furthermore, if very large inoculum sizes (>1,000 viruses) coincide with the application of highly efficient prophylaxis (>99%), the model assumptions may be violated.
Module IV, which estimates from the transmitter's viral load the distribution of viruses entering a target‐cell compartment, is obviously not drug‐specific. A noteworthy feature is that the stochastic nature of HIV‐1 transmission is explicitly taken into account. To our knowledge there is currently no model making this connection and thus there is currently no model linking TasP in the transmitter population with PrEP in the exposed population at risk. To this end, the model is readily integrable into epidemiological models.
We generally assumed, akin to other studies,42 that the concentrations of NRTI‐TP in peripheral mononuclear blood cells (PBMC) serve as a good surrogate measurement for HIV target cells after different modes of transmission. While some authors state concentrations of NRTI‐TP in rectal/mucosal cell homogenates,48 these samples usually contain a large fraction of HIV insusceptible cells over which the concentrations are averaged, in contrast to PBMC consisting mainly of HIV‐1 susceptible cells.49 It is important to acknowledge that NRTI‐TP concentrations are likely different in different cell types, since they are taken up by active transport and require intracellular phosphorylation. Due to experimental difficulties, only few studies have extracted actual target cells (CD4 ) from relevant anatomic sites and subsequently measured NRTI‐TP concentrations. These studies40 indicate that PBMC cells, CD4 cells from relevant anatomical sites and from the peripheral blood contain similar concentrations of NRTI‐TP after oral administration, arguing that the use of the PBMC surrogate measurement is justifiable. However, more research is needed to quantify NRTI‐TP concentrations in target cells derived from anatomical target sites. Note also that NRTI‐TP measurements may depend on the sampling design and sample processing,50 strongly arguing for standardization in NRTI‐TP quantification.
The success of PrEP with TDF+FTC has delivered a proof of concept and motivated the exploration of other PrEP candidates.4 Suitable PrEP compounds require an excellent safety profile, efficaciousness, and cost‐effectiveness. Moreover, they should not contribute to the spread of drug resistance and be robust to imperfect adherence. The latter point is currently addressed by the development of long‐acting injectable compounds and PrEP “on demand.” The presented modular system pharmacology pipeline is a useful tool to screen and optimize suitable PrEP candidates and PrEP strategies by integrating the diverse scales which determine PrEP efficacy.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
M.v.K. and S.D. acknowledge financial support from the BMBF e:Bio junior research group “Systems Pharmacology & Disease Control,” grant number 031A307. V.S. acknowledges financial support through the BMBF funded research consortium PrevOP‐OVERLOAD, grant number 01EC1408. The funders had no role in study design, data collection, or analysis, decision to publish, or preparation of the article.
Conflict of Interest
No conflicts of interest to declare.
Author Contributions
S.D. and M.v.K. wrote the article and designed the research. S.D., V.S., and M.v.K. performed the research and analyzed the data.
References
- 1. Chun, T.W , Moir, S. & Fauci, A.S HIV reservoirs as obstacles and opportunities for an HIV cure. Nat. Immunol. 16, 584–589 (2015). [DOI] [PubMed] [Google Scholar]
- 2. UNAIDS . AIDS by the Numbers 2015. <http://www.unaids.org/en/resources/documents/2015/AIDS_by_the_numbers_2015>. Accessed 31 Mar 2016. Technical report, 2015.
