

We discovered how the coupling of Na+ and Ca2+ electrochemical gradients regulates cross talk of surface membrane and Ca2+ clocks to regulate automaticity via the coupled-clock system; our numerical model simulations detected “a window” of system parameters for this effect on normal automaticity that borders on abnormal automaticity.
Keywords: cardiac automaticity, sodium, calcium, electrochemical driving forces
Abstract
Coupling of an intracellular Ca2+ clock to surface membrane ion channels, i.e., a “membrane clock, ” via coupling of electrochemical Na+ and Ca2+ gradients (ENa and ECa, respectively) has been theorized to regulate sinoatrial nodal cell (SANC) normal automaticity. To test this hypothesis, we measured responses of [Na+]i, [Ca2+]i, membrane potential, action potential cycle length (APCL), and rhythm in rabbit SANCs to Na+/K+ pump inhibition by the digitalis glycoside, digoxigenin (DG, 10–20 μmol/l). Initial small but significant increases in [Na+]i and [Ca2+]i and reductions in ENa and ECa in response to DG led to a small reduction in maximum diastolic potential (MDP), significantly enhanced local diastolic Ca2+ releases (LCRs), and reduced the average APCL. As [Na+]i and [Ca2+]i continued to increase at longer times following DG exposure, further significant reductions in MDP, ENa, and ECa occurred; LCRs became significantly reduced, and APCL became progressively and significantly prolonged. This was accompanied by increased APCL variability. We also employed a coupled-clock numerical model to simulate changes in ENa and ECa simultaneously with ion currents not measured experimentally. Numerical modeling predicted that, as the ENa and ECa monotonically reduced over time in response to DG, ion currents (ICaL, ICaT, If, IKr, and IbNa) monotonically decreased. In parallel with the biphasic APCL, diastolic INCX manifested biphasic changes; initial INCX increase attributable to enhanced LCR ensemble Ca2+ signal was followed by INCX reduction as ENCX (ENCX = 3ENa − 2ECa) decreased. Thus SANC automaticity is tightly regulated by ENa, ECa, and ENCX via a complex interplay of numerous key clock components that regulate SANC clock coupling.
NEW & NOTEWORTHY
We discovered how the coupling of Na+ and Ca2+ electrochemical gradients regulates cross talk of surface membrane and Ca2+ clocks to regulate automaticity via the coupled-clock system; our numerical model simulations detected “a window” of system parameters for this effect on normal automaticity that borders on abnormal automaticity.
the primary determinants of automaticity of sinoatrial nodal cells (SANCs) lie within mechanisms that regulate the slope of the spontaneous diastolic depolarization (DD). Although classical perspectives had posited SANC automaticity to be regulated exclusively by surface membrane ion channels, recent evidence indicates that a coupled-clock system operates within SANCs to regulate normal automaticity (52, 54). Specifically, the sarcoplasmic reticulum (SR), functioning as a Ca2+ clock, and the ensemble of sarcolemmal ion channels and transporters as a “membrane clock” (M clock), comprise a coupled system that ensures robust, but flexible, SANC function (16, 51). Because M clock component functions regulate [Ca2+]i, the oscillatory substrate of the Ca2+ clock, the M clock indirectly regulates spontaneous local Ca2+ release (LCR), generated via ryanodine receptors of the Ca2+ clock. LCR activation of NCX is a crucial factor in clock coupling. Both the characteristics and rhythmicity of spontaneous LCRs beneath the SANC surface membrane during DD and the average action potential cycle length (APCL) and its variability (APCLV) inform on the fidelity of clock coupling within the system (19, 52, 54, 57). Thus the coupling of Na+ and Ca2+ electrochemical driving forces, Vm − ENa and Vm − ECa, respectively, can be envisioned as the major regulator of the clock coupling that regulates SANC normal automaticity (16, 19, 25).
Although there have been numerous prior studies of changes of [Na+]i, [Ca2+]i, Vm, ENa, and ECa effects on contraction and abnormal automaticity, evoked by digitalis glycosides (DG) in ventricular cells, and on both normal or abnormal automaticity in isolated SAN tissue (39, 41) and Purkinje fibers (11, 20, 21, 29–31, 35), how DG-induced changes in Vm, ENa, and ECa regulate automaticity of individual SANCs remains unexplored.
We employed a high DG concentration (10–20 μmol/l) to effect Na+/K+ pump inhibition that would accelerate time-dependent increases in [Na+]i and [Ca2+]i and cause a reduction in maximum diastolic potential (MDP), leading to changes in ENa and ECa (11) in a relatively short time (25 min), enabling comparison of changes in the experimentally measured parameters at varying times following DG exposure to a specific [Na+]i at those specific times (11) and create a model simulation linked to a specific [Na+]i increase. We measured membrane potential and spontaneous APCL, the APCL waveform, APCLV, [Na+]i, the cytosolic Ca2+ transient induced by an action potential (AP), and LCR characteristics in subsets of single, isolated rabbit SANCs. Employing a coupled-clock SANC numerical model (26), we simulated, in silico, simultaneous effects of the experimentally measured increase in [Na+]i over time, attributable to blockade of Na+/K+ ATPase by DG on MDP, ENa, ECa, and ENCX, spontaneous APCL, ion currents (If, INaK, ICaL, ICaT, IbNa, INCX, and IKr), LCRs, SR Ca2+ pumping, systolic and diastolic cytosolic Ca2+ (Cacyt), and Ca2+ in the submembrane space (Casub). This combined experimental and numerical modeling approach revealed how Na+ and Ca2+ electrochemical gradients in single, isolated SANCs regulate cross talk between surface membrane M and Ca2+ clock functions and their coupling to effect changes in SANC APCL.
MATERIALS AND METHODS
SANC isolation.
Spontaneously beating SANCs were isolated from male New Zealand white rabbits (Charles River Laboratories) weighing 1.8−2.5 kg, as previously described (47). Rabbits were deeply anesthetized in accordance with NIH guidelines for the care and use of animals, protocol no. 034LCS2016, which was approved by the NIH Animal Care and Use Committee. The heart was removed quickly and placed in the solution containing (in mmol/l): 130.0 NaCl, 24.0 NaHCO3, 1.2 NaH2PO4, 1.0 MgCl2, 1.8 CaCl2, 4.0 KCl, and 5.6 glucose equilibrated with 95% O2-5% CO2 (pH 7.4 at 35.5°C). The sinoatrial node (SA) region was cut into small strips (∼1.0 mm wide) perpendicular to the crista terminalis (CT) and excised (47). The final SA node preparation consisted of SA node strips attached to the small portion of CT. The SA node preparation was washed twice in Ca2+-free solution containing (in mmol/l): 140.00 NaCl, 5.40 KCl, 0.50 MgCl2, 0.33 NaH2PO4, 5.00 HEPES, and 5.50 glucose (pH = 6.9); the preparation was incubated at 35.5°C for 30 min in the same solution with addition of elastase type IIA (0.6 mg/ml; Sigma), collagenase type 2 (0.6 mg/ml; Worthington), and 0.1% bovine serum albumin (Sigma). The SA node preparation was washed in modified Kraftbruhe (KB) solution, containing (in mmol/l): 70.0 potassium glutamate, 30.0 KCl, 10.0 KH2PO4, 1.0 MgCl2, 20.0 taurine, 10.0 glucose, 0.3 EGTA, and 10.0 HEPES (titrated to pH 7.4 with KOH); it was kept at 4°C for 1 h in KB solution containing 50 mg/ml polyvinylpyrrolidone (Sigma). Single cells were dispersed from the SA node preparation by gentle pipetting in the KB solution and stored at 4°C.
All experiments were performed at 35.5°C. Temperature was controlled by an Analog TC2BIP 2/3Ch Bipolar Temp controller from Cell MicroControls.
[Na+]i measurements in intact single SANCs.
[Na+]i was assessed by an IonOptix Myocam system (Ion-Optix) in spontaneously beating intact SANCs. [Na+]i was measured with Na+-sensitive fluorescent dye benzofuran isophthalate (SBFI-AM; Molecular Probes) at 35.5°C. SANCs were loaded with 10 μmol/l of SBFI-AM and 10% Pluronic F-127 in Tyrode solution for 90–110 min at room temperature. Pluronic F-127 was used to facilitate loading. SANCs were washed in normal Tyrode solution for 20 min to ensure deesterification of the SBFI-AM to SBFI and placed in a temperature-controlled bath (at 35.5°C) on an inverted fluorescence microscope equipped with an LWD Fluor ×40 lens (Olympus). [Na+]i was measured at baseline and every minute following DG (10 μmol/l) exposure by exposing spontaneously beating SANCs to the light, emitted by a 75-W mercury xenon lamp, passed through either a 340-nm or 380-nm band-pass filter (bandwidths were ± 15 nm) at 200 Hz. Emitted light was collected by a Hamamatsu photomultiplier tube. The ratio of fluorescence (340/380) represents the level of [Na+]i. After each measurement of [Na+]i, the F340/F380 ratios were calibrated (Fig. 1) using a dual-point approach (at 150 mmol/l and 0 mmol/l extracellular [Na+], [Na+]o) in the presence of amphotericin (100 μmol/l) and monensin (100 μmol/l), as described previously (7, 10). Background fluorescence was corrected before the ratio was calculated. Autofluorescence was undetectable. The method provides a good fit to the data over the range of [Na+]i studied.
Fig. 1.
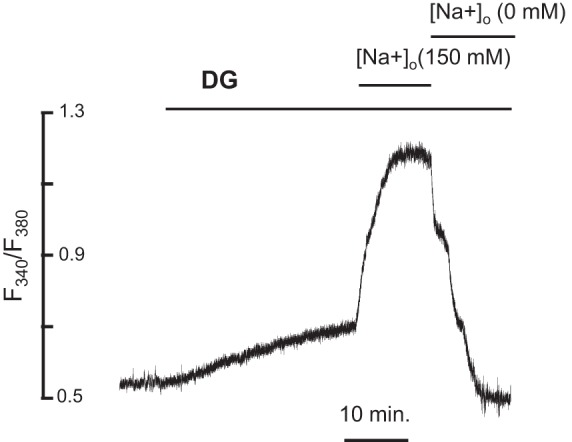
[Na+]i measurements. Representative example of [Na+]i measurement in a representative cell before and in the presence of digoxigenin (DG). F340/F380 was calibrated at the end of each experiment by sequentially applying solutions (see materials and methods) containing 150 mmol/l and 0 mmol/l extracellular sodium ([Na+]o) in the presence of DG (10 μmol/l). Background fluorescence was corrected before the ratio was calculated. The short horizontal lines indicate the onset and the duration of the superfusion of the cells with calibration solutions containing 150 mM and 0 mM [Na+]o in the presence of DG (10 μmol/l). The superfusion with normal Tyrode solution before calibration was 10 min.
Measurement of systolic and diastolic Ca2+ by Indo-1 in intact SANCs.
We measured time-dependent changes in systolic and diastolic [Ca2+]i in response to DG via a calibrated ratiometric Ca2+ indicator, Indo-1, as described before (55). SANCs were placed in a chamber on an inverted fluorescence microscope stage (Zeiss IM-35), were loaded with 14 μmol/l Indo-1 AM (Molecular Probes) for 15 min at room temperature, and were subsequently superfused with Tyrode solution for 20 min to remove excess indicator. To allow full deesterification, the bath temperature was slowly increased to 35.5°C. The apparatus to detect Indo-1 fluorescence was described previously (38) except that a ×63/1.4 N.A. oil UV fluorglycerin-immersion objective (Zeiss) was used. Indo-1 fluorescence was corrected for background, and autofluorescence was determined in every experiment by subtracting averaged signals from cells not loaded with Indo-1 (n = 5). [Ca2+]i was calculated as previously described (55).
