

We illustrate sex dimorphisms in crossbridge kinetics where male hypertrophic cardiomyopathy hearts displayed an increased while female hypertrophic cardiomyopathy hearts displayed a decreased tension cost. In addition, we have found sex- and mutation-dependent differences in cardiac remodeling at the morphometric, histological, and cellular level.
Keywords: sex/gender, crossbridge cycle
Abstract
Familial hypertrophic cardiomyopathy (HCM) is a disease of the sarcomere and may lead to hypertrophic, dilated, restrictive, and/or arrhythmogenic cardiomyopathy, congestive heart failure, or sudden cardiac death. We hypothesized that hearts from transgenic HCM mice harboring a mutant myosin heavy chain increase the energetic cost of contraction in a sex-specific manner. To do this, we assessed Ca2+ sensitivity of tension and crossbridge kinetics in demembranated cardiac trabeculas from male and female wild-type (WT) and HCM hearts at an early time point (2 mo of age). We found a significant effect of sex on Ca2+ sensitivity such that male, but not female, HCM mice displayed a decrease in Ca2+ sensitivity compared with WT counterparts. The HCM transgene and sex significantly impacted the rate of force redevelopment by a rapid release-restretch protocol and tension cost by the ATPase-tension relationship. In each of these measures, HCM male trabeculas displayed a gain-of-function when compared with WT counterparts. In addition, cardiac remodeling measured by echocardiography, histology, morphometry, and posttranslational modifications demonstrated sex- and HCM-specific effects. In conclusion, female and male HCM mice display sex dimorphic crossbridge kinetics accompanied by sex- and HCM-dependent cardiac remodeling at the morphometric, histological, and cellular level.
NEW & NOTEWORTHY
We illustrate sex dimorphisms in crossbridge kinetics where male hypertrophic cardiomyopathy hearts displayed an increased while female hypertrophic cardiomyopathy hearts displayed a decreased tension cost. In addition, we have found sex- and mutation-dependent differences in cardiac remodeling at the morphometric, histological, and cellular level.
cardiomyopathies critically underlie heart failure and can be characterized as hypertrophic, dilated, restrictive, and/or arrhythmogenic (33). Many of these cardiomyopathies are genetically linked with a reported frequency of ∼1:500 in the general population. The R403Q point mutation in β-myosin heavy chain (MyHC) is the first identified mutation leading to hypertrophic cardiomyopathy (HCM) with a heritable component (23). During the progression of cardiac disease, as occurs in R403Q hearts, the myocardium undergoes cellular and molecular remodeling, including a changing metabolic and energetic landscape (18, 25), such that the failing heart can be characterized as energy starved (26). The molecular underpinnings of the metabolic derangements reside in changes in the mediators of ATP generation, utilization, and delivery initiated by mutant sarcomeric proteins. Given the extremely high demand for ATP in the heart (26), small derangements in basal ATP concentration or its utilization could lead to metabolic and energetic deficiencies (36, 47). An early study linked HCM with such an energy stress where NMR on murine male R403Q hearts reveals decreased levels of phosphocreatine and increased levels of inorganic phosphate compared with WT littermates (51).
It is now clear that the dynamics of cardiac contraction and relaxation during HCM are governed by downstream mechanisms, particularly the kinetics and energetics of the crossbridge cycle (52). In vitro analysis of R403Q myosin kinetics yields inconsistent results such as reduced (11) or enhanced (40) actin filament velocity and reduced (53) or enhanced (58) actin-activated ATPase. On the other hand, human myofibril or multicellular R403Q samples consistently show accelerated tension generation and increased ATP hydrolysis rates (5, 57). At the very least, the presence of the R403Q mutation alters myofilament function and ATP utilization at the level of the sarcomere. An important question that remains is how energetic perturbations in crossbridge kinetics, as a result of inheritable mutations, become integrated with cellular signaling cascades leading to HCM (49, 59).