- 3. Mouquet, H. & Nussenzweig, M.C. HIV: Roadmaps to a vaccine. Nature 496, 441–442 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Boffito, M. , Jackson, A. & Asboe, D. Pharmacology lessons from chemoprophylaxis studies. Clin. Infect. Dis. 59(supp. 1), S52–S54 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Cohen, M.S. et al Prevention of HIV‐1 infection with early antiretroviral therapy. N. Engl. J. Med. 365, 493–505 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Attia, S. , Egger, M. , Müller, M. , Zwahlen, M. & Low, N. Sexual transmission of HIV according to viral load and antiretroviral therapy: systematic review and meta‐analysis. AIDS 23, 1397–1404 (2009). [DOI] [PubMed] [Google Scholar]
- 7. Duwal, S. , Winkelmann, S. , Schütte, C. & von Kleist, M. Optimal treatment strategies in the context of ‘treatment for prevention’ against HIV‐1 in resource‐poor settings. PLoS Comput. Biol. 11, e1004200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yousef, K.P. et al Inferring HIV‐1 transmission dynamics in Germany from recently transmitted viruses. J. Acquir. Immune. Defic. Syndr., accepted, 2016. [DOI] [PubMed] [Google Scholar]
- 9. Grant, R.M. et al Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N. Engl. J. Med. 363, 2587–2599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Plosker, G.L. Emtricitabine/tenofovir disoproxil fumarate: a review of its use in HIV‐1 pre‐exposure prophylaxis. Drugs 73, 279–291 (2013). [DOI] [PubMed] [Google Scholar]
- 11. Cottrell, M.L. et al A translational pharmacology approach to predicting HIV pre‐exposure prophylaxis outcomes in men and women using tenofovir disoproxil fumarate +/‐ emtricitabine. J. Infect. Dis. 214, 55–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Painter, G.R. , Almond, M.R. , Mao, S. & Liotta, D.C. Biochemical and mechanistic basis for the activity of nucleoside analogue inhibitors of HIV reverse transcriptase. Curr. Top. Med. Chem. 4, 1035–1044 (2004). [DOI] [PubMed] [Google Scholar]
- 13. Bazzoli, C. et al Intracellular pharmacokinetics of antiretroviral drugs in HIV‐infected patients, and their correlation with drug action. Clin. Pharmacokinet. 49, 17–45, 2010. [DOI] [PubMed] [Google Scholar]
- 14. García‐Lerma, J.G. et al Natural substrate concentrations can modulate the prophylactic efficacy of nucleotide HIV reverse transcriptase inhibitors. J. Virol. 85, 6610–6617 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. von Kleist, M. , Metzner, P. , Marquet, R. & Schütte, C. HIV‐1 polymerase inhibition by nucleoside analogs: cellular‐ and kinetic parameters of efficacy, susceptibility and resistance selection. PLoS Comput. Biol. 8, e40382 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Madrasi, K. , Burns, R.N. , Hendrix, C.W. , Fossler, M.J. & Chaturvedula, A. Linking the population pharmacokinetics of tenofovir and its metabolites with its cellular uptake and metabolism. CPT Pharmacometrics Syst. Pharmacol. 3, e147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duwal, S. & von Kleist, M. Top‐down and bottom‐up modeling in system pharmacology to understand clinical efficacy: An example with NRTIs of HIV‐1. Eur. J. Pharmaceut. Sci. 2016; e‐pub ahead of print 2016. doi: 10.1016/j.ejps.2016.01.016. [DOI] [PubMed] [Google Scholar]
- 18. Duwal, S. , Schütte, C. & von Kleist, M. Pharmacokinetics and pharmacodynamics of the reverse transcriptase inhibitor tenofovir and prophylactic efficacy against HIV‐1 infection. PLoS One 7, e40382 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Borroto‐Esoda, K. et al In vitro evaluation of the anti‐HIV activity and metabolic interactions of tenofovir and emtricitabine. Antivir. Ther. 11, 377–384 (2006). [PubMed] [Google Scholar]
- 20. von Kleist, M. & Huisinga, W. Pharmacokinetic‐pharmacodynamic relationship of NRTIs and its connection to viral escape: an example based on zidovudine. Eur. J. Pharm. Sci. 36, 532–543 (2009). [DOI] [PubMed] [Google Scholar]
- 21. Keele, B.F. et al Identification and characterization of transmitted and early founder virus envelopes in primary HIV‐1 infection. Proc. Natl. Acad. Sci. U. S. A. 105, 7552–7557 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baeten, J.M. et al Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N. Engl. J. Med. 367, 399–410 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Molina, J.M. et al On‐demand preexposure prophylaxis in men at high risk for HIV‐1 infection. N. Engl. J. Med. 373, 2237–2246 (2015). [DOI] [PubMed] [Google Scholar]
- 24. McCormack, S. et al Pre‐exposure prophylaxis to prevent the acquisition of HIV‐1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open‐label randomised trial. Lancet 387, 53–60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. von Kleist, M. , Menz, S. & Huisinga, W. Drug‐class specific impact of antivirals on the reproductive capacity of HIV. PLoS Comput. Biol. 6, e1000720 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frank, M. et al Quantifying the impact of nevirapine‐based prophylaxis strategies to prevent mother‐to‐child transmission of HIV‐1: a combined pharmacokinetic, pharmacodynamic, and viral dynamic analysis to predict clinical outcomes. Antimicrob. Agents Chemother. 55, 5529–5540 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duwe, S. et al Frequency of genotypic and phenotypic drug‐resistant HIV‐1 among therapy‐naive patients of the german seroconverter study. J. Acquir. Immune Defic. Syndr. 26, 266–273 (2001). [DOI] [PubMed] [Google Scholar]