AP recordings and AP waveform analysis in single intact SANCs.
Spontaneous APs were recorded by a perforated patch-clamp technique with 0.05 mmol/l of β-escin added to the electrode solution that contained (in mmol/l): 120 K-gluconate, 5 NaCl, 3 Mg-ATP, 5 HEPES, 20 KCl, 3 Na2ATP (pH adjusted to 7.2 with KOH) (47). SANCs were continuously superfused with normal Tyrode solution containing (in mmol/l): 140.0 NaCl, 5.4 KCl, 5.0 HEPES, 2.0 MgCl2, 1.8 CaCl2, and 10.0 glucose (pH 7.4). APs were recorded by a standard zero-current-clamp technique (Axopatch 200B; Molecular Devices). Experiments were terminated following ∼25 min of DG exposure.
The APCL and AP characteristics were analyzed via a customized program (4, 24). The APCL was measured either as the interval between AP upstrokes (maximum dV/dt, V/s), or, in a subset of cells, as the interval between AP-induced Ca2+ transient. Other measured AP parameters also include MDP, AP overshoot (MaxV), AP amplitude, AP upstroke (dV/dtmax), AP duration measured at 90% of repolarization time (APD90), time from MDP to the onset of nonlinear DD (TNLDD), and time from previous maximum AP to MDP (tMDP). Beat-to-beat variations (percent change) in spontaneous APCLs and other selected AP parameters were assessed in each cell by the coefficient of variation (CV), calculated as standard deviation divided by its mean (CV = SD/mean·100).
The average [Na+]i was assigned to each cell as its [Na+]i at a specific time point during continuous AP recordings when APCL reached reference points following DG exposure (short APCL, back-to-base APCL, above-base APCL, and grossly dysrhythmic APCL).
Local Ca2+ signal imaging in single intact SANCs.
Ca2+ transients and spontaneous diastolic LCRs were recorded in intact SANCs superfused with the normal Tyrode solution (see above) at baseline and in the presence of DG (10–20 μmol/l) by confocal microscopy (Zeiss LSM-410 inverted confocal microscope; Carl Zeiss). We used a 488-nm argon ion laser for excitation of fluo-4 AM (10 μmol/l), and the fluorescence signal was collected at wavelengths of >515 nm in line-scan mode oriented along the cell periphery (46).
Local Ca2+ signal imaging in permeabilized SANCs.
To determine whether DG directly affects the SR Ca2+ cycling, we measured LCRs in permeabilized (with saponin, 0.01%) SANCs at 100 nmol/l [Ca2+]i, as previously described (36). LCRs were measured by confocal microscopy in the recording solution containing (in mmol/l): 100.00 DL-aspartic acid potassium salt, 25.00 KCl, 10.00 NaCl, 3.00 MgATP, 0.81 MgCl2, 20.00 Hepes, 0.50 EGTA, 10.00 phosphocreatine; this was in addition to 5.00 U/ml creatine phosphokinase (pH 7.2 adjusted with KOH). Fluo-4 salt (30 μmol/l) and Ca2+ were added to the recording solutions. The final free [Ca2+]i = 100 nmol/l was calculated by a computer program (WinMAXC 2.50, Stanford University).
Analysis of diastolic local Ca2+ signals.
Fluo-4 measurements of LCRs in intact SANCs are presented as F/F0 (normalized Ca2+ signal). LCR spatial size was indexed as the full width at the half-maximum amplitude (FWHM), and its duration was characterized as the full duration at half maximum amplitude (FDHM). LCR events were normalized per 100 μm of the line-scan image and during a 1-s time interval (36). The Ca2+ signal of an individual LCR was estimated as follows: LCR = FWHM × FDHM(F/F0 − 1)/2. The Ca2+ signal of the LCR ensemble within a cell was calculated as the sum of all Ca2+ signals of individual LCRs (36). All images were processed with IDL software (6.1., Research Systems).
In silico numerical modeling of DG effects.
A coupled-clock model of rabbit SANCs (26) was employed to explore various responses to the experimentally measured time-dependent increase in [Na+]i during blockade of Na+/K+ ATPase current. In these simulations, we assumed that, in the presence of DG, the remaining Na+/K+ ATPase activity (and INaK density) is 30% of baseline (before drug application), as previously reported (49). The time-dependent increase in [Na+]i was simulated by clamping [Na+]i using a slow ramp (0.075 mmol/l per s) from 8 mmol/l to 14 mmol/l and simultaneously simulating at a given [Na+]i dynamic changes in Vm, INCX, If, ICaL, ICaT, IKr, INaK, IbNa, LCRs, Casub (submembrane Ca2+), Cacyt (cytosolic Ca2+), and diastolic SR Ca2+ release flux (jCarel).
Experimental design.
The increase in [Na+]i and other variables was monitored over time following exposure to a high concentration of DG (10–20 μmol/l), to effect a wide-range [Na+]i increase within a relatively brief period of time, about 25 min. The high concentration of DG (20 μmol/l) has been used in a similar experimental approach in ventricular myocytes (1). In our study, we tested DG-induced effects on spontaneous APCL and the APCL waveform, [Na+]i, the systolic and diastolic [Ca2+], and spontaneous LCR in subsets of single, isolated rabbit SANCs via different techniques (patch clamp, confocal microscopy, Indo-1, and IonOptix Myocam system). The time of ∼25 min was chosen based on the time window of the most reliable AP recording. Specifically, during continuous AP recordings, each perforated patch-clamp experiment has three phases: phase 1, when GigaOhm seal is established and β-escin is perforating the membrane patch (within about 4–5 min) to allow electrical access into the cell; phase 2, when perforation is completed and AP can be reliably measured; phase 3, when AP recording becomes unstable as the GigaOhm seal starts to deteriorate after 25–30 min in spontaneously beating SANCs. To have comparable measurements between different experiments, we used ∼25–30 min as an end point for the experiment.
Drugs.
DG (C23H34O5) was obtained from Sigma.
Statistics.
Data are presented as means ± SE (n = number of SANCs). P < 0.05 is considered statistically significant. A linear mixed-effects model with a Dunnett's post hoc adjustment to the P value to compare each time point with the baseline was used to test changes of AP-induced Ca2+ transients and systolic and diastolic [Ca2+]i measured by Indo-1 in response to DG. A linear mixed-random effects model for repeated measures (44) followed by Bonferroni adjustment of the P values for multiple comparisons were used to test changes of AP waveform and local Ca2+ signals in response to DG, measured by patch clamp and confocal microscopy, respectively. The two-sample paired t-test for means was used to compare [Na+]i before and after DG exposure in intact single SANCs.
RESULTS
DG-induced changes in [Na+]i.
Changes in [Na+]i measured in a subset of SANCs (n = 7), at baseline and every minute following 10 μmol/l DG exposure, are presented in Fig. 2. By employing a high DG concentration to effect an increase in [Na+]i in a relatively short time (20–25 min), we could relate changes in the experimentally measured parameters at varying times following DG exposure to a specific [Na+]i at those times (11) and create model simulations linked to a specific [Na+]i increase. The average [Na+]i increased exponentially over the time following DG exposure (P < 0.05) (Fig. 2A). Note that the rate at which [Na+]i increased over time following DG exposure varied among cells (Fig. 2B).
Fig. 2.

[Na+]i increases in response to DG in spontaneously beating sinoatrial nodal cells (SANCs). A: average [Na+]i change at baseline and every minute following 10 μmol/l DG exposure in spontaneously beating SANCs (n = 7). The arrow indicates the time when superfusion with DG started (2-sample paired t-test for means was used to compare [Na+]i before and after DG exposure; see details in Table 2). B: example of [Na+]i change in 3 different SANCs.
Effect of DG on spontaneous APCL and MDP.
Figure 3 illustrates a continuous recording of time-dependent effects of DG on the spontaneous APCL (Fig. 3A, arrow indicates the onset of DG exposure) and MDP (Fig. 3B) in the SANC. Examples of the AP waveform in this cell at baseline (before DG) and at varying times following DG administration are illustrated in Fig. 4.
Fig. 3.

Time-dependent effect of DG on spontaneous action potential cycle length (APCL) and maximum diastolic potential (MDP) in a single SANC. A and B: examples of continuous recordings of time-dependent effects of DG (10 μmol/l) on the spontaneous APCL and MDP. The arrow indicates the time when superfusion with DG started. C: Poincaré plots, which display each APCL as a function of preceding APCL at baseline and at selected times (at shortest APCL, back to baseline APCL, prolonged above baseline APCL, grossly dysrhythmic APCL) following DG exposure in the same cell presented in A. D: comparison of the average beat-to-beat variation of APCL and the average coefficient of variation of APCL (APCLV) of the same cell in A at baseline and during DG exposure at the same times as in C.
Fig. 4.

Alterations in the action potential (AP) waveform in response to DG. A: representative examples of AP recordings at baseline and at selected times (at minimum APCL, APCL prolongation back to baseline, APCL prolongation above baseline, and grossly dysrhythmic APCL) following DG (10 μmol/l) exposure in the same cell. B: overlay of the AP waveforms (synchronized at the AP overshoot) at baseline and during DG response in the same cell presented in A (colors in B match colors in A).
The earliest response to DG was a reduction in APCL (Fig. 3A). The time to achieve the minimum APCL following DG exposure varied among cells (Table 2). On average, APCL decreased to 61.7 ms (18%) from 349.9 ± 14.8 ms at baseline to 288.2 ± 11.1 (P < 0.05, Table 1) and was accompanied by a small (5%) but significant increase in [Na+]i, from 8.4 ± 0.4 mmol/l at baseline to 8.9 ± 0.1 mmol/l at the shortest APCL (P < 0.05, Table 2). At the time at which the APCL reached its nadir in each cell, MDP was reduced (Fig. 3). On average, MDP was reduced from −58.2 ± 1.4 mV at baseline to −55.0 ± 2.0 mV at the time when average APCL reached its nadir (Table 1).
Table 2.