The direct impact of posttranslational modifications (PTM) are key to understanding the modifications that occur in the short- and long-term development of HCM progression (12). However, using phosphorylation status of sarcomeric proteins as a tool to bridge sarcomeric mechanics to molecular signaling has proved difficult (4, 7, 30). For example, we were unable to directly attribute the observed pattern of myofilament function in R403Q hearts to a particular pattern of myofilament phosphorylation (32). What is evident from studies using the R403Q HCM model is that, although the primary defect may reside in the sarcomere, the development of the HCM phenotype depends on the interaction of the initiated signaling pathways, environmental stressors, and individual genotype, especially sex/gender.
Despite an increasing appreciation and knowledge regarding sex dimorphisms in the pathophysiology of HCM, many inconsistencies plague the cellular and molecular mechanisms underlying these differences. It is well known that premenopausal women show consistently better outcomes with many forms of cardiac disease, including hypertension, myocardial infarction, and HCM (17, 56). Also, sex differences become more apparent with age in HCM, which highlights its progressive nature (17). In separate R403Q murine models, both males and females develop hypertrophy at four months, and, whereas males begin to show left ventricular dilation and systolic impairment, females do not (22, 37).
As mentioned above, human R403Q samples show an increase in the rate of ATP hydrolysis (5, 57), suggesting an energetic stress imparted by the R403Q mutation. In this study, we tested the hypothesis that the R403Q mutation will alter crossbridge kinetics, including ATP utilization, in a sex-independent manner. To determine the early cues leading to cardiac pathology, we chose an early time point (2 mo of age) where male and female HCM mice show only early signs of pathology. In addition, we sought to elucidate whether differences in the patterning of PTM among male and female R403Q hearts underlies any of the sex dimorphisms.
MATERIALS AND METHODS
Animal Model
The experimental murine model has been detailed previously and consisted of male mice heterozygous for the mutant rodent α-myosin transgene (55). The transgene coding region contains a point mutation, R403Q, and a deletion of 59 amino acids in the actin-binding site bridged by the addition of 9 nonmyosin amino acids. Wild-type (WT) littermates were used as controls for the familial hypertrophic cardiomyopathy mice. Mice were genotyped from a tail sample by the University of Arizona Genetics Core Facility before inclusion in the study. To confirm equivalent levels of transgene expression, total RNA was isolated from the left ventricles of WT and R403Q HCM hearts (female and male) using the RNeasy Midi Kit RNA isolation kit (Qiagen) according to the manufacturer's protocol, and cDNA was generated using the NCode miRNA First-Strand cDNA Synthesis Kit (Invitrogen). Equivalent transgene expression in male and female HCM mice was confirmed by RT-PCR using primers specific for sequences spanning the transgene (Fig. 1). Previous work by our group determined a 10–12% expression of the mutant protein in HCM mice (55). All experiments were performed using protocols that adhered to guidelines of and approved by the Institutional Animal Care and Use Committee at the University of Arizona and to 2011 National Institutes of Health guidelines for the care and use of laboratory animals.
Fig. 1.
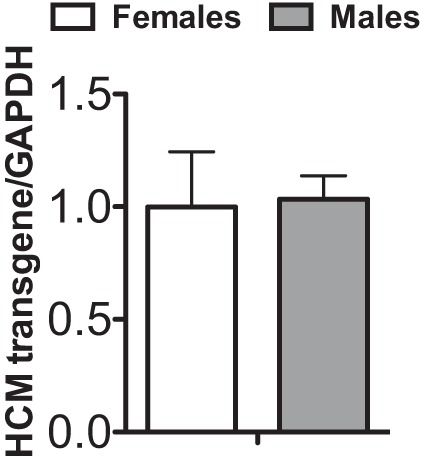
Transgene expression in hypertrophic cardiomyopathy (HCM) female and male hearts. Bar graph representation of HCM transgene expression normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) following RT-PCR using a unique primer set spanning the transgene. Our group has previously shown that this results in 10–12% replacement of the native peptide (55).
