

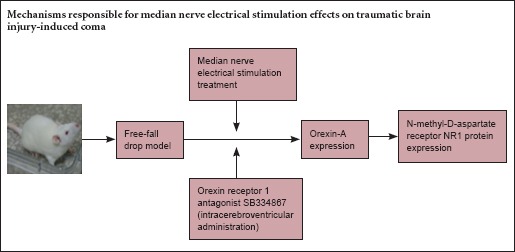
Keywords: nerve regeneration, NR1, median nerve electrical stimulation, traumatic brain injury, coma, wakefulness, orexin-A, prefrontal cortex, neural regeneration
Abstract
Electrical stimulation of the median nerve is a noninvasive technique that facilitates awakening from coma. In rats with traumatic brain injury-induced coma, median nerve stimulation markedly enhances prefrontal cortex expression of orexin-A and its receptor, orexin receptor 1. To further understand the mechanism underlying wakefulness mediated by electrical stimulation of the median nerve, we evaluated its effects on the expression of the N-methyl-D-aspartate receptor subunit NR1 in the prefrontal cortex in rat models of traumatic brain injury-induced coma, using immunohistochemistry and western blot assays. In rats with traumatic brain injury, NR1 expression increased with time after injury. Rats that underwent electrical stimulation of the median nerve (30 Hz, 0.5 ms, 1.0 mA for 15 minutes) showed elevated NR1 expression and greater recovery of consciousness than those without stimulation. These effects were reduced by intracerebroventricular injection of the orexin receptor 1 antagonist SB334867. Our results indicate that electrical stimulation of the median nerve promotes recovery from traumatic brain injury-induced coma by increasing prefrontal cortex NR1 expression via an orexin-A-mediated pathway.
Introduction
Traumatic brain injury (TBI) is a major cause of disability and mortality. TBI-induced coma has devastating effects on victims and their families, and is a considerable economic burden on society (Galing et al., 2014; Plummer et al., 2016). There are multiple therapeutic options for TBI-induced coma, including photostimulation, speech, and various drug therapies, but the effects of these interventions are limited. Therefore, new treatments are needed. The median nerve can be thought of as a “portal” from the periphery to the central nervous system. Electrical stimulation of the median nerve (MNS) is a simple, inexpensive and noninvasive technique that has been shown to improve recovery from brain injury and hasten awakening from coma (Cooper et al., 2003), but the mechanisms responsible for its effects are incompletely understood.
MNS might act by affecting neurotransmitter levels (Cooper et al., 2005; Lei et al., 2015). Ikonomidou et al. (2000) suggested that the excitatory neurotransmitter glutamate promotes neuronal survival after TBI by activating N-methyl-D-aspartate receptors (NMDARs), which are ionotropic glutamate receptors. NMDARs are important in neurodevelopment, tissue remodeling, dendritic spine formation and synaptic plasticity (Monti and Contestabile, 2000; Thomas and Huganir, 2004; Hardingham, 2009; Shohami and Biegon, 2014). Intrathalamic application of the selective NMDAR antagonist DL-2-amino-5-phosphono-pentanoic acid promotes sleep and reduces wakefulness in adult cats (Juhász et al., 1990). However, whether NMDAR is involved in MNS-mediated wakefulness remains to be elucidated.
Our previous study suggested that MNS upregulates the expression of orexin-A and its receptor, orexin receptor 1 (OX1R), in the prefrontal cortex following TBI (Feng et al., 2015). Orexinergic neurons interact with NMDARs in the locus coeruleus-cerebrocortical projection system (Tose et al., 2009). Moreover, orexin-A is strongly associated with glutamate in the prefrontal cortex and can enhance glutamatergic neurotransmission (Zhang et al., 2007).
Here, we investigate the effects of MNS on protein expression of the NR1 subunit of the NMDAR, and explore the possible mechanisms underlying the wake-promoting effect of MNS using the selective OX1R antagonist SB334867.
Materials and Methods
Ethics statement
Animal experiments were approved by the Animal Care and Use Committee of Nanchang University (Nanchang, China), and performed in accordance with the NMDAR of Health Guide for the Care and Use of Laboratory Animals. All precautions were taken to minimize suffering and the number of animals used.
Animals
We used 155 specific pathogen-free adult Sprague-Dawley rats (males and females), weighing 250−300 g. The rats were obtained from the Institute of Laboratory Animals of Huazhong University of Science and Technology, China (license No. SCXK (E) 2010-0009) and housed in the Laboratory Animal Center, First Affiliated Hospital of Nanchang University, China, under controlled temperature and light conditions and with food and water continuously available.