Changes of APCL and [Na+]i in the individual SANCs in response to DG
| Cell No. | Baseline APCL, ms | Short APCL, ms | [Na+]i, mM | DG Time, min | Back to Baseline APCL, ms | [Na+]i, mM | DG Time, min | Above Baseline APCL, ms | [Na+]i, mM | DG Time, min | Grossly Dysrhythmic APCL, ms | [Na+]i, mM | DG Time, min |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 361.0 | 268.3 | 8.7 | 2.0 | 360.0 | 13.1 | 10.0 | 413.0 | 18.2 | 17.0 | 782.1 | 18.2 | 21.0 |
| 2 | 278.7 | 214.9 | 8.7 | 2.0 | 278.0 | 13.1 | 10.0 | 346.4 | 18.2 | 15.0 | 801.4 | 18.2 | 19.0 |
| 3 | 305.4 | 281.3 | 8.7 | 2.0 | 305.0 | 9.6 | 5.0 | 328.9 | 11.5 | 7.5 | |||
| 4 | 348.3 | 285.6 | 8.7 | 2.0 | 347.5 | 13.1 | 10.0 | 400.0 | 18.2 | 13.5 | 630.1 | 18.2 | 16.5 |
| 5 | 334.9 | 313.9 | 9.3 | 4.0 | 334.9 | 15.3 | 12.0 | 379.4 | 18.2 | 14.0 | |||
| 6 | 407.3 | 330.7 | 8.7 | 2.0 | 406.1 | 10.1 | 6.0 | 472.8 | 11.5 | 8.0 | 485.9 | 13.8 | 11.0 |
| 7 | 367.1 | 333.9 | 8.9 | 3.0 | 367.6 | 10.1 | 6.0 | 451.0 | 13.1 | 14.0 | 751.3 | 16.6 | 17.0 |
| 8 | 423.0 | 562.1 | 9.6 | 5.0 | 596.4 | 11.5 | 8.0 | ||||||
| 9 | 434.1 | 303.7 | 8.7 | 1.8 | |||||||||
| 10 | 339.6 | 426.0 | 13.1 | 10.0 | 741.4 | 18.2 | 15.0 | ||||||
| 11 | 313.2 | 282.7 | 8.7 | 2.0 | |||||||||
| 12 | 286.5 | 266.8 | 9.6 | 4.5 | 286.0 | 11.5 | 7.5 | ||||||
| 349.9 ± 14.8 | 288.1 ± 11.1* | 8.9 ± 0.1§ | 2.5 ± 0.3 | 335.6 ± 15.5† | 12.0 ± 0.7§ | 8.3 ± 0.9 | 420.0 ± 23.5*†‡ | 14.6 ± 1.2† | 11.6 ± 1.4 | 684.1 ± 43.9 | 16.4 ± 1.0 | 15.4 ± 1.7 |
Applicable values are means ± SE. [Na+]i was assigned to each cell from the average [Na+]i curve (Fig. 2) at the time point when each cell APCL reached reference points following DG exposure (short APCL, back to base APCL, above base APCL, grossly dysrhythmic APCL). The 2-sample paired t-test for means was used to compare [Na+]i before and after DG exposure.
P < 0.05 vs. baseline;
P < 0.05 vs. short;
P < 0.05 vs. back to baseline via a linear mixed-random effects model to analyze APCL repeated measurements (with Bonferroni adjustments for P values);
P < 0.05 vs. baseline [Na+]i = 8.4 ± 0.4 mM. APCLs recorded during grossly dysrhythmic APCLs were excluded from statistical comparison (see details in Table 1).
Table 1.
AP waveform parameters in intact SANCs at baseline and during DG exposure
| Baseline (n = 12) | Short (n = 10)b | Back to Baseline (n = 8)c | Prolonged Above Baseline (n = 9)d | Grossly Dysrhythmica (n = 7)e | |
|---|---|---|---|---|---|
| After DG, min | 0 | 2.5 ± 0.3 | 8.3 ± 0.9 | 11.6 ± 1.4 | 15.4 ± 1.7 |
| APCL, ms | 349.9 ± 14.8 | 288.2 ± 11.1* | 335.6 ± 15.5† | 420.0 ± 24.0*†‡ | 684.0 ± 43.9 |
| MaxV, mV | 35.2 ± 2.1 | 32.8 ± 2.3 | 32.0 ± 3.0 | 26.3 ± 3.1* | 23.5 ± 3.7 |
| MDP, mV | −58.2 ± 1.4 | −55.0 ± 2.0 | −54.4 ± 2.7 | −47.8 ± 2.8*†‡ | −49.8.0 ± 1.0 |
| Amplitude, mV | 93.6 ± 2.7 | 87.8 ± 2.3 | 86.7 ± 4.1 | 74.1 ± 5.3*†‡ | 73.3 ± 3.3 |
| dV/dtmax, V/s | 10.3 ± 1.2 | 8.8 ± 0.8 | 8.7 ± 1.0 | 5.7 ± 1.2* | 4.8 ± 1.1 |
| APD90, ms | 185.9 ± 9.1 | 150.7 ± 6.0* | 166.4 ± 7.3† | 212.5 ± 13.2†‡ | 374.4 ± 17.4 |
| TNLDD, ms | 184.7 ± 15.0 | 132.2 ± 7.8* | 176.8 ± 12.4† | 252.8 ± 24.0*†‡ | 546.3 ± 68.7 |
| tMDP, ms | 150.2 ± 10.0 | 134.6 ± 7.8 | 139.5 ± 7.4 | 150.5 ± 7.8 | 182.8 ± 13.4 |
Values are means ± SE.
P < 0.05 vs. baseline;
P < 0.05 vs. short;
P < 0.05 vs. back to baseline.
Only relatively stable action potential (AP) cycle lengths (APCLs) were used for statistical comparison by a linear mixed-effects model to analyze repeated measurements (Bonferroni adjustment was made to the P values).
Parameters recorded during grossly dysrhythmic APCLs were excluded from statistical comparison.
This pattern of average APCL reduction in response to digoxigenin (DG) was observed in 10 out of 12 sinoatrial nodal cells (SANCs). Two other SANCs showed only a prolongation of APCL above baseline during the experiment.
Two of 10 SANCs in which APCL was initially reduced failed to achieve the pre-DG baseline, and their average APCL remained shorter than the original pre-DG baseline value during the remaining experiment.
Nine SANCs exhibited this behavior, 7 of 8 SANCs that previously had returned to pre-DG baseline and the 2 SANCs that exhibited only APCL prolongation during the experiment. One from 8 SANCs that reached the pre-DG baseline value failed to prolong above baseline during the remaining experimental time.
These marked grossly dysrhythmic APCLs were registered in 7 from 9 SANCs in which APCL was originally prolonged at earliest of DG response. In 2 other SANCs, APCL was prolonged but remained relatively rhythmic.
MDP, maximum diastolic potential; APD90, AP duration measured at 90% of repolarization time; TNLDD, time from MDP to the onset of nonlinear diastolic depolarization.
Achievement of the shortest APCL in a given cell was followed by gradual prolongation of APCL toward the original baseline in that cell (Fig. 3B). The time to return to the pre-DG baseline APCL varied among SANCs in which APCL initially became reduced in response to DG and was, on average, 8.3 ± 0.6 min after DG exposure (Table 2). At this time, when the original pre-DG average APCL was achieved, the average [Na+]i increased to 12.0 ± 0.7 mmol/l (Table 2), and the average MDP was −54.4 ± 2.7 mV (Table 1).
Following achievement of the pre-DG APCL baseline, APCL continued to prolong, and MDP became further reduced (Fig. 3, A and B). The average APCL increased by 70 ms above baseline (P < 0.05, Table 1), and average MDP decreased from −58.2 ± 1.4 mV at baseline to 47.8 ± 2.8 mV (P < 0.05, Table 1). At this time, the average [Na+]i increased to 14.6 ± 1.2 mmol/l (Table 2). The time to achieve this response to DG varied among SANCs; on average it was 11.6 ± 1.4 min (Table 2). Although there was significant prolongation in APCL (P < 0.05), gross arrhythmias were still not observed during this stage of the DG response.
As the time following DG exposure increased, the stage of prolonged but still relatively stable APCLs was followed by marked MDP depolarization and by gross prolongation of APCL (Figs. 3 and 4). On average, the time to reach this stage of the DG response varied among SANCs and was 15.4 ± 1.7 min (Table 2), and the average APCL at this stage became prolonged to 334.2 ms above baseline (P < 0.05, Table 1), the average MDP decreased further to 49.6 ± 1.1 mV (P < 0.05, Table 1), and the average [Na+]i increased to 15.4 ± 1.7 mmol/l.
AP waveform parameters.
In addition to APCL and MDP, other measured parameters of the AP waveform in response to DG varied over time (Table 1). Figure 4 illustrates the AP waveform of the cell presented in Fig. 3. When APCL achieved the pre-DG baseline level, the AP amplitude began to decrease (Fig. 4, green traces) and continued to decrease further (Fig. 4, blue and pink traces) as the time of DG exposure increased (Table 2). On average, the AP amplitude among cells was significantly reduced from 93.6 ± 2.7 mV at baseline to 74.1 ± 5.3 mV (P < 0.05, Table 1) as the time of DG exposure increased, on average, to 11.6 ± 1.4 min. Note that the biphasic change in APCL as the time in DG increased is paralleled by biphasic changes in the TNLDD and APD90 (Table 1). In contrast, the MDP, AP amplitude, AP upstroke (dV/dtmax), and AP overshoot (MaxV) monotonically decreased over time in response to DG. Thus the effects of DG on SANC automaticity included an initial reduction in APCL, MDP, and other AP waveform characteristics including TNLDD (Table 1). This phase was followed by gradual prolongation of APCL, TNLDD, APD90, and a progressive reduction of MDP, AP amplitude, MaxV, and dV/dtmax; grossly dysrhythmic APCL evolved in 7 out of 12 cells (Tables 1 and 2). These time-dependent effects of DG on APCL and MDP occurred commensurately with graded, time-dependent increases in [Na+]i (Fig. 2). Of note, there was substantial heterogeneity in the times to achieve hallmarks of the various stages of [Na+]i, [Ca2+]i, and APCL in response to DG.
APCLV in response to DG.
APCLV was calculated as a beat-to-beat variation in spontaneous APCLs during continuous AP recordings and reported as CV (%). Variability in the APCL, which informs on SANC clock coupling (52, 57), emerged during exposure to DG, even before the advent of grossly dysrhythmic APCL. The APCLV slightly decreased as APCL decreased in the cell in Fig. 3 during the early response to DG and then increased when APCL prolonged back to baseline and above baseline values with longer time of DG exposure (Fig. 3D). In the cell in Fig. 3, cycle-to-cycle APCLV at baseline and during exposure to DG are also revealed in a Poincaré plot (Fig. 3C). Specifically, in the Poincaré plot, in which each AP beating interval (APCL) is plotted against its predecessor, the scattering between two successive APCLs reflects the response to DG. Poincaré plots of the cell described in Fig. 3A revealed four different stages of changes over time following DG exposure; when the APCL became reduced following DG exposure, the dispersion among APCLs decreased (red markers in Fig. 3C); then, as the APCL prolonged back to pre-DG baseline, dispersion among APCLs then slightly increased (green markers in Fig. 3C) vs. baseline (black markers in Fig. 3C); as the APCL became prolonged above baseline as time to DG exposure increased, APCLV increased further (blue markers in Fig. 3C); when the APCL became grossly dysrhythmic, the dispersion of subsequent APCLs increased dramatically (pink markers in Fig. 3C, inset). The average APCLV among cells along with CV of other AP parameters at baseline and during the reference points following DG exposure, noted above, are listed in Table 3.
Table 3.
CV of AP waveform parameters
| Baseline (n = 12) | Short (n = 10)b | Back to Baseline (n = 8)c | Prolonged Above Baseline (n = 9)d | Grossly Dysrhythmica (n = 7)e | |
|---|---|---|---|---|---|
| After DG, min | 0 | 2.5 ± 0.3 | 8.3 ± 0.9 | 11.6 ± 1.4 | 15.4 ± 1.7 |
| APCL, % | 3.5 ± 0.3 | 3.1 ± 0.3 | 5.1 ± 1.4 | 8.8.1 ± 1.5*†‡ | 29.2 ± 2.9* |
| MaxV, % | 2.1 ± 0.4 | 2.7 ± 0.5 | 5.6 ± 3.2 | 10.5 ± 3.0*† | 37.4 ± 12.5* |
| MDP, % | 1.0 ± 0.1 | 1.7 ± 0.5 | 1.7 ± 0.5 | 3.8 ± 1.8* | 4.0 ± 0.7* |
| Amplitude, % | 1.2 ± 0.2 | 1.9 ± 0.4 | 2.5 ± 1.3 | 5.4 ± 1.7* | 10.3 ± 2.3* |
| dV/dtmax, % | 4.8 ± 0.7 | 5.7 ± 1.0 | 7.0 ± 3.0 | 11.9 ± 2.5* | 29.5 ± 6.1* |
| APD90, % | 5.2 ± 0.7 | 3.8 ± 0.4 | 4.8 ± 0.8 | 14.9 ± 4.1*†‡ | 72.3 ± 11.1* |
| TNLDD, % | 8.6 ± 0.7 | 10.3 ± 1.6 | 11.9 ± 2.0 | 33.8 ± 8.0*†‡ | 133.3 ± 21.8* |
| tMDP, % | 4.0 ± 0.2 | 4.5 ± 0.2 | 4.6 ± 0.4 | 7.4 ± 0.7*†‡ | 39.5 ± 16.5* |
Values are means ± SE. Coefficients of beat-to-beat variation (CVs, percent of change) of AP waveform parameters were compared by a linear mixed-random effects model for repeated measures with Bonferroni adjustment of the P values.