Preparation of Cardiac Fibers
Mice were deeply anesthetized using isoflurane, and hearts were rapidly excised and retrogradely perfused in a modified Krebs-Henseleit (KH) solution (16). The KH solution contained the following (in mM): 0.5 EDTA, 10.0 d-glucose, 1.5 pyruvic acid, 0.7 CaCl2, 25 NaHCO3, 118.5 NaCl, 5.0 KCl, 1.2 MgSO4, 2.0 NaH2PO4, and 20 mM 2,3-butanedione monoxime. The pH of the KH solution was adjusted to 7.35–7.4 using NaHCO3 before perfusion. Free-standing unbranched trabecula fibers were extracted from the right ventricle and placed in an ice-cold standard relaxing solution containing 1% Triton X-100 overnight for chemical demembranation. The remaining ventricular tissue was snap-frozen in liquid nitrogen for biochemical interrogation.
Free Ca2+ Concentration Solutions and Compositions
Each trabecula was bathed in relaxing/preactivating/activating solutions. The ionic strength was kept at 180 mmol/l by adding the appropriate amount of potassium propionate. The pH was adjusted to 7.0 at 15°C with KOH. Ca-EGTA is made by mixing equimolar amounts of CaCl2 and EGTA. In addition, each solution contained (in mM): 5.0 NaN3, 10.0 phosphoenolpyruvate, 0.2 A2P5, 1.0 oligomycin, 0.8 mg/ml pyruvate kinase, and 0.05 mg/ml lactate dehydrogenase. The following cocktail of protease inhibitors was added: 4 ul/ml protease inhibitor cocktail (P8340; Sigma), 1 mM DTT, 7.2 mg/ml N-p-tosyl-l-phenylalanine chloromethyl ketone (TPCK), 7.2 mg/ml Nα-tosyl-l-lysine chloromethyl ketone hydrochloride (TLCK), and 0.1 mM phenylmethylsulfonyl fluoride (PMSF).
Ca2+ Sensitivity of Tension
Demembranated fibers were attached using aluminum T-clips to a high-speed-length controller (model 315C; 0.25-ms step response; Aurora Scientific) and a silicon strain gauge force transducer (model kx801; Kronex Technologies). Both the length controller and the force transducer are attached to X-Y-Z manipulators mounted on a movable microscope stage that is temperature controlled (20 ± 0.1°C). Fiber cross-sectional area was measured directly through a microscope reticle. Force was recorded by a personal computer equipped with an analog-to-digital converter using custom LabView software for off-line analysis. The sarcomere length (SL) was measured directly by a He-Ne laser as previously described (13). All procedures were performed at a SL of 2.2 μm. Ca2+-dependent force is determined by a series of preactivating-activating-relaxation cycles. A range of free Ca2+ concentration ([Ca2+]) in the activating solution is achieved by mixing in appropriate an amount of relaxing solution. Each individual Ca2+-force relationship was fit to the Hill equation: Frel = [Ca2+]n/(EC50n +[Ca2+]n). In the equation above, Frel is the force relative to maximum Ca2+-saturated force, EC50 is the [Ca2+] at which force is half-maximal, and n is slope of the Ca2+-force relationship (Hill coefficient). Fibers were allowed to reach steady-state tension and then rapidly slackened by 15% of total fiber length. The difference between steady-state tension and slacked tension determined total tension. Active tension at each [Ca2+] is the difference between total tension and relaxed passive tension.
Rate of Tension Redevelopment
Once the fiber has reached steady-state force generation, a rapid release of 15% of the muscle length ensues. This is quickly (5 ms) followed by a rapid stretch 15% beyond the original fiber length. Immediately following the stretch (1 ms), the fiber is returned to the original length and allowed to regenerate force. This rapid release/restretch serves to remove myosin from actin, including weakly bound states. This regeneration of force can be fit by a monoexponential, and the rate constant, ktr, can be calculated as F = (Fo − Fres)(1 − ektrt) + Fres, where Fo is the steady state isometric force, and Fres is the residual force from which redevelopment of tension occurs; ktr (s−1) is therefore the summed rate of crossbridges entering (fapp) and exiting (gapp) a force-generating state. This protocol was then applied to maximal and submaximal activations.