Establishment of TBI-induced coma model
Rats were allocated to four groups: stimulated (TBI + MNS), antagonist (TBI + MNS + SB334867), TBI (TBI alone) and control no TBI groups. Eleven rats died during TBI-induced coma and were replaced so that each group contained 36 rats.
TBI models were established using the weight-drop method (Feeney et al., 1981). Briefly, rats in the TBI, stimulated, and antagonist groups were anesthetized by ether inhalation (concentration ≥ 99%), then allowed to breathe air spontaneously. A midline longitudinal incision was made in the scalp, and the skin was retracted to expose the skull. A cross was marked 2 mm left of the midline and 1 mm anterior to the coronal suture, using a syringe needle. When toe-pinch reflex was restored, a 400-g cylindrical impact hammer (Guangyuan Forging Co., Ltd., Nanchang, Jiangxi Province, China) was dropped onto the marked cross from a height of 40–44 cm (depending on body weight), resulting in a concave fracture of the skull. Control rats underwent anesthesia and skin incision, but no impact injury. The incision was disinfected and closed, and the rats were placed in recovery cages. We used six classical levels of consciousness (Stephens and Levy, 1994) to define comatose rats as those remaining at levels V and VI for 30 minutes or more; these were used in the subsequent procedures.
Consciousness evaluation
We classified consciousness into six degrees based on sensory and motor function (Stephens and Levy, 1994): I, animal freely engaged in normal activity; II, decreased activity; III, decreased activity with impaired motor coordination; IV, righting reflex could be elicited, animals could not stand up; V, no righting reflex, but reaction to noxious stimuli remained; VI, no reaction to noxious stimuli. The first evaluation of consciousness was performed after TBI to determine whether the rats were suitable for the subsequent procedure. Rats in the TBI, stimulated and antagonist groups were evaluated again 1 hour after treatment. Evaluation criteria were inversely correlated with neurological outcome. Blinded evaluation was carried out throughout the experiment. After the final evaluation, the rats were returned to their home cages and provided with sufficient food and water until euthanasia.
Intracerebroventricular administration of SB334867
Comatose rats were pretreated with gentamicin (0.1 mL/100 g; intramuscular injection in the thigh) and anesthetized with 10% chloral hydrate (0.3 mL/100 g; intraperitoneal injection) 1 hour before surgery. The rats were positioned in a stereotaxic frame (ZS-B/S; Beijing Zhongshi Dichuang Science and Technology Development Co., Ltd., Beijing, China), and the guide cannula was mapped to the following coordinates: 1.0 mm posterior to bregma, 1.5 mm lateral to the midline, 4.5 mm ventral to the skull surface (Feng et al., 2015). An injection catheter was inserted into the cerebral ventricle of rats in the antagonist group under sterile conditions. The OX1R inhibitor SB334867 (10 mg/kg; Tocris Bioscience, Ellisville, MO, USA) was dissolved in 60:40 dimethyl sulfoxide/saline solution and administered in a volume of 5 μL (Feng et al., 2015). One hour later, rats were prepared for MNS.
MNS
After TBI and the first evaluation of consciousness, MNS was performed on the right foreleg, using a low-frequency electrical stimulator (ES-420; ITO Physiotherapy & Rehabilitation, Tokyo, Japan). An acupuncture needle was inserted (depth: 1 mm; angle: 45°) 5 mm lateral to the midpoint of the volar wrist crease between the two tendons and was connected to the stimulator. Ipsilateral thumb twitch was observed. The stimulation parameters were as follows: frequency, 30 Hz; pulse width, 0.5 ms; electrical current, 1.0 mA; total stimulation time, 15 minutes (Okazaki et al., 2008). The TBI group underwent an identical procedure to the stimulated group, but without any current output.