P < 0.05 vs. baseline;
P < 0.05 vs. short;
P < 0.05 vs. back to base.
CVs during grossly dysrhythmic APCLs were compared by paired t-test vs. baseline only.
This pattern of average APCL reduction in response to DG was observed in 10 out of 12 SANCs. Two other SANCs showed only a prolongation of APCL above baseline during the experiment.
Two of 10 SANCs in which APCL was initially reduced failed to achieve the pre-DG baseline, and their average APCL remained shorter than the original pre-DG baseline value during the remaining experiment.
Nine SANCs exhibited this behavior, 7 of 8 SANCs that previously had returned to pre-DG baseline and the 2 SANCs that exhibited only APCL prolongation during the experiment. One from 8 SANCs that reached the pre-DG baseline value failed to prolong above baseline during the remaining experimental time.
These marked grossly dysrhythmic APCLs were registered in 7 from 9 SANCs in which APCL was originally prolonged at earliest of DG response. In 2 other SANCs, APCL was prolonged but remained relatively rhythmic.
Strikingly noticeable is that, when the APCL became prolonged at an averaged time of 11.6 ± 1.4 min (Table 2), the CVs of all AP parameters were all significantly increased above baseline values (Table 3), even though APCLs at that point still were not grossly dysrhythmic (Figs. 3 and 4, blue plots).
Histogram distributions of all individual APCLs in all cells in response to DG are illustrated in Fig. 5. Note that the histogram distribution of APCLs (Fig. 5) shifts to the left of baseline when the average APCL is reduced; the distribution shifts back to baseline when the average pre-DG APCL is achieved; then, at longer times following DG exposure, the distribution of APCLs becomes skewed far off to the right, even in the absence of grossly dysrhythmic APCL; this distribution becomes very broad when grossly dysrhythmic APCLs occur.
Fig. 5.

Distributions of spontaneous APCLs over the time course of the DG response. Distribution of individual spontaneous APCLs, measured during continuous APCL recordings at baseline and at times as in Fig. 4 following DG (10 μmol/l) exposure (at shortest APCL, APCL prolongation back to baseline, APCL prolongation above baseline, and grossly dysrhythmic APCL) (n = 13,363 APCLs).
Diastolic Ca2+ and systolic Ca2+ transients.
Diastolic [Ca2+]i and peak [Ca2+]i and the AP-induced Ca2+ transient (peak [Ca2+]i-diastolic [Ca2+]i) were measured with the ratiometric Ca2+ indicator, Indo-1 AM. Figure 6A shows Indo-1 AM recordings in single SANCs at baseline (Fig. 6A, top) and during DG exposure (Fig. 6A, middle and bottom). Average data for diastolic and peak [Ca2+]i and their differences (the Ca2+ transient) measured at baseline and at 5-min intervals following DG exposure are illustrated in Fig. 6B and listed, along with other parameters of Ca2+ transient, in Table 4. The average diastolic [Ca2+]i, AP-induced peak [Ca2+]i, and Ca2+ transient amplitude (peak-diastolic) increased as a function of time following exposure to DG. The rates at which these increases occurred following exposure to DG varied among cells, similar to the time-dependent increase in [Na+]i. The time controls for the data in Table 4 did not significantly change (Table 5).
Fig. 6.

Spontaneous AP-induced Ca2+ transients, systolic and diastolic [Ca2+]i measured by Indo-1 in spontaneously beating SANCs in the presence of DG. A: representative examples of AP-induced Ca2+ transients recorded in normal Tyrode solution by Indo-1 at baseline (top), and at 3 and 10 min following DG exposure (middle and bottom, respectively). B: average changes in the amplitude of systolic, diastolic [Ca2+]i, and AP-induced Ca2+ transient at baseline and during a 20-min superfusion with 10 μmol/l DG (P < 0.05 vs. baseline via a linear mixed-effects model with a Dunnett's post hoc test for the P values; n = 5).
Table 4.
Average characteristics of spontaneous AP-induced Ca2+ transients, systolic and diastolic [Ca2+]i measured by Indo-1 in the presence of DG (10 μmol/l)
| Time in DG, min | 0 | 1 | 3 | 5 | 10 | 15 | 20 |
|---|---|---|---|---|---|---|---|
| Systolic Ca2+, nM | 293.2 ± 19.4 | 331.9 ± 20.1 | 368.6 ± 23.2* | 398.6 ± 18.0* | 410.4 ± 31.0* | 412.6 ± 25.6* | 423.4 ± 21.0* |
| Diastolic Ca2+, nM | 124.3 ± 8.9 | 128.8 ± 10.9 | 139.2 ± 12.7* | 149.2 ± 11.5* | 157.7 ± 11.2* | 161.1 ± 12.9* | 161.8 ± 11.6* |
| Ca2+ transient, nM | 168.9 ± 22.6 | 203.1 ± 16.1 | 229.3 ± 15.2 | 249.4 ± 16.7* | 252.5 ± 30.1* | 251.5 ± 28.0* | 261.6 ± 23.0* |
| T-Pc, ms | 116.1 ± 9.4 | 98.3 ± 5.1 | 109.1 ± 3.8 | 107.2 ± 9.9 | 111.3 ± 5.3 | 110.5 ± 6.8 | 136 ± 18.6 |
| T-90c, ms | 287.2 ± 16.1 | 262.6 ± 25.7 | 295.4 ± 18.1 | 301.7 ± 19.6 | 320.5 ± 13.4 | 333.0 ± 16.5* | 365.2 ± 19.1* |
Values are means ± SE. A linear mixed-effects model with a Dunnett's post hoc adjustment to the P value was used to compare each time point in DG with the baseline (0 min). T-Pc, time to peak; T-90c, time to decay.
P < 0.05 vs. baseline (n = 5).
Table 5.
Average characteristics of spontaneous AP-induced Ca2+ transients, systolic and diastolic [Ca2+]i measured by Indo-1 in time control cells in the absence of DG
| Time, min | 0 | 1 | 3 | 5 | 10 | 15 | 20 |
|---|---|---|---|---|---|---|---|
| Systolic Ca2+, nM | 311.5 ± 26.2 | 317.2 ± 23.6 | 323.9 ± 38.1 | 331.5 ± 24.7 | 324.0 ± 30.7 | 334.0 ± 27.7 | 331.2 ± 28.1 |
| Diastolic Ca2+, nM | 126.2 ± 8.0 | 126.7 ± 7.1 | 126.6 ± 5.8 | 127.3 ± 7.4 | 130.0 ± 7.6 | 131.3 ± 8.8 | 135.3 ± 8.3 |
| Ca2+ transient, nM | 197.2 ± 13.3 | 209.6 ± 16.6 | 202.3 ± 18.3 | 196.2 ± 16.6 | 200.0 ± 15.1 | 207.8 ± 16.2 | 204.5 ± 15.0 |
| T-Pc, ms | 107 ± 7.9 | 105 ± 10.4 | 106.9 ± 5.2 | 100.9 ± 4.2 | 108.2 ± 2.8 | 107.6 ± 8.0 | 114.4 ± 6.4 |
| T-90c, ms | 291.8 ± 18.4 | 291.5 ± 21.9 | 316.3 ± 24.1 | 292.2 ± 28.6 | 302.3 ± 20.3 | 317.8 ± 23.1 | 281.5 ± 22.5 |
Values are means ± SE. Time controls were recorded in different cells (n = 5), P > 0.05.
Confocal imaging of spontaneous local diastolic ryanodine receptor (RyR) activation was measured as LCRs in SANCs with intact sarcolemmal function. Figure 7, left, illustrates representative examples of confocal line-scan images of SANCs loaded with the Ca2+-sensitive fluorescent dye fluo-4 AM at baseline and at different times following DG exposure. Note that, unlike the APCL, which can be recorded continuously during DG exposure, recordings of fluo-4 fluorescence cannot be continuous because of bleaching of the probe. In other words, short recording of LCRs (asterisks in Fig. 7, A–C) and AP-induced Ca2+ transients were necessary. The times at which we recorded LCRs and AP-induced Ca2+ transients were guided by the average times to achieve reference points following DG exposure during continuous AP recordings (Table 2).
Fig. 7.

Effect of DG on spontaneous local diastolic Ca2+ releases (LCRs) in intact, spontaneously beating SANCs. A–C: representative examples of confocal line-scan images of SANCs loaded with the Ca2+-sensitive fluorescent dye fluo-4 AM (10 μmol/l) in the same cell at baseline and in response to DG (10 μmol/l) at 2 min (B) and 8 min (C). LCRs are marked with asterisks. D and E: the average change (% of baseline) of AP-induced Ca2+ transient cycle length, a faithful proxy of the APCL, and LCR period at baseline and at time points shown in A of DG (10–20 μmol/l) response. F–J: average change (% of baseline) of LCR characteristics at baseline (n = 16 SANCs) and at time points shown in A of DG response (in 7 of 16 SANCs, both reduction and prolongation of APCL were recorded in response to DG; 6 of 16 SANCs exhibited only reduced APCL; 3 of 16 SANCs showed only prolonged APCL in response to DG during the experimental time; *P < 0.05 via linear mixed-effects model for repeated measurements with Bonferroni adjustment for the P values).
Figure 7, right, illustrates the average change in the spontaneous AP-induced Ca2+ transient cycle length, the faithful proxy of the APCL (57), LCR period, and other LCR characteristics, measured in SANCs loaded with the Ca2+ indicator, fluo-4 AM, at baseline and during DG exposure. Responses of the spontaneous AP-induced Ca2+ transient cycle length (Fig. 7E) to DG were similar to those described above for APCL (Table 1). On average, the LCR period (Fig. 7D, Table 6) was reduced by 20.0% at 3.0 ± 0.4 min following DG exposure, from 307.2 ± 15.0 ms at baseline to 251.3 ± 14.2 ms (P < 0.05). The average spontaneous AP-induced Ca2+ transient cycle length (Fig. 7E, Table 6) at this time was reduced by 17.7%, from 364.6 ± 15.1 ms at baseline to 309.7 ± 15.7 ms (P < 0.05). At longer (9.5 ± 2.3 min) times of DG exposure, the average spontaneous AP-induced Ca2+ transient cycle length (Fig. 7E, Table 6) prolonged above its baseline by 33.8%, from 364.6 ± 15.1 ms at baseline to 464.8 ± 49.7 ms (P < 0.05). The average LCR period (Fig. 7D, Table 6) at this time was increased by 28.2% above its baseline, from 307.2 ± 15.0 ms at baseline to 373.7 ± 34.7 ms (P < 0.05).
Table 6.