ATPase Activity
The ATPase activity of the skinned trabecula is measured on-line by means of an enzyme-coupled assay as previously described (13, 15). Briefly, a broad-spectrum light source is projected in the muscle bath and is split using a dichroic mirror toward a 340- and 400-nm wavelength detector. Formation of ADP by the muscle is stoichiometrically coupled first to the synthesis of pyruvate and ATP from phosphoenolpyruvate. This reaction is catalyzed by the enzyme pyruvate kinase and leads to the synthesis of lactate, a reaction that is then catalyzed by the enzyme lactate dehydrogenase and during which NADH is oxidized to NAD+. NADH absorbs light at 340 nm and is subsequently added dropwise to each activating solution to reduce the signal sufficiently for the subsequent fiber-induced reaction; 400-nm wavelength is insensitive to NADH and serves as a reference signal. Therefore, the activation of a fiber produces a signal proportional to the amount of ATP consumed (i.e., amount of NADH oxidized). Changes in the 340-nm wavelength due to NADH oxidation are calibrated by injecting 0.5 nmol of ADP in a stepwise fashion.
PSR Staining and Determination of Collagen Content
Hearts were excised, weighed, and then fixed in 10% formalin overnight, paraffin embedded, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E) or picrosirius red (PSR). The PSR-stained tissue was visualized under polarized light (ZEISS Axio Imager M1) to detect birefringence of collagen fibers. Three fields were chosen randomly from each heart sample. The images were quantified by a semiautomated imaging analysis program (AxioVision). A color threshold was defined in such a way to detect mature collagen. The area of birefringence was normalized by the total area of interest and used as an indicator of collagen content (10, 28)
High-Resolution Cardiac Ultrasound
High-resolution cardiac ultrasound was performed using a Visual Sonics Vevo 2100 high-resolution imaging system (Visual Sonics, Toronto, ON, Canada). Briefly, the chest of animals was dipilatated with a chemical hair remover, and anesthesia was maintained by 1% isoflurane with oxygen. Body temperature was maintained using a heated platform. Respiratory rates and electrocardiograms were monitored throughout the study. Two-dimensional M-mode echocardiographic images were obtained from the parasternal short-axis views at the level of the midventricles. Cardiac chamber dimensions and the left ventricular wall thickness were measured. Interventricular septum, left ventricular posterior wall thickness (LVPWT), and internal dimension (LVIDd) were measured from the M-mode images. Relative wall thickness [(LVPWT/LVIDd) × 2] was calculated from the M-mode measurements. Data were analyzed off-line using Vevo 2100 analytic software. The data were obtained in triplicate and averaged.
Mouse Cardiac Samples for SDS-PAGE
A small portion of right or left ventricular tissue was broken off using a liquid nitrogen-cooled mortar and pestel (5–12 mg). The cardiac tissue was homogenized in 50% high-urea buffer (8 M urea, 2 M thiourea, 3% SDS, 0.05 M Tris·HCl, 0.075 M dithiothreitol, and 0.03% bromphenol blue) and 50% glycerol with protease inhibitors [25 μl/1 ml Protease Inhibitor Cocktail (P8340; Sigma), 0.2 M PMSF, 17 mM TPCK, 16 mM TLCK, 0.1 M NaP2O7, 0.1 M Na3VO4, and 0.5 M NaF]. After homogenization, samples were centrifuged at maximum speed for 30 s, and sample buffer was added to bring the final volume to 20× the cardiac tissue mass. Samples were incubated at 95°C for 5 min, and then aliquots were frozen at −80°C.
Separation of MyHC Isoforms
Frozen cardiac samples were prepared as detailed for SDS-PAGE to determine relative cardiac MyHC content as previously described (28). Gels were silver stained and analyzed using a Umax PowerLook 1120 flatbed scanner with 1,200 × 2,400 dots/in. and 3.7 maximum dynamic range of light intensity. To determine relative MyHC content, several dilutions of each sample were analyzed to stay in the linear densitometric range. A linear relationship was determined for each MyHC isoform, allowing the relative MyHC content of each isoform to be extrapolated. Soleus muscle (consisting of primarily β-MyHC and type IIa MyHC) was used as a standard expression of both isoforms.