- 28. Quinn, T.C. et al Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai Project Study Group. N. Engl. J. Med. 342, 921–929 (2000). [DOI] [PubMed] [Google Scholar]
- 29. Bartmeyer, B. et al Prevalence of transmitted drug resistance and impact of transmitted resistance on treatment success in the german HIV‐1 seroconverter cohort. PLoS One 5, e12718 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Menéndez‐Arias, L. Mechanisms of resistance to nucleoside analogue inhibitors of HIV‐1 reverse transcriptase. Virus Res. 134, 124–146 (2008). [DOI] [PubMed] [Google Scholar]
- 31. Menéndez‐Arias, L. Molecular basis of human immunodeficiency virus drug resistance: an update. Antiviral Res. 85, 210–231 (2010). [DOI] [PubMed] [Google Scholar]
- 32. Rath, B.A. et al In vitro HIV‐1 evolution in response to triple reverse transcriptase inhibitors & in silico phenotypic analysis. PLoS One 8, e61102 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weber, J. et al Diminished replicative fitness of primary human immunodeficiency virus type 1 isolates harboring the K65R mutation. J. Clin. Microbiol. 43, 1395–1400 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nicol, M.R. et al Models for predicting effective HIV chemoprevention in women. J. Acquir. Immune Defic. Syndr. 68, 369–376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grant, R.N. et al Results of the iPrEx open‐label extension (iPrEx OLE) in men and transgender women who have sex with men: PrEP uptake, sexual practices, and HIV incidence. AIDS 20–25 (2014). [Google Scholar]
- 36. Royce, R.A. , Seña, A. , Vates, Jr, M. & Cohen, M.S. Sexual transmission of HIV. N. Engl. J. Med. 336, 1072–1078 (1997). [DOI] [PubMed] [Google Scholar]
- 37. Boily, M.C. et al Heterosexual risk of HIV‐1 infection per sexual act: systematic review and meta‐analysis of observational studies. Lancet Infect. Dis. 9, 118–129 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sagaon‐Teyssier, L. et al Uptake of PrEP and condom and sexual risk behavior among MSM during the anrs IPERGAY trial. AIDS Care. 28, 48–55 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Velter, A. et al HIV prevalence and sexual risk behaviors associated with awareness of HIV status among men who have sex with men in Paris, France. AIDS Behav. 17, 1266–1278 (2012). [DOI] [PubMed] [Google Scholar]
- 40. Louissaint, N.A. et al Single dose pharmacokinetics of oral tenofovir in plasma, peripheral blood mononuclear cells, colonic tissue, and vaginal tissue. AIDS Res. Hum. Retroviruses 29, 1443–1450 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baeten, J.M. et al Single‐agent tenofovir versus combination emtricitabine plus tenofovir for pre‐exposure prophylaxis for HIV‐1 acquisition: an update of data from a randomised, double‐blind, phase 3 trial. Lancet Infect. Dis. 14, 1055–1064 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anderson, P.L. et al Emtricitabine‐tenofovir concentrations and pre‐exposure prophylaxis efficacy in men who have sex with men. Sci. Transl. Med. 4, 151ra125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hendrix, C.W. Exploring concentration response in HIV pre‐exposure prophylaxis to optimize clinical care and trial design. Cell 155, 515–518 (2013). [DOI] [PubMed] [Google Scholar]
- 44. Abraham, B.K. & Gulick, R. Next‐generation oral preexposure prophylaxis. Curr. Opin. HIV AIDS 7, 600–606 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lewis, W. , Day, B.J. & Copeland, W.C. Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nat. Rev. Drug Discov. 2, 812–822 (2003). [DOI] [PubMed] [Google Scholar]
- 46. Anderson, P.L. , Kakuda, T.N. & Lichtenstein, K.A. The cellular pharmacology of nucleoside‐ and nucleotide‐analogue reverse‐transcriptase inhibitors and its relationship to clinical toxicities. Clin. Infect. Dis. 38, 743–753 (2004). [DOI] [PubMed] [Google Scholar]
- 47. Smith, A.J. & Scott, W.A. The influence of natural substrates and inhibitors on the nucleotide‐dependent excision activity of HIV‐1 reverse transcriptase in the infected cell. Curr. Pharm. Des. 12, 1827–1841 2006. [DOI] [PubMed] [Google Scholar]
- 48. Trezza, C.R. & Kashuba, A.D.M. Pharmacokinetics of antiretrovirals in genital secretions and anatomic sites of HIV transmission: Implications for HIV prevention. Clin. Pharmacokinet. 53, 611–624 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bisset, L.R. et al Reference values for peripheral blood lymphocyte phenotypes applicable to the healthy adult population in Switzerland. Eur. J. Haematol. 72, 203–212 (2004). [DOI] [PubMed] [Google Scholar]
- 50. Durand‐Gasselin, L. et al Evidence and possible consequences of the phosphorylation of nucleoside reverse transcriptase inhibitors in human red blood cells. Antimicrob. Agents Chemother. 51, 2105–2111 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information