Changes in LCR period and APCL in each individual SANC, measured by confocal microscopy at baseline and during DG exposure
| Cell No. | Baseline LCR Period, ms | APCL, ms | Short LCR Period, ms | APCL, ms | Time in DG, min | Prolonged LCR Period, ms | APCL, ms | Time in DG, min |
|---|---|---|---|---|---|---|---|---|
| 1 | 242.6 | 291.0 | 207.9 | 241.1 | 1 | 264.7 | 335.8 | 5 |
| 2 | 337.7 | 391.0 | 230.6 | 271.0 | 2 | 544.4 | 582.1 | 11 |
| 3 | 449.1 | 490.6 | 294.2 | 336.0 | 1 | 558.0 | 660.9 | 4 |
| 4 | 315.4 | 371.2 | 194.0 | 233.0 | 3 | |||
| 5 | 283.6 | 323.0 | 344.9 | 433.0 | 4 | |||
| 6 | 338.3 | 400.4 | 250.7 | 345.6 | 3 | |||
| 7 | 326.6 | 405.1 | 284.8 | 330.5 | 5 | |||
| 8 | 371.2 | 427.1 | 333.5 | 388.3 | 3 | |||
| 9 | 256.2 | 292.0 | 261.6 | 306.2 | 7 | |||
| 10 | 268.2 | 334.1 | 260.1 | 321.9 | 4 | 282.2 | 344.8 | 6 |
| 11 | 227.6 | 289.9 | 305.7 | 327.0 | 4 | |||
| 12 | 219.8 | 291.7 | 200.4 | 270.9 | 3 | 455.4 | 769.9 | 20 |
| 13 | 314.9 | 388.8 | 291.8 | 365.1 | 3 | 352.8 | 443.5 | 9 |
| 14 | 348.6 | 417.5 | 305.0 | 394.4 | 4 | 367.0 | 444.5 | 25 |
| 15 | 272.6 | 319.8 | 257.5 | 294.7 | 2 | |||
| 16 | 343.0 | 401.1 | 156.4 | 233.4 | 5 | |||
| 307.2 ± 15.0 | 364.6 ± 15.1 | 251.±14.2* | 309.7 ± 15.7* | 3 ± 0.4 | 373.7 ± 34.7† | 464.8 ± 40.7† | 9.5 ± 2.3 |
Applicable values are means ± SE. Time was in minutes to achieve different stages of DG response.
P < 0.05 vs. baseline;
P < 0.05 vs. baseline and short via linear mixed-effects model to analyze repeated measurements (Bonferroni adjustment was made to the P values).
LCR, local diastolic Ca2+ release.
The average changes in other LCR parameters (size, duration, and amplitude of the Ca2+ signal of individual LCRs and of the LCR ensemble) at the aforementioned times during DG exposure are illustrated in Fig. 7, right. When the average spontaneous AP-induced Ca2+ transient cycle length and LCR period became reduced following DG exposure, as LCR size and LCR duration became increased (Fig. 7, F and G; P < 0.05), the Ca2+ signal of individual LCRs and the Ca2+ signal of LCR ensemble became larger (Fig. 7, H and I, P < 0.05). At longer times of DG exposure (9.5 ± 2.3 min), when the average LCR size decreased and the Ca2+ signal of individual LCRs and the Ca2+ signal of LCR ensemble became reduced, the spontaneous AP-induced Ca2+ transient cycle length and LCR period became prolonged. The average number of LCRs per cell (Fig. 7J) at this time decreased by about 40%, from 8.4 ± 0.3 s/100 μm at baseline to 4.7 ± 1.1 s/100 μm (P < 0.05), and LCRs became almost undetectable because of an increase of diastolic [Ca2+]i (Tables 4 and 5).
Figure 8A illustrates histogram distributions of APCLs (taken as the intervals between the peaks of 2 adjacent AP-triggered Ca2+ transients), and Fig. 8B illustrates histograms of the LCR period (measured as the time between the rapid upstroke of the prior AP-triggered Ca2+ transient and the onset of an LCR in diastole), measured in SANCs loaded with the Ca2+ indicator, fluo-4 AM, when APCL was reduced and prolonged in response to DG. Note that the histogram distributions of the LCR period and APCLs exhibited similar patterns in response to DG as distribution of APCL during continuous AP recordings (Fig. 5). Specifically, histograms of both the LCR period and APCLs when the average APCL and LCR period became reduced are skewed to the left of baseline and then shifted to the right when the average APCL and LCR period became prolonged with longer time of DG exposure.
Fig. 8.

Relationship and histograms of APCLs and LCR period at baseline and during DG exposure in intact SANCs by confocal microscopy. A and B: distributions of APCLs and LCR period in SANCs loaded with the Ca2+ indicator, fluo-4 AM (10 μmol/l), at baseline and times of DG exposure as in Fig. 7 (n = 347 events from n = ∼10–16 SANCs). C: relationship between individual LCR periods (measured as the time between the rapid upstroke of the prior AP-triggered Ca2+ transient and the onset of an LCR in diastole) and APCL (taken as the interval between the peaks of 2 adjacent AP-triggered Ca2+ transients) in SANCs loaded with the Ca2+ indicator, fluo-4 AM, at baseline and during DG exposure conforms to a line of identity (y = 0.9x + 73.1; r2 = 0.8). D: relationship between the average APCL and the average LCR period in each cell for data shown in A and B. Data shown as a percent change of baseline (y = 1.2x − 11.6; r2 = 0.9).
Relationship of DG-induced changes in the LCR period and changes in APCL.
Figure 8C illustrates all individual APCLs plotted against all LCR periods (n = 347 events) at baseline and at the time when APCL was reduced and prolonged during DG exposure. Figure 8D illustrates the relationship of the average APCL (percent change) to the average LCR period (percent change) among SANCs (n = ∼10–16 SANCs for each data point) during these times of DG exposure. As has been reported previously for a wide range of interventions that alter APCL (45, 51), a single monotonic function subtends all the points at baseline and during DG exposure even though the AP-induced Ca2+-transient cycle lengths and LCR periods exhibited biphasic changes from baseline, an early reduction followed by prolongation.
LCR characteristics in permeabilized SANCs.
To test whether DG has a direct effect on SR Ca2+ cycling that generates LCRs in the absence of Na+/K+ pump function, and in the absence of other surface membrane channel functions, we permeabilized individual SANCs with saponin and recorded LCRs in a physiological buffer with a free [Ca2+]i of 100 nmol/l. Unlike intact SANCs (Fig. 7), following exposure to DG in permeabilized SANCs, there was no significant change in LCR characteristics (data not shown), as shown recently in permeabilized ventricular myocytes (1).
Numerical model simulations.
To get mechanistic insights into how [Na+]i relegates the pacemaker cell system, we performed numerical model simulations within a broad [Na+]i range from 6–14 mmol/l that included the landmarks of APCL changes, following DG exposure (as discussed above). It is important to note that we did not simulate [Na+]i changes; rather [Na+]i was an independent parameter in the model. Model simulations were performed at each given [Na+]i via a slow [Na+]i ramp clamp (0.075 mM/s). The slow ramp allowed the model to attain steady state for each [Na+]i. We also tested slower ramps, as slow as 0.01 mm/s, i.e., 0.6 mm/min, similar to the experimentally measured rate of [Na+]i change, but this did not change the results. Thus we performed numerical modeling to simulate how Na+-induced changes in cytosolic Ca2+, LCRs, and MDP regulate the APCL, i.e., SANC automaticity.
It is important to note that, although DG inhibits INaK, this is a minor factor in model simulations. Rather it is the DG-induced [Na+]i rise that determines the model behavior observed experimentally. In our model equations, we have reduced the density of INaK in the presence of DG to 37.5% of control (parameter INaKmax from 2.88 to 1.08 pA/pF), in line with experimental data of INaK blockade by DG within the range of 24% to 38% reported previously for atrial responses at 36.5°C (49). Although our results are presented for the 37.5% of the initial INaKmax, we also tested model behavior with stronger INaKmax inhibition (to 25% of the initial value), but this did not change the results.
The model reproduced experimental results.
Model stimulations reproduced the biphasic average APCL changes (Fig. 9A, blue plot) observed experimentally (Fig. 3), as [Na+]i increased within the experimentally measured continuum of [Na+]i (Fig. 2). Examples of the APCL change over the continuum of [Na+]i are illustrated in Fig. 9, C–F. The biphasic effect consisted of APCL reduction from 335 ms at 8 mmol/l [Na+]i (at baseline, before DG, Fig. 9C) to a minimum of 300 ms at an [Na+]i of 11.3 mmol/l (Fig. 9D); this was followed by an increase in APCL to return to the baseline level of 335 ms at 12.8 mmol/l (Fig. 9E). As [Na+]i increased further, the simulated APCL prolonged above baseline (Fig. 9F); the AP firing subsequently became dysrhythmic and ceased as [Na+]i increased higher than 13.2 mmol/l (Fig. 9A, blue plot), in close agreement with the experimental data (Figs. 3 and 4).
Fig. 9.

Numerical model simulations reproduce experimental effects of the DG-induced increase in [Na+]i on SANC APCLs and Ca2+ transients. These effects occur concomitantly with a slow rise in [Na+]i from 6 mmol/l to 14 mmol/l on the key system characteristics. A: APCL (blue plot) and MDP (dark red plot). B: systolic and diastolic cytosolic Ca2+ concentration (Cacyt, red and blue plot, respectively). C–F: representative APs (Vm) illustrated for 4 different [Na+]i within the entire simulation range: baseline, minimum APCL, APCL prolongation back to baseline, and APCL prolongation above baseline. G–L: cytosolic Ca2+ transients (Cacyt) selected at reference points noted above for Vm.
Our model simulations also reproduced MDP changes observed experimentally (Fig. 3B); as [Na+]i increased from baseline to 13 mmol/l, the MDP became progressively depolarized from −61.7 mV to −50.0 mV (Fig. 9A, dark red plot). Also, the model simulations reproduced experimentally observed changes of Ca2+ transients; the peak of the systolic Ca2+ signal amplitude (dark red curve in Fig. 9B) and diastolic [Ca2+]i levels both increased (blue curve in Fig. 9B and simulated cytosolic Ca2+ transients in Fig. 9, G–L) as [Na+]i increased, similar to that measured with Indo-1 (Fig. 6).
Insights via numerical simulations of electro-driving forces.
As we increased [Na+]i in the model, MDP depolarized and intracellular levels of [Na+]i and [Ca2+]i increased. These factors ought to influence Na+ and Ca2+ equilibrium potentials ENa = (RT/F)ln[Na+]o/[Na+]i and ECa = (RT/F)ln[Ca2+]o/[Ca2+]subspace and, importantly, their respective electrochemical driving forces Vm − ENa, Vm − ECa. The driving forces for Na+ and Ca2+ obviously regulate respective selective Ca2+ and Na+ ion currents. We performed numerical model simulations of the driving forces to evaluate the scale of this regulation (Fig. 10A), taking advantage of model predictions of Ca2+ in the submembrane space (i.e., the space where Ca2+ is released by RyR and interacts with the NCX) (Fig. 11F), rather than using bulk cytosolic [Ca2+]i measured experimentally by Indo-1 (Fig. 6).
Fig. 10.