ProQ Diamond Phosphoprotein Stain
Briefly, the compositions of SDS-PAGE gels were as follows: stacking gel: 5% polyacrylamide (29:1 acrylamide-bisacrylamide), 0.1% SDS, 0.1% ammonium persulfate (APS), 0.1% TEMED, and 0.125 M Tris, pH 6.8; resolving gel: 12% acrylamide (29:1 acrylamide-bisacrylamide), 0.1% SDS, 0.1% APS, 0.06% TEMED, and 0.375 M Tris, pH 8.8. Gels were run using the Criterion system in ice-cold running buffer (2.5 mM Tris, 19 mM glycine, and 0.35 mM SDS) and at constant current (30 mA/gel). Gels were then fixed and stained according to the manufacturer's instructions (Invitrogen). Following ProQ Diamond staining, gels were stained for total protein content using Coomassie Brillant Blue. Optical densities of phosphorylated proteins were quantified using LabImage one-dimensional (1D) software and were normalized to their respective Coomassie-stained total protein bands.
The 97-kDa band was analyzed by LC-MS/MS (Arizona Proteomics Consortium) using Sequest (version 1.3.0.339; Thermo Fisher Scientific, San Jose, CA). Scaffold (version Scaffold_4.4.1.1; Proteome Software, Portland, OR) was used to validate the identity of this band with a >99.0% probability and contained at least seven identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm.
Western Blot Analysis
SDS-PAGE was used to separate myofibrillar samples. After transfer [polyvinylidene fluoride (PVDF) membrane], total protein was measured with Ponceau S stain (Sigma). Antibodies were used to probe for total cardiac troponin I (cTnI, 1:5,000; AbCam, Cambridge, UK), phospho (p)-cTnI-Ser23/24 (1:1,000; Cell Signaling, Beverly, MA), p-cTnI-Ser150 (1:200; AbCam, Cambridge, UK), total cardiac myosin-binding protein C [cMyBP-C, 1:1,000; Western, rabbit (AbCam) or PhosTag, mouse (Santa Cruz Biotechnology)], and p-cMyBP-C-Ser282 (1:2,500; Enzo Life Sciences, East Farmingdale, NY) (19). Protein optical densities were quantified using LabImage 1D software and normalized to total protein to adjust for alterations in loading parameters. Normalized optical densities from p-cTnI-Ser23/24, p-cTnI-Ser150, and p-cMyBP-C-Ser282 were divided by the total cTnI and total cMyBP-C optical density. In the case of phospho-Ser150 cTnI, the secondary antibodies were conjugated with fluorescent dyes with infrared excitation spectra and were used for detection. Infrared Western blots were analyzed using the Odyssey Infrared Imaging System (Li-Cor Biosciences).
Phospho-Affinity SDS-PAGE
Phospho-affinity SDS-PAGE (PhosTag) was carried out using either precast SuperSep PhosTag (50 μM), 10% gel (Wako Pure Chemical Industries, Osaka, Japan), or hand-cast PhosTag gels. The compositions of hand-cast gels are as follows: 5% polyacrylamide (stacking gel: 29:1 acrylamide-bisacrylamide), 0.1% SDS, 0.1% APS, 0.1% TEMED, and 0.125 M Tris, pH 6.8; 11% acrylamide (resolving gel: 29:1 acrylamide-bisacrylamide), 0.1% SDS, 0.15% APS, 0.3% TEMED, 0.4 M Tris, pH 8.8, 0.2 mM MnCl2, and 0.1 mM Mn-PhosTag acrylamide. Approximately 30 μg of total protein were loaded on each lane. Gels were run using the Criterion system on ice, in cold running buffer (2.5 mM Tris, 19 mM glycine, and 0.35 mM SDS). Proteins were transferred to PVDF membranes using the Trans-Blot system (Bio-Rad). Membranes were probed using the above antibodies. Total protein was measured by SYPRO Ruby stain (Bio-Rad). Human recombinant cardiac troponin I (hcTnI, kindly provided by Dr. Jil Tardiff) and mouse cardiomyocytes (skinned, 1% Triton) treated with protein kinase A (PKA catalytic subunit; 1 U/μl) were used as relative standards to identify different phosphospecies populations. hcTnI was used to identify maximally migrating, and therefore minimally phosphorylated, cTnI phosphospecies; PKA-treated cardiomyocytes were used to identify minimally migrating, and therefore highly phosphorylated, cTnI phosphospecies.