Western blot assay
At 6, 12, and 24 hours after injury, six rats from each group were sacrificed by intraperitoneal injection of 10% chloral hydrate. Brains were carefully removed, the prefrontal cortex was quickly dissected on ice, and tissue was homogenized using a Tissue Protein Extraction Kit (CW0891; Beijing Kangwei Biotechnology Co., Ltd., Beijing, China). After centrifugation, the supernatant was collected and assayed to determine protein concentration. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (8% gel) was used to separate proteins (80 V from the stacking gel to the separating gel, then 110 V). The proteins were transferred to polyvinylidene fluoride membranes. The membranes were incubated with rabbit anti-NR1 polyclonal antibody (1:200; Wuhan Boster Biotechnology Co., Ltd., Wuhan, Hubei Province, China) and mouse anti-β-actin monoclonal antibody (1:500; Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China) overnight at 4°C, and washed in Tris buffered saline with Tween 20. The membranes were then incubated in peroxidase-conjugated AffiniPure goat anti-rabbit IgG (1:5,000; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.) at room temperature for 1 hour. β-Actin was used as an internal reference. The optical density of the bands was measured using Quantity One software (Bio-Rad, Hercules, CA, USA) and the value for NR1 was normalized to that for β-actin in the same sample.
Immunohistochemistry
Six rats from each group were decapitated at 6, 12, and 24 hours after injury for immunohistochemistry. The rats were deeply anesthetized by intraperitoneal injection of 10% chloral hydrate and perfused through the heart with 4% paraformaldehyde. The brains were carefully removed, and 40-μm coronal sections were cut. The sections were rinsed in phosphate buffered saline (PBS), and treated with 0.3% hydrogen peroxide (H2O2) for 30 minutes, followed by three 5-minute washes in PBS, before being incubated with normal goat serum for 20 minutes. The sections were then incubated with polyclonal rabbit anti-NMDAR1 antibody (1:100; Wuhan Boster Biotechnology Co., Ltd.) overnight at 4°C. After rinsing in PBS, the sections were incubated with a biotinylated goat anti-rabbit antibody (ready-to-use; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.) at 37°C. Protein expression was visualized using diaminobenzidine in a peroxidase reaction, and quantified as a function of the percentage and staining intensity of immunoreactive cells (Soslow et al., 2000), as follows: A × B, where A represents the percentage of positive cells (0–4, where 0 = 0–1%, 1 = 1–10%, 2 = 10–50%, 3 = 50–80%, 4 = 80–100%) and B represents the intensity of staining (0–3, where 0 = no significant staining, 1 = weak staining, 2 = moderate staining, 3 = strong staining).
Statistical analysis
Western blot data are expressed as the mean ± SD, and immunohistochemistry data as the mean rank. SPSS 17.0 software (SPSS, Chicago, IL, USA) was used for one-way analysis of variance (for western blot analysis) and Kruskal-Wallis H test (for immunohistochemistry). Statistical significance was set at P < 0.05.
Results
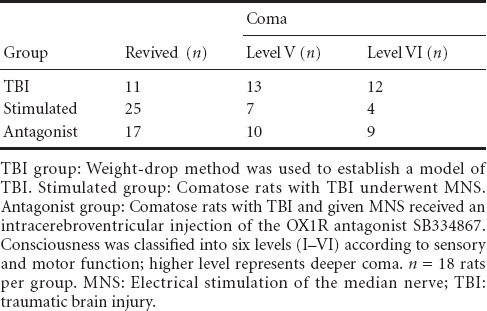
MNS promoted recovery of consciousness in rats with TBI-induced coma
Eleven (31%) of 36 comatose rats in the TBI group awoke. Thirteen and 12 remained at levels V and VI, respectively. Twenty-five rats (69%) awoke in the stimulated group, and eleven remained comatose (seven at level V; four at level VI). Seventeen rats (47%) awoke in the antagonist group (ten remained at level V, and nine at level VI) (Table 1).
Table 1.
Effect of MNS on the recovery of consciousness in rats with TBI-induced coma
MNS upregulated NR1 expression in the prefrontal cortex of rats with TBI-induced coma
Western blot assay revealed significant differences in NR1 expression in the prefrontal cortex between the four groups at 6, 12 and 24 hours (antagonist group < TBI group < control group < stimulated group) (P < 0.05). A within-group comparison showed that the level of NR1 expression was as follows: 6 hours < 12 hours < 24 hours in the control group (P < 0.05), TBI group (P < 0.05) and stimulated group (P > 0.05), and 6 hours < 24 hours < 12 hours in the antagonist group (P < 0.05; Figure 1).
Figure 1.