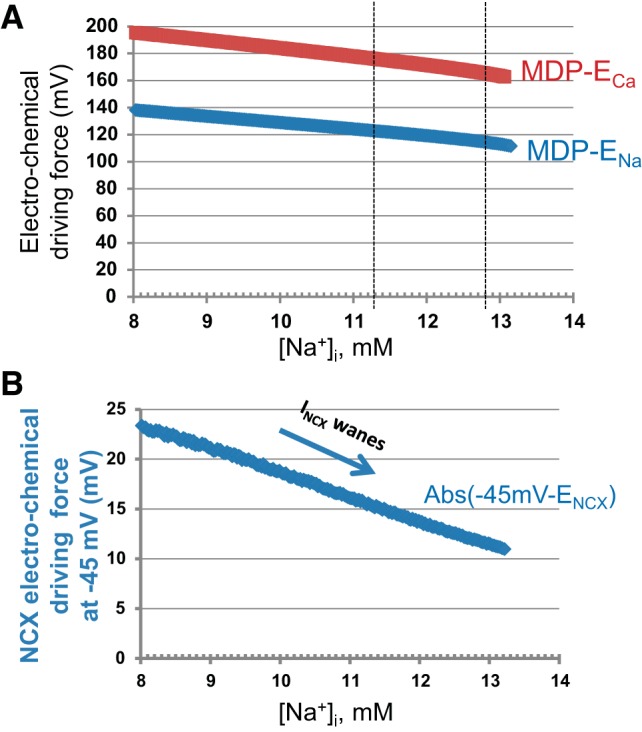
Model simulations of the electro-chemical driving forces in response to increase in [Na+]i. A: changes of the electro-chemical driving force for Ca2+ (MDP − ECa, red plot) and Na+ (MDP − ENa, blue plot). B: changes of the electro-chemical driving force for INCX at −45 mV (−45 mV − ENCX).
Fig. 11.

Numerical model simulations provide mechanistic insights into INCX biphasic change that underlie the biphasic regulation of AP firing rate with [Na+]i rising from 8 to 14 mmol/l. A: INCX amplitude changes at a key diastolic membrane potential of −45 mV, as a function of [Na+]i. B: representative INCX waveforms at baseline, minimum APCL, APCL prolongation back to baseline, and APCL prolongation above baseline. C: illustration of the positive INCX regulation by submembrane Ca2+ (i.e., by ensemble of LCR Ca2+ signal). D–F: representative examples of simulated waveforms of free sarcoplasmic reticulum (SR) Ca2+ load, SR Ca2+ release flux, and submembrane Ca2+ concentration directly interacting with NCX. Colors representing different [Na+]i match colors in Fig. 9. The overlapped traces are synchronized at the AP overshoot (vertical dashed lines).
As [Na+]i increases, the shifts in ENa and ECa, combined with MDP depolarization, indeed reduce Na+ and Ca2+ driving forces (Fig. 10A). However, the scale of the changes in each driving force alone is rather small, remaining within about 15% over 8.0–13.2 mmol/l of [Na+]i increase. Thus it is difficult to expect a substantial regulation of a selective calcium current (ICaL) or a selective sodium current (IbNa) via just its driving force decrease alone.
On the other hand, ENCX, the reversal potential for INCX, is determined, not just by either driving force alone, but by the difference between Ca2+ and Na+ gradients (2); ENCX = 3ENa − 2ECa. This ion-driving mechanism makes a crucial exceptional difference for how INCX changes in response to DG compared with other ion currents. A crucial submembrane Ca2+ level is that attained at the ICaL activation threshold before the AP upstroke (at about −45 mV). Thus, when these factors are taken into consideration in calculating the change in the driving force for NCX at −45 mV over the range of [Na+]i from 8.0 to 13.2 mM, the change in ENCX is indeed substantially decreasing, by more than 50% (from 23.3 to 10.9 mV, Fig. 10B).
Further insights from numerical simulations of ion currents and Ca2+ dynamics.
Additional simulations provided insights for Na+ regulation of intrinsic Ca2+-, voltage-, and time-dependent ion current changes during AP firing that cannot be accessed experimentally (Figs. 11 and 12).
Fig. 12.

Insights gleaned using numerical model simulations on Na+ regulation of the cell membrane ion currents (via direct and indirect effects). Plots of continuous membrane current changes (left) for over the entire range of [Na+]i from 8 to 14 mmol/l illustrated with their representative waveforms (right) at the 4 key [Na+]i. Colors representing different [Na+]i match colors in Fig. 9. The overlapped traces are synchronized at the AP overshoot (vertical dashed lines). A: L-type Ca2+ current (ICaL). B: T-type Ca2+ current (ICaT). C: rapid delayed rectifier K+ current (IKr). D: funny current (If). E: Na+ background current (IbNa). F: Na+/K+ ATPase current (INaK, remaining after INaK inhibition by DG).
The simulated changes of INCX during diastole (at −45 mV, Fig. 11, A and B) showed the same biphasic behavior as for experimentally observed and model-simulated APCL. As [Na+]i increased from 8.0 to 10.5 mmol/l, the magnitude of the diastolic INCX in the model simulations increased and then decreased as [Na+]i continued to increase further to 13.2 mmol/l (Fig. 11B). The biphasic change in INCX occurs because of contributions of the two opposing forces; an increase in Casub amplifies INCX (Fig. 11C), but the concurrent increase in [Na+]i reduces INCX via a decrease in the NCX driving force (Vm − ENCX) (Fig. 10B). The Na+-induced increase in Casub in the model (Fig. 11F) is linked to a larger SR Ca2+ load (Fig. 11D), resulting in a larger diastolic SR Ca2+ release flux (jCarel) (Fig. 11E), reflecting LCR ensemble Ca2+ signal (Fig. 7I). Furthermore, that the average LCR period is initially reduced in the model simulations (blue curve, Fig. 11E) is in good agreement with our experimental results, which demonstrate that the LCR period becomes initially reduced following DG exposure (Figs. 7 and 8, Table 6). As [Na+]i increased further, diastolic jCarel activated earlier, but its time course during DD (i.e., before AP-induced transient) became substantially prolonged, resulting in the integrated release occurring later in time, which was in line with the prolonged LCR period found experimentally at a longer time of DG exposure (Figs. 7 and 8).
The simulated peak l-type Ca2+ current (ICaL) substantially decreases (by a factor of about 3) as [Na+]i increases (Fig. 12A), attributable, in part, to the concomitant collapse of the Ca2+-driving force (dark red curve in Fig. 10A). The major factor of ICaL decline, however, is linked to the Ca2+-induced Ca2+ inactivation as submembrane Ca2+ rises (Casub, Fig. 11F). Specifically, the respective scaling factor (fCa) of ICaL at the time of the ICaL peak is 0.50 at 8 mmol/l [Na+]i, but it is reduced to 0.24 at 13 mmol/l [Na+]i. Both T-type Ca current (ICaT) and the rapid delayed rectifier current (IKr) also decrease (Fig. 12, B and C) as MDP depolarizes, and AP amplitude concomitantly collapses. Both If and IbNa (Fig. 12, D and E) decrease because of the reduction in the Na+-driving force (Vm − ENa, Fig. 10A). However, the reduction of If is much greater than that of IbNa because of If voltage-dependent kinetics; as MDP depolarizes (dark red plot in Fig. 9A), If voltage-dependent activation is concomitantly reduced (because If is activated by membrane hyperpolarization). The amplitude of If, however, is determined by the square of the If gating variable y in our model. Capturing this factor in our simulations, we found that this specific y2 factor (at its peak) is substantially reduced in the model from 1.7% to 1.1% at 8 and 13 mmol/l [Na+]i, respectively, indicating its major importance for the If decline in addition to the If decrease attributable to change in the driving force for Na+. The Na+/K+-ATPase (INaK) outward current (partially inhibited by DG) monotonically increased as [Na+]i increased (Fig. 12F).
DISCUSSION
Using a combination of experimental and numerical modeling approaches, we determined how the coupling of Na+ and Ca2+, electrochemical gradients within the pacemaker cell coupled-clock system, regulates automaticity of single SANCs. We measured responses of [Na+]i, [Ca2+]i, membrane potential, and APCL and rhythm in rabbit SANCs to Na+/K+ pump inhibition by the digitalis glycoside, DG.
Our results indicate that even a small increase in [Na+]i perturbs SANC automaticity via impacting numerous coupled-clock mechanisms that include, not only direct [Na+]i and [Ca2+]i changes (i.e., their gradients and respective driving forces), but also complex indirect effects attributable to clock coupling involving SR and sarcolemmal mechanisms. Specifically, DG, acting directly on the sarcolemmal Na+/K+ pump, not only produces a progressive increase in [Na+]i and MDP depolarization, but also indirectly perturbs Ca2+ clock functionality (reflected in LCR changes) and induces alterations in ICaL, ICaT, If, INCX, INaK, IbNa, and IKr, which result from the ensemble of the aforementioned DG effects.
Response to DG when [Na+]i minimally increases.
A reduction in APCL is the initial response to DG that emerges in the context of a minimal [Na+]i gain. The average 18% (P < 0.05) reduction in APCL in rabbit SANCs in response to DG occurs when [Na+]i increases (on average) only by 5% (P < 0.05). This effect is indicative of high gain of [Na+]i regulation of SANC automaticity as in ventricular myocytes (3, 11, 21, 22, 34). The average amplitude of Ca2+ transient increases, accompanied by the slight increase of diastolic [Ca2+]i. We interpreted the acceleration of AP firing during the initial response to DG to be an extension of normal automaticity, i.e., a positive chronotropic response, analogous to the positive inotropic response to the small increase in [Na+]i that alters SR Ca2+ loading in ventricular myocytes (5, 21, 22). In addition to a reduction of the average APCL that accompanied the initial small increase in [Na+]i from 8.4 ± 0.4 mmol/l at baseline to 8.9 ± 0.1 mmol/l in the present study during early DG responses, the LCR period becomes reduced, and the average LCR size, the Ca2+ signal of the LCR ensemble, becomes increased. Furthermore, acceleration of DD occurs earlier during this stage of DG response, manifested by a reduced time to nonlinear DD part (Table 1, parameter TNLDD).
The early reduction in APCL in response to DG (Fig. 3) was strongly correlated with the reduction in the LCR period (Figs. 7 and 8). We interpret the reductions in APCL and the LCR period, the increase in LCR size and Ca2+ signal of LCR ensemble, and changes in other AP waveform parameters to indicate an increase in coupling of clock molecules that comprise the coupled-clock system (18, 26, 50). Because of clock coupling, direct and specific effects (pharmacological or genetic) on any single molecule of either clock inevitably impact the functions of all molecules that regulate Vm, ENa, and ECa within the coupled-clock system. For example, the prolongation of the spontaneous SANC APCL by ivabradine, a specific If (funny current) blocker, had been exclusively attributed, in early studies, to its direct reduction in If (6, 53). More recent studies, however, clearly indicate that, because the ivabradine-induced APCL prolongation induced [Ca2+]i reduction, ivabradine effects on APCL also indirectly involve a reduction in the Ca2+ clock function; thus the net APCL prolongation by ivabradine reflects a reduction in clock coupling (54). Therefore, the ivabradine effect on SANC APCL extends far beyond its direct effect on surface membrane ion channels, like DG in the present study.
Our experiments in permeabilized SANC bathed in physiological free [Ca2+]i of 100 nmol/l demonstrated that DG was without significant effect on LCR characteristics, as shown recently in ventricular myocytes (1). Thus DG effects on the LCR period and APCL are due to a direct effect of Na+/K+ pump inhibition that induces an increase in [Na+]i and reduction Ca2+ extrusion via NCX and therefore to changes in [Na+]i and [Ca2+]i.