Data and Statistical Analysis
Results are presented as means ± SE. A two-way or one-way ANOVA with the Bonferroni's post hoc test, where appropriate, was used to determine differences between means. Multiple-linear regression was also used to determine the relationship between ktr and tension. P values <0.05 were considered significant.
RESULTS
Muscle Mechanics
Ca2+ sensitivity of tension development.
A recent study reports that the R403Q mutation does not impact Ca2+-sensitive tension development in demembranated cardiac fibers when compared with controls from either sex in 10- to 20-wk-old mice, when R403Q mice exhibit no observable HCM pathology (22, 38, 39). We previously showed that cardiac trabeculas from 10-mo-old female HCM mice were significantly more sensitive to Ca2+ than WT controls, whereas male HCM trabeculas were not different from WT fibers (32). Therefore, we determined the Ca2+ sensitivity of tension for HCM and WT males and females. Figure 2 displays the Ca2+ sensitivity of tension for WT and HCM females (Fig. 2A) and males (Fig. 2B). The solid and dashed lines indicate the best fit of the Ca2+ force data to a modified Hill equation whose parameters are summarized in Table 1. Ca2+ sensitivity of tension determined in females was not impacted by HCM. On the other hand, Ca2+ sensitivity of tension was less sensitive (right shifted) in HCM males compared with WT males. Moreover, while Ca2+ sensitivity was similar among WT female, HCM female, and WT male fibers, HCM male trabeculas were less sensitive to Ca2+ than trabeculas from all other groups. In addition, we concluded that neither HCM nor sex impacted length sensitivity by also measuring Ca2+ sensitivity of tension at SL of 2.0 μm for each group (ΔEC50) (Table 1). Cooperativity, as indicated by the Hill coefficient, and maximum tension generated was not different for the groups studied.
Fig. 2.
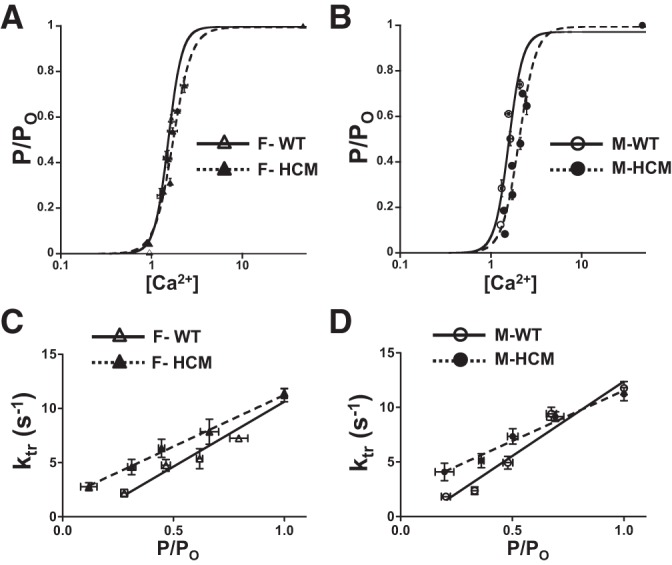
Ca2+ sensitivity of tension and rate of tension redevelopment. A: Ca2+ sensitivity of tension in demembranated cardiac trabeculas from wild-type (WT) females (F) (n = 4) and HCM females (n = 6). B: Ca2+ sensitivity of tension in demembranated cardiac trabeculas from WT males (M) (n = 4) and HCM males (n = 6). C: rate of tension redevelopment (ktr) in demembranated cardiac trabeculas from WT females (n = 4) and HCM females (n = 6). D: ktr in demembranated cardiac trabeculas from WT males (n = 4) and HCM males (n = 6). ktr plotted as a function of normalized tension; data plots represent binned values. Sarcomere length (SL) was set to 2.2 μm, and all data were normalized to saturating Ca2+ (maximum) tension.