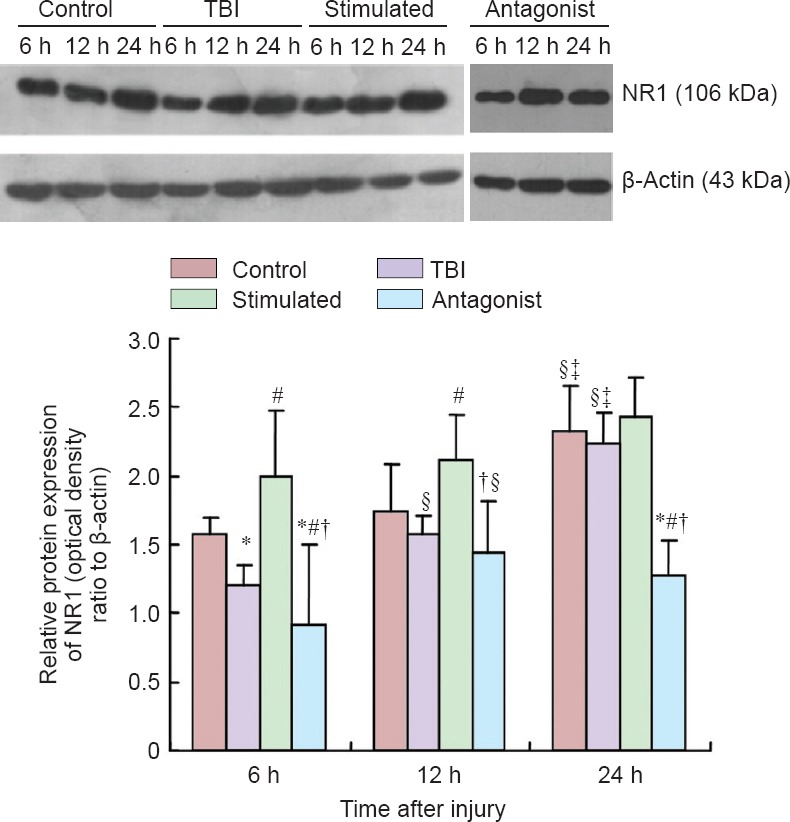
Effect of MNS on NR1 expression in the prefrontal cortex of rats with TBI-induced coma (western blot assay).
Control group: Sham-operated rats (skin incision but no TBI). TBI group: Weight-drop method was used to establish a model of TBI. Stimulated group: Comatose rats with TBI underwent MNS. Antagonist group: Comatose rats with TBI and given MNS received an intracerebroventricular injection of the OX1R antagonist SB334867. Data are expressed as the mean ± SD (n = 6 rats per time point per group). *P < 0.05, vs. control group; #P < 0.05, vs. TBI group; †P < 0.05, vs. stimulated group; §P < 0.05, vs. 6 h; ‡P < 0.05, vs. 12 h (one-way analysis of variance). MNS: Electrical stimulation of the median nerve; TBI: traumatic brain injury; h: hours.
Influence of MNS on NR1 immunoreactivity in the prefrontal cortex after TBI
Significant differences were observed in NR1 immunoreactivity between the four groups (A × B in the control, TBI, stimulated and antagonist groups was 39.94, 28.97, 58.22 and 18.86, respectively; χ2= 36.825;P < 0.001). The following trend was observed in NR1 expression: antagonist group < TBI group < control group < stimulated group (P < 0.001). There were no significant differences in NMDAR NR1 subunit expression between 6, 12 and 24 hours within the same group (A × B = 30.63, 37.79 and 41.08, respectively;P > 0.05; Figure 2).
Figure 2.

Effect of MNS on NR1 immunoreactivity in the prefrontal cortex of rats with TBI-induced coma (× 400).
Control group: Sham-operated rats (skin incision but no TBI). TBI group: Weight-drop method was used to establish a model of TBI. Stimulated group: Comatose rats with TBI underwent MNS. Antagonist group: Comatose rats with TBI and given MNS received an intracerebroventricular injection of the OX1R antagonist SB334867. Brown cells (red arrows) are NR1-immunoreactive. Positive immunostaining for NR1 was found in the cytoplasm, cell membrane, and nucleus of neurons in the prefrontal cortex. MNS: Electrical stimulation of the median nerve; TBI: traumatic brain injury; h: hours.