The initial reduction in APCL to DG observed experimentally is predicted by our simulations of a coupled-clock pacemaker model (Fig. 9D) (26). The initial increases in [Na+]i and [Ca2+]i accelerate the kinetics of SR Ca2+ cycling, leading to a reduction in the average period of spontaneous LCRs, an increase in the amplitude of the ensemble of LCR Ca2+ signal, which occurs at an early time, leading to early and stronger INCX (Fig. 11B, blue plots), and to a reduction in the APCL. This is in good agreement with our experimental results (Fig. 7, D, E, and I). It is important to note that the APCL reduction happens despite a decrease in all major currents, including INCX, as ECa, ENa, and ENCX and their respective driving forces all decrease, indicating the complex nature of this APCL reduction effect, in which diastolic LCRs play the major role.
Please note the occurrence of these changes that accompanied a minimal increase in [Na+]i by DG is also a manifestation of a tight clock coupling. The model simulation of [Ca2+]i and other parameters contrasts strikingly with a prior simulation of a pacemaker function, in which an increase in [Na+]i to 15 mM was required for [Ca2+]i to impact on automaticity (27), which was interpreted to indicate that [Ca2+]i is implicated in a dysrhythmic behavior. Thus [Ca2+]i is implicated only in abnormal automaticity.
Transition from initial APCL reduction to APCL prolongation.
The initial experimentally measured reduction of the average APCL was followed by APCL prolongation back to baseline values, which was accompanied by an [Na+]i increase to 12.0 ± 0.7 mmol/l (42%). The subsequent APCL prolongation above baseline to 420.0 ± 24.0 ms was accompanied by a further increase in [Na+]i to 14.6 ± 1.2 mmol/l (73%) and a marked (2.5-fold) increase in APCLV (Table 3). The average LCR period became prolonged and less synchronized, and LCR occurrence became reduced (Figs. 7 and 8). As has been reported previously for a wide range of interventions that alter APCL (45, 51), a single monotonic function subtends all the points at baseline and during DG exposure, even though the APCLs and LCR periods exhibited biphasic changes from baseline, an early reduction followed by prolongation. Numerous prior studies have demonstrated that changes in APCL in response to different perturbations are linked to concurrent changes in LCR periods (i.e., times from the prior AP-induced Ca2+ transient upstrokes to the subsequent diastolic LCR occurrence) and in other LCR characteristics (18, 19, 54). The effects of DG, however, on the AP-induced Ca2+ transient cycle length and spontaneous LCRs during DD have not been described previously. As the LCR ensemble Ca2+ signal wanes and is delayed (Fig. 11E), the decrease in driving force for INCX (i.e., Vm − ENCX) (Fig. 10B) becomes the dominating factor in APCL prolongation (Fig. 9A).
We interpreted the gradual prolongation of the average APCL and LCR period, the reduction in LCR size and Ca2+ signal of LCR ensemble, the prolongation of the average TNLDD and the MDP depolarization, increase in the variability of APCL and AP waveform parameters, and a further reduction in ENa − ECa that accompanies [Na+]i and [Ca2+]i accumulations to reflect a reduction in coupling of clock molecules within the coupled-clock system.
Grossly dysrhythmic APCLs as [Na+]i and [Ca2+]i further increase, and MDP markedly depolarizes.
As the average [Na+]i increased to 15.4 ± 1.7 mmol/l, the MDP became markedly reduced, APCLs became grossly dysrhythmic (684.0 ± 43.9 ms), APCLV increased to 8.3-fold above baseline (Table 3), and LCR became almost undetectable as diastolic [Ca2+]i increased (Figs. 6 and 11F, Table 4). We interpreted this gross arrhythmic APCL stage of the response to DG to reflect marked clock uncoupling (41). The steep relationships between the degree of clock coupling and increase in [Na+]i and reduction in ENa − ECa (or change in 3ENa − 2ECa) would appear to be an underpinning of the well-recognized narrow therapeutic:toxic ratio of DG compounds (31, 37).
Stereotyped responses of excitable cardiac tissue and cells to DG.
The pattern of APCL responses of single SANCs in the present study over a range of increases of [Na+]i and [Ca2+]i MDP depolarization, and a progressive reduction in ENa and ECa over time following DG exposure, bears a striking resemblance to that measured in intact sinus node (SAN) tissue in response to a high DG concentration (39, 41). Specifically, in isolated SAN preparation, a response to a high DG concentration includes an initial reduction in APCL followed by a APCL prolongation and, with further time, marked arrhythmic behavior (DG toxicity emerges) (39, 41). In studies in SAN preparations in which incremental concentrations of DG rather than a single high DG dose are employed, the early reduction in APCL is not detected, i.e., only APCL prolongation is observed (32); in contrast, in single atrioventricular nodal cells in which a single DG dose was significantly higher than in the present study, only APCL reduction was detected followed by cell contracture at a longer time of DG exposure (13). Although, in these studies in multicellular SAN preparations, changes of [Na+]i were not monitored and APCL changes over time were not related to [Na+]i, ENa − ECa, or ENCX, the biphasic response to DG was observed. In the present study in single SANCs, the time course of [Na+]i and [Ca2+]i gain, MDP depolarization, and biphasic changes in APCL following DG exposure was heterogeneous among cells. However, because similar time-dependent changes in response to DG occur both in SAN tissue and single, isolated SANCs in the present study, this indicates that the pattern of changes in APCL is not an artifact related to isolation of SANCs from SAN tissue.
DG-induced heterogeneity of excitability in intact SAN tissue has been implicated in DG-induced SAN dysrhythmias (39, 41). The present study also discovered AP cycle variability, not only in APCL, but also in other parameters of the AP waveform (Table 3). The cycle-to-cycle variability within individual SANCs residing within SAN tissue may be implicated in DG-induced dysrhythmia in SAN tissue. The pattern of APCL changes over time as [Na+]i increases in the present study following DG exposure also mimics the contractile responses of the intact heart, ventricular tissue, or isolated ventricular myocytes (VM); an increase in AP-induced phasic contraction initially occurs in response to small increase in [Na+]i, followed by reduction in contraction (17, 34). As [Na+]i and [Ca2+]i continue to increase, and at longer times of exposure to DG as higher [Na+]i and [Ca2+]i levels are achieved, the amplitude of phasic contractions declines, spontaneous diastolic Ca2+ oscillations emerge, their summation elevates diastolic force, and abnormal action potential and arrhythmias occur (17). Thus, in response to a small increase in [Na+]i and [Ca2+]i induced by DG, the VM, isolated SAN tissue, and single SANCs all exhibit an initial positive inotropic, contractile, or chronotropic responses, respectively; at higher [Na+]i and [Ca2+]i, marked dysrhythmic response occurs in the VM, SAN tissue, and SANC. The full-spectrum stereotyped response patterns to DG can be obtained in any cardiac cells in the heart over short experimental times in response to a high DG concentration (as in the present study), or over longer times during exposure to DG at incremental (low to high) DG concentrations.
In summary, SANC automaticity is tightly regulated by ENa, ECa, and ENCX via a complex interplay of numerous key pacemaker mechanisms that regulate SANC clock coupling.
Limitations.
It is important to note that the aim of the study was to explore how complex, interrelated changes in [Na+]i and [Ca2+]i regulate the coupled-clock pacemaker system, rather than regulation of [Na+]i homeostasis per se. Therefore, [Na+]i was not simulated by the model, but the model was used to predict simultaneous changes in ion currents and ENa, ECa, and ENCX based on submembrane Ca2+ concentration changes rather than measured experimentally cytoplasmic [Ca2+]i. Although our model closely reproduced essential features of the experimental results, a limitation of the model is that it is a common pool model type that reproduces only LCR ensemble Ca2+ signal rather than individual LCR Ca2+ signal. A more detailed examination of submembrane signals and local electrochemical driving forces will require future studies to use local Ca2+ SANC models, such as those recently developed (40).
Other intracellular responses that accompany the reduction in ENa and ECa that were not measured in the present study merit investigation in future studies. These include Ca2+ activation of adenylyl cyclase (AC) and calcium/calmodulin-dependent protein kinase II (CAMKII), leading to changes in cAMP and phosphorylation states of Ca2+ cycling proteins, which impacts AP firing rate, and protein kinase C (PKC) activation, which has been implicated in the DG-evoked positive inotropic response in rat right atrial cells (33). Additionally, although not measured in the present study, the Na+/K+ pump is regulated by phospholemman phosphorylation (28), involving negative regulation by type 1 protein phosphatase inhibitor (12). When phosphorylated in ventricular myocytes by PKA and PKC, phospholemman stimulates the Na+/K+ pump to tightly regulate [Na+]i and [Ca2+]i. During β-adrenergic receptor stimulation of ventricular cells, an increase of phospholemman phosphorylation stimulates the Na+/K+ pump and limits an increase [Na+]i, thus limiting [Ca2+]i increase to [Ca2+]i overload and thus preventing propensity to arrhythmias in the heart (28).
Changes in SANC energy metabolism linked to AC cAMP, PKA, and CAMKII that have recently been addressed in prototypic numerical pacemaker models (56) are not addressed in the present study. Likewise, changes in mitochondrial [Ca2+]i accumulation (55) that occur in response to DG (because the mitochondrial uniporter competes with SERCA2 for Ca2+ via modulating the SR Ca2+ clock function) are not addressed in the present study. An increase in intracellular reactive oxygen species (ROS), induced by Ca2+-dependent or -independent DG effects on RyR, leads to changes in spontaneous RyR activation and SR Ca2+ load in ventricular myocytes (14). Furthermore, an increased [Na+]i reduces mitochondrial [Ca2+]i (8, 15, 23, 48), resulting in an increase of cytosolic NADH/NAD+ redox potential that alters sarcolemmal NCX activity independently of mitochondrial respiration by inhibiting INCX via NADH-induced cytosolic ROS and may also be involved in the SANC responses to DG observed in the present study. Finally, intracellular pH becomes reduced when Na+/K+ pump is inhibited (9, 42, 43), and this certainly impacts on numerous coupled-clock functions experimentally measured or numerically modeled in our study.
GRANTS
This work was supported by the Intramural Research Program of the NIH, National Institute on Aging.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.S. and E.G.L. conception and design of research; S.S., V.A.M., Y.Y., R.B., and D.Y. performed experiments; S.S., Y.Y., and R.B. analyzed data; S.S., V.A.M., and E.G.L. interpreted results of experiments; S.S., Y.Y., V.A.M., and R.B. prepared figures; S.S., V.A.M., and E.G.L. drafted manuscript; S.S., V.A.M., Y.Y., H.A.S., and E.G.L. edited and revised manuscript; S.S., V.A.M., Y.Y., R.B., D.Y., T.M.V., H.A.S., and E.G.L. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address for D. Yaeger: Department of Biology, Willamette University, Salem, OR.
REFERENCES
- 1.Altamirano J, Li Y, DeSantiago J, Piacentino V 3rd, Houser SR, Bers DM. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J Physiol 575: 845–854, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht, The Netherlands: Kluwer Academic, 2001. [Google Scholar]
- 3.Bers DM, Barry WH, Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res 57: 897–912, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Bogdanov KY, Maltsev VA, Vinogradova TM, Lyashkov AE, Spurgeon HA, Stern MD, Lakatta EG. Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ Res 99: 979–987, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Brody TM, Akera T. Relations among Na+, K+-ATPase activity, sodium-pump activity, transmembrane sodium movement, and cardiac contractility. Fed Proc 36: 2219–2224, 1977. [PubMed] [Google Scholar]
- 6.Bucchi A, Baruscotti M, DiFrancesco D. Current-dependent block of rabbit sino-atrial node I(f) channels by ivabradine. J Gen Physiol 120: 1–13, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi HS, Wang DY, Noble D, Lee CO. Effect of isoprenaline, carbachol, and Cs+ on Na+ activity and pacemaker potential in rabbit SA node cells. Am J Physiol Heart Circ Physiol 276: H205–H214, 1999. [DOI] [PubMed] [Google Scholar]
- 8.Cooper LL, Li W, Lu Y, Centracchio J, Terentyeva R, Koren G, Terentyev D. Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J Physiol 591: 5895–5911, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deitmer JW, Ellis D. Interactions between the regulation of the intracellular pH and sodium activity of sheep cardiac Purkinje-fibers. J Physiol 304: 471–488, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 105: 2543–2548, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Eisner DA, Lederer WJ, Vaughanjones RD. The quantitative relationship between twitch tension and intracellular sodium activity in sheep cardiac Purkinje-fibers. J Physiol 355: 251–266, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Armouche A, Wittkopper K, Fuller W, Howie J, Shattock MJ, Pavlovic D. Phospholemann-dependent regulation of the cardiac Na/K-ATPase activity is modulated by inhibitor-1 sensitive type-1 phosphatase. FASEB J 25: 4467–4475, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Hancox JC, Levi AJ. Actions of the digitalis analog strophanthidin on action potentials and L-type calcium current in single cells isolated from the rabbit atrioventricular node. Br J Pharmacol 118: 1447–1454, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho HT, Stevens SCW, Terentyeva R, Carnes CA, Terentyev D, Gyorke S. Arrhythmogenic adverse effects of cardiac glycosides are mediated by redox modification of ryanodine receptors. J Physiol 589: 4697–4708, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kennedy DJ, Vetteth S, Xie M, Periyasamy SM, Xie Z, Han C, Basrur V, Mutgi K, Fedorov V, Malhotra D, Shapiro JI. Ouabain decreases sarco(endo)plasmic reticulum calcium ATPase activity in rat hearts by a process involving protein oxidation. Am J Physiol Heart Circ Physiol 291: H3003–H3011, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Lakatta EG. Intracellular Ca2+ cycling is a general regulator of how fast and how strong the heart beats. Dialog Cardiovasc Med 15: 253–278, 2010. [Google Scholar]
- 17.Lakatta EG, Lappe DL. Diastolic scattered light fluctuation, resting force and twitch force in mammalian cardiac muscle. J Physiol 315: 369–394, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ Res 106: 659–673, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lakatta EG, Vinogradova TM, Maltsev VA. The missing link in the mystery of normal automaticity of cardiac pacemaker cells. Ann NY Acad Sci 1123: 41–57, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lederer WJ, Tsien RW. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibres. J Physiol 263: 73–100, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee CO. 200 years of digitalis—the emerging central role of the sodium-ion in the control of cardiac force. Am J Physiol Cell Physiol 249: C367–C378, 1985. [DOI] [PubMed] [Google Scholar]
- 22.Levi AJ, Boyett MR, Lee CO. The cellular actions of digitalis glycosides on the heart. Prog Biophys Mol Biol 62: 1–54, 1994. [DOI] [PubMed] [Google Scholar]
- 23.Liu T, Brown DA, O'Rourke B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. J Mol Cell Cardiol 49: 728–736, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyashkov AE, Juhaszova M, Dobrzynski H, Vinogradova TM, Maltsev VA, Juhasz O, Spurgeon HA, Sollott SJ, Lakatta EG. Calcium cycling protein density and functional importance to automaticity of isolated sinoatrial nodal cells are independent of cell size. Circ Res 100: 1723–1731, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Maltsev VA, Lakatta EG. Dynamic interactions of an intracellular Ca2+ clock and membrane ion channel clock underlie robust initiation and regulation of cardiac pacemaker function. Cardiovasc Res 77: 274–284, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Maltsev VA, Lakatta EG. Synergism of coupled subsarcolemmal Ca2+, clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am J Physiol Heart Circ Physiol 296: H594–H615, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noble D. Modelling the heart: insights, failures and progress. Bioessays 24: 1155–1163, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Pavlovic D, Fuller W, Shattock MJ. Novel regulation of cardiac Na pump via phospholemman. J Mol Cell Cardiol 61: 83–93, 2013. [DOI] [PubMed] [Google Scholar]
- 29.Reuter H, Henderson SA, Han TY, Ross RS, Goldhaber JI, Philipson KD. The Na+-Ca2+ exchanger cardiac is essential for the action of glycosides. Circ Res 90: 305–308, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Rosen MR. Cellular electrophysiology of digitalis toxicity. J Am Coll Cardiol 5: 22A–34A, 1985. [DOI] [PubMed] [Google Scholar]
- 31.Rosen MR, Gelband H, Merker C, Hoffman BF. Mechanisms of digitalis toxicity. Effects of ouabain on phase four of canine Purkinje fiber transmembrane potentials. Circulation 47: 681–689, 1973. [DOI] [PubMed] [Google Scholar]
- 32.Satoh H. Ouabain actions on the spontaneous activity and ionic currents in rabbit sinoatrial node cells. Gen Pharmacol 25: 1591–1598, 1994. [DOI] [PubMed] [Google Scholar]
- 33.Satoh H, Narushima I, Nakashima T. Possible involvement of protein kinase C in the modulation of inotropic and chronotropic effects induced by ouabain in rat right atrial muscles. J Pharm Pharmacol 49: 925–929, 1997. [DOI] [PubMed] [Google Scholar]
- 34.Shattock MJ, Ottolia M, Bers DM, Blaustein MP, Boguslavskyi A, Bossuyt J, Bridge JH, Chen-Izu Y, Clancy CE, Edwards A, Goldhaber J, Kaplan J, Lingrel JB, Pavlovic D, Philipson K, Sipido KR, Xie ZJ. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J Physiol 593: 1361–1382, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheu SS, Fozzard HA. Transmembrane Na+ and Ca2+ electrochemical gradients in cardiac muscle and their relationship to force development. J Gen Physiol 80: 325–351, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sirenko S, Yang D, Li Y, Lyashkov AE, Lukyanenko YO, Lakatta EG, Vinogradova TM. Ca2+-dependent phosphorylation of Ca2+ cycling proteins generates robust rhythmic local Ca2+ releases in cardiac pacemaker cells. Sci Signal 6: ra6, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Souza FC, Marques EB, Barros RBM, Scaramello CBV. Study of digoxin use in a public health unit. An Acad Bras Cienc 87: 1033–1040, 2015. [DOI] [PubMed] [Google Scholar]
- 38.Spurgeon HA, Stern MD, Baartz G, Raffaeli S, Hansford RG, Talo A, Lakatta EG, Capogrossi MC. Simultaneous measurement of Ca2+, contraction, and potential in cardiac myocytes. Am J Physiol Heart Circ Physiol 258: H574–H586, 1990. [DOI] [PubMed] [Google Scholar]
- 39.Steinbeck G, Bonke FI, Allessie MA, Lammers WJ. The effect of ouabain on the isolated sinus node preparation of the rabbit studied with microelectrodes. Circ Res 46: 406–414, 1980. [DOI] [PubMed] [Google Scholar]
- 40.Stern MD, Maltseva LA, Juhaszova M, Sollott SJ, Lakatta EG, Maltsev VA. Hierarchical clustering of ryanodine receptors enables emergence of a calcium clock in sinoatrial node cells. J Gen Physiol 143: 577–604, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takayanagi K, Jalife J. Effects of digitalis intoxication on pacemaker rhythm and synchronization in rabbit sinus node. Am J Physiol Heart Circ Physiol 250: H567–H578, 1986. [DOI] [PubMed] [Google Scholar]
- 42.Vaughan-Jones RD, Eisner DA, Lederer WJ. Effects of changes of intracellular pH on contraction in sheep cardiac Purkinje-fibers. J Gen Physiol 89: 1015–1032, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol 46: 318–331, 2009. [DOI] [PubMed] [Google Scholar]
- 44.Verbeke G, Molenberghs G. Linear Mixed Models for Longitudinal Data Introduction. New York, NY: Springer-Verlag New York, 2000. [Google Scholar]
- 45.Vinogradova TM, Lakatta EG. Regulation of basal and reserve cardiac pacemaker function by interactions of cAMP-mediated PKA-dependent Ca2+ cycling with surface membrane channels. J Mol Cell Cardiol 47: 456–474, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vinogradova TM, Sirenko S, Lyashkov AE, Younes A, Li Y, Zhu WZ, Yang DM, Ruknudin AM, Spurgeon H, Lakatta EG. Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+, releases. Circ Res 102: 761–769, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Vinogradova TM, Zhou YY, Bogdanov KY, Yang DM, Kuschel M, Cheng HP, Xiao RP. Sinoatrial node pacemaker activity requires Ca2+/calmodulin-dependent protein kinase II activation. Circ Res 87: 760–767, 2000. [DOI] [PubMed] [Google Scholar]
- 48.Wasserstrom JA. Are we ready for a new mechanism of action underlying digitalis toxicity? J Physiol 589: 5015–5015, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto S, Akera T, Brody TM. Positive inotropic action of digoxigenin and sodium pump inhibition: effects of enhanced sodium influx. J Pharmacol Exp Ther 213: 105–109, 1980. [PubMed] [Google Scholar]
- 50.Yaniv Y, Ahmet I, Liu J, Lyashkov AE, Guiriba TR, Okamoto Y, Ziman BD, Lakatta EG. Synchronization of sinoatrial node pacemaker cell clocks and its autonomic modulation impart complexity to heart beating intervals. Heart Rhythm 11: 1210–1219, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yaniv Y, Lakatta EG, Maltsev VA. From two competing oscillators to one coupled-clock pacemaker cell system. Front Physiol 6: 28, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yaniv Y, Lyashkov AE, Sirenko S, Okamoto Y, Guiriba TR, Ziman BD, Morrell CH, Lakatta EG. Stochasticity intrinsic to coupled-clock mechanisms underlies beat-to-beat variability of spontaneous action potential firing in sinoatrial node pacemaker cells. J Mol Cell Cardiol 77: 1–10, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yaniv Y, Maltsev VA, Ziman BD, Spurgeon HA, Lakatta EG. The “funny” current (I-f) inhibition by ivabradine at membrane potentials encompassing spontaneous depolarization in pacemaker cells. Molecules 17: 8241–8254, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yaniv Y, Sirenko S, Ziman BD, Spurgeon HA, Maltsev VA, Lakatta EG. New evidence for coupled clock regulation of the normal automaticity of sinoatrial nodal pacemaker cells: Bradycardic effects of ivabradine are linked to suppression of intracellular Ca2+ cycling. J Mol Cell Cardiol 62: 80–89, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yaniv Y, Spurgeon HA, Lyashkov AE, Yang D, Ziman BD, Maltsev VA, Lakatta EG. Crosstalk between mitochondrial and sarcoplasmic reticulum Ca2+ cycling modulates cardiac pacemaker cell automaticity. PLoS One 7: e37582, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yaniv Y, Spurgeon HA, Ziman BD, Lyashkov AE, Lakatta EG. Mechanisms that match ATP supply to demand in cardiac pacemaker cells during high ATP demand. Am J Physiol Heart Circ Physiol 304: H1428–H1438, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yaniv Y, Stern MD, Lakatta EG, Maltsev VA. Mechanisms of beat-to-beat regulation of cardiac pacemaker cell function by Ca(2+) cycling dynamics. Biophys J 105: 1551–1561, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]