Discussion
Coma is a serious complication following TBI, and remains a challenge in neurological rehabilitation (Kabadi and Faden, 2014). MNS effectively speeds recovery from coma, but its mechanisms remain unclear. The followings have been proposed (Cooper et al., 2005; Lei et al., 2015): (1) stimulation of the ascending reticular activating system, a complex within the brainstem, which is essential for wakefulness; (2) strengthening of the functional connections between the thalamus and the cortex; (3) increasing of cerebral blood flow, thus improving the blood supply in the penumbra; (4) upregulation of brain-derived neurotrophic factor, which plays an important role in neuroplasticity; (5) alteration of neurotransmitter secretion across the brain.
Glutamate is the primary excitatory neurotransmitter and activates NMDARs, which are composed of two obligatory NR1 subunits and two regulated NR2 subunits (Nagy, 2008). The NR1 subunit is essential for a functional NMDAR and is mainly present at synaptic locations, mediating fast neurotransmission (Dingledine et al., 1999; Cull-Candy et al., 2001). NR1 is widely distributed across the brain, including in the prefrontal cortex, hippocampus, thalamus, striatum, cerebellum and brainstem (Monyer et al., 1994; Sun et al., 2013). The neuroprotective effects of NMDAR activity were originally discovered in 1994 (Gould et al., 1994). Ikonomidou et al. (1999) suggested that blocking NMDAR activity using MK-801 could increase the density of degenerating neurons in layer II of the frontal cortex. Adams et al. (2004) showed that transgenic knock-out and pharmacological blockade of the NMDA receptor increased cell death in the ventrobasal nucleus of the thalamus. These observations suggest that the NMDAR plays an essential role in neuroprotection.
In the present study, we established a weight-drop model of TBI, and evaluated consciousness behaviorally. More rats recovered from TBI-induced coma after MNS than those with no intervention, consistent with a previous study (Zhong et al., 2015). Furthermore, NR1 levels were significantly greater in the stimulated group than in the TBI group at 6 and 12 hours, but not at 24 hours. This may be because we focused on a single aspect of MNS, the wake-promoting actions, which decreased gradually with time. These data support the view that upregulation of NMDAR NR1 protein expression plays an important role in the promotion of wakefulness by MNS.
In a rat model of closed head injury, Biegon et al. (2004) showed that the NMDAR hyperactivation was short-lived (< 1 hour) and was followed by long-lasting hypofunction, suggesting a decrease in NMDAR activity between 1 and 24 hours post-injury. Dhawan et al. (2010) described a marked loss of NMDA receptors a few hours after injury in various brain trauma and ischemia models, with the loss lasting 24 hours or more. These observations are consistent with our findings, which show that NR1 expression in the TBI group was lower than in the control group. The mechanisms responsible for this phenomenon may be attributed to brain injury-induced ribosome depolymerization and inhibition of NR1 protein synthesis, resulting in a reduction in NMDAR expression. MNS appears to be closely related to NR1 upregulation following TBI.
Orexin-A is involved in wakefulness. Orexin-producing neurons project widely to various areas of the brain, such as the prefrontal cortex (Yan et al., 2012). Glutamatergic neurons in the brainstem project to the thalamus and regulate cortical activity. The glutamatergic neurons of the cerebral cortex are not just a target of the arousal system, but themselves contribute to regulation of arousal (Shi and Yu, 2013). A close and reciprocal anatomical action between glutamatergic and orexinergic neurons has been suggested. Aluisio et al. (2014) demonstrated a functional interaction between the OX1R and NMDAR using MK-801-induced glutamate release, showing that inhibition of the OX1R interferes with glutamatergic function in the mouse frontal cortex. These observations agree with our immunohistochemistry and western blot results, which show that rats pretreated with the OX1R antagonist SB334867 had significantly lower NR1 expression in the prefrontal cortex after MNS than rats that received MNS without the antagonist. These results were also consistent with our consciousness evaluations. In our previous study, we showed that MNS upregulates orexin-A expression following TBI (Feng et al., 2015). Therefore, orexin-A may be involved in the regulation of NR1 expression following MNS.
Western blot assay demonstrated that NR1 expression in the control group increased with time. We considered this to represent the physiological secretion cycle. The sampling times for NMDA NR1 protein (6, 12 and 24 hours) may be related to the physiological secretion cycle. NMDAR NR1 expression in the TBI group showed a significantly increasing trend over time, indicating that rats recovered gradually after TBI. However, the different pattern observed in the antagonist group across three time points indicates that OX1R antagonism is more effective in the early stages after TBI.
The present study examined only one effect of MNS, and the effects of stimulation with different parameters and treatment periods on TBI-induced coma in rats remain to be explored. However, our results highlight a relationship between MNS, NR1, and orexin-A after TBI, showing that MNS elicits NMDA NR1 increase via orexin-A. Additionally, orexin-A is closely related to other neurotransmitters associated with wakefulness, which may determine whether orexin-A acts as an arousal “switch”; this pathway warrants further investigation.
In conclusion, we used MNS intervention to investigate NR1 expression in the prefrontal cortex of rats with TBI-induced coma, and evaluated the functional interaction between orexin-A and NR1 after microinjecting OX1R antagonist SB334867 into the cerebral ventricle. Our study offers the first evidence indicating that the promotion of wakefulness by MNS is due, at least in part, to the upregulation of NR1 expression via orexin-A, and provides new insight into the wake-promoting mechanism of MNS in the treatment of traumatic coma.
Footnotes
Funding: This study was supported by the National Natural Science Foundation of China, No. 81260295; the Natural Science Foundation of Jiangxi Province of China, No. 20132BAB205063.
Conflicts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded and stringently reviewed by international expert reviewers.
Copyedited by Slone-Murphy J, Rave W, Yu J, Qiu Y, Li CH, Song LP, Zhao M
References
- Adams SM, de Rivero Vaccari JC, Corriveau RA. Pronounced cell death in the absenc- e of NMDA receptors in the developing somatosensory thalamus. J Neurosci. 2004;24:9441–9450. doi: 10.1523/JNEUROSCI.3290-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluisio L, Fraser I, Berdyyeva T, Tryputsen V, Shireman BT, Shoblock J, Lovenberg T, Dugovic C, Bonaventure P. Pharmacological or genetic orexin1 receptor inhibition attenuates MK-801 induced glutamate release in mouse cortex. Front Neurosci. 2014;8:107. doi: 10.3389/fnins.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegon A, Fry PA, Paden CM, Alexandrovich A, Tsenter J, Shohami E. Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: Implications for treatment of neurological and cognitive deficits. Proc Natl Acad Sci U S A. 2004;101:5117–5122. doi: 10.1073/pnas.0305741101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EB, Cooper JB. Electrical treatment of coma via the median nerve. Acta Neurochir Suppl. 2003;87:7–10. doi: 10.1007/978-3-7091-6081-7_2. [DOI] [PubMed] [Google Scholar]
- Cooper EB, Scherder EJ, Cooper JB. Electrical treatment of reduced consciousness: experience with coma and Alzheimer's disease. Neuropsychol Rehabil. 2005;15:389–405. doi: 10.1080/09602010443000317. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- Dhawan J, Benveniste H, Nawrocky M, Smith SD, Biegon A. Transient focal ischemia results in persistent and widespread neuroinflammation and loss of glutamate NMDA receptors. Neuroimage. 2010;51:599–605. doi: 10.1016/j.neuroimage.2010.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–62. [PubMed] [Google Scholar]
- Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 1981;211:67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhong YJ, Wang L, Wei TQ. Resuscitation therapy for traumatic brain injury-induced coma in rats: mechanisms of median nerve electrical stimulation. Neural Regen Res. 2015;10:594–598. doi: 10.4103/1673-5374.155433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garling RJ, Watts LT, Sprague S, Fletcher L, Jimenez DF, Digicaylioglu M. Does progesterone show neuroprotective effects on traumatic brain injury through increasing phosphorylation of Akt in the hippocampus? Neural Regen Res. 2014;9:1891–1896. doi: 10.4103/1673-5374.145355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Cameron HA, McEwen BS. Blockade of NMDA receptors increases cell death and birth in the developing rat dentate gyrus. J Comp Neurol. 1994;340:551–565. doi: 10.1002/cne.903400408. [DOI] [PubMed] [Google Scholar]
- Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans. 2009;37:1147–1160. doi: 10.1042/BST0371147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Stefovska V, Turski L. Neuronal death enhanced by N-methyl-D-aspartate antagonists. Proc Natl Acad Sci U S A. 2000;97:12885–12890. doi: 10.1073/pnas.220412197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Juhász G, Kékesi K, Emri Z, Soltesz I, Crunelli V. Sleep-promoting action of excitatory amino acid antagonists: a different role for thalamic NMDA and non-NMDA receptors. Neurosci Lett. 1990;114:333–338. doi: 10.1016/0304-3940(90)90586-x. [DOI] [PubMed] [Google Scholar]
- Kabadi SV, Faden AI. Selective CDK inhibitors: promising candidates for future clinical traumatic brain injury trials. Neural Regen Res. 2014;9:1578–1580. doi: 10.4103/1673-5374.141779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J, Wang L, Gao G, Cooper E, Jiang J. Right median nerve electrical stimulation for acute traumatic coma patients. J Neurotrauma. 2015;32:1584–1589. doi: 10.1089/neu.2014.3768. [DOI] [PubMed] [Google Scholar]
- Monti B, Contestabile A. Blockade of the NMDA receptor increases developmental apoptotic elimination of granule neurons and activates caspases in the rat cerebellum. Eur J Neurosci. 2000;12:3117–3123. doi: 10.1046/j.1460-9568.2000.00189.x. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Nagy J. Alcohol related changes in regulation of NMDA receptor functions. Curr Neuropharmacol. 2008;6:39–54. doi: 10.2174/157015908783769662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki Y, Morimoto T, Sawai H. Parameters of optic nerve electrical stimulation affecting neuroprotection of axotomized retinal ganglion cells in adult rats. Neurosci Res. 2008;61:129–135. doi: 10.1016/j.neures.2008.01.016. [DOI] [PubMed] [Google Scholar]
- Plummer S, Van den Heuvel C, Thornton E, Corrigan F, Cappai R. The neuroprotective properties of the amyloid precursor protein following traumatic brain injury. Aging Dis. 2016;7:163–179. doi: 10.14336/AD.2015.0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YF, Yu YQ. The roles of glutamate in sleep and wakefulness. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2013;42:583–590. [PubMed] [Google Scholar]
- Shohami E, Biegon A. Novel approach to the role of NMDA receptors in traumatic brain injury. CNS Neurol Disord Drug Targets. 2014;13:567–573. doi: 10.2174/18715273113126660196. [DOI] [PubMed] [Google Scholar]
- Song CH, Chen XW, Xia JX, Yu ZP, Hu ZA. Modulatory effects of hypocretin-1/orexin-A with glutamate and γ-aminobutyric acid on freshly isolated pyramidal neurons from the rat prefrontal cortex. Neurosci Lett. 2006;399:101–105. doi: 10.1016/j.neulet.2006.01.065. [DOI] [PubMed] [Google Scholar]
- Soslow RA, Dannenberg AJ, Rush D, Woerner BM, Khan KN, Masferrer J, Koki AT. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer. 2000;89:2637–2645. doi: 10.1002/1097-0142(20001215)89:12<2637::aid-cncr17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Stephens JR, Levy RH. Effects of valproate and citrulline on ammonium-induced encephalopathy. Epilepsia. 1994;35:164–171. doi: 10.1111/j.1528-1157.1994.tb02928.x. [DOI] [PubMed] [Google Scholar]
- Sun H, Jia N, Guan L, Su Q, Wang D, Li H, Zhu Z. Involvement of NR1, NR2A different expression in brain regions in anxiety-like behavior of prenatally stressed offspring. Behav Brain Res. 2013;257:1–7. doi: 10.1016/j.bbr.2013.08.044. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tose R, Kushikata T, Yoshida H, Kudo M, Furukawa K, Ueno S, Hirota K. Interaction between orexinergic neurons and NMDA receptors in the control of locus coeruleus-cerebrocortical noradrenergic activity of the rat. Brain Res. 2009;1250:81–87. doi: 10.1016/j.brainres.2008.10.041. [DOI] [PubMed] [Google Scholar]
- Yan J, He C, Xia JX, Zhang D, Hu ZA. Orexin-A excites pyramidal neurons in layer 2/3 of the rat prefrontal cortex. Neurosci Lett. 2012;520:92–97. doi: 10.1016/j.neulet.2012.05.038. [DOI] [PubMed] [Google Scholar]
- Zhang CQ, Xia JX, Chen PH, Hu ZA. Regulation of glutamate current by orexin A on pyramidal neurons in rat prefrontal cortex. Disan Junyi Daxue Xuebao. 2007;29:922–924. [Google Scholar]
- Zhong YJ, Feng Z, Wang L, Wei TQ. Wake-promoting actions of median nerve stimulation in TBI-induced coma: an investigation of orexin-A and orexin receptor 1 in the hypothalamic region. Mol Med Rep. 2015;12:4441–4447. doi: 10.3892/mmr.2015.3898. [DOI] [PubMed] [Google Scholar]