Table 1.
Mean values for Ca2+ sensitivity parameters and tension redevelopment
| F-WT | F-HCM | M-WT | M-HCM | |
|---|---|---|---|---|
| Ca2+ sensitivity of tension | ||||
| n | 4 | 6 | 4 | 6 |
| EC50, μM | 1.53 ± 0.08 | 1.71 ± 0.08 | 1.62 ± 0.09 | 2.08 ± 0.09* |
| pCa50, μM | 5.82 ± 0.02 | 5.77 ± 0.02 | 5.79 ± 0.02 | 5.68 ± 0.02* |
| Hill coefficient | 5.93 ± 1.7 | 4.18 ± 0.40 | 4.22 ± 0.44 | 4.19 ± 0.36 |
| ΔEC50 | 0.63 ± 0.07 | 0.54 ± 0.08 | 0.55 ± 0.09 | 0.36 ± 0.08 |
| ΔpCa50 | 0.15 ± 0.01 | 0.12 ± 0.03 | 0.13 ± 0.01 | 0.07 ± 0.01 |
| Maximum tension, mn/mm2 | 32.9 ± 2.3 | 39.5 ± 5.1 | 34.3 ± 3.2 | 39.6 ± 4.7 |
| Rate of tension redevelopment | ||||
| n | 11 | 9 | 10 | 6 |
| Maximum ktr, s−1 | 11.6 ± 0.4 | 11.2 ± 0.6 | 13.2 ± 1.5 | 11.1 ± 0.6 |
| Linear fit, s−1·P−1·P0−1 | 12.0 ± 1.0 | 9.5 ± 1.2 | 13.3 ± 1.0* | 9.3 ± 1.1* |
| R2 | 0.98 | 0.99 | 0.97 | 0.93 |
Values are means ± SE; n, no. of subjects. P/P0, relative tension [total tension (P)/maximum tension (P0)]. Average Hill fit parameters of the Ca2+ sensitivity of tension and rate of tension redevelopment (ktr) obtained from wild-type (WT) and hypertrophic cardiomyopathy (HCM) female (F) and male (M) cardiac trabeculas at a sarcomere length (SL) of 2.2 μm. Hill equation: Frel = [Ca2+]n/(EC50n + [Ca2+]n), where Frel is relative force, EC50 is [Ca2+] at which force is half-maximal, and n is the slope of the Ca2+-force relationship (Hill coefficient). As a measure of length dependent activation, the ΔEC50 and ΔpCa50 were determined from SL 2.2 and 2.0 μm. ktr was determined for each experimental group. In addition, ktr was plotted against relative tension and fit by linear regression. A 2-way ANOVA followed by Bonferroni's post hoc test compared differences between groups.
P < 0.05 from WT counterpart.
Rate of tension redevelopment.
The rate of tension redevelopment (ktr) is a measure of the sum of cross bridges entering and exiting a force-generating state using a simplified two-state model (5). ktr at maximum tension was not significantly different for any group studied. However, given that maximum tension is generated at relatively nonphysiological levels of Ca2+, ktr was also measured at submaximal tension. The tension-ktr relationship was then fit by linear regression. We found a significant impact of the HCM transgene on the tension-ktr relationship (Fig. 2, C and D). Interestingly, HCM males (Fig. 2D) showed a significantly decreased (more shallow) slope compared with WT males, whereas female HCM and WT (Fig. 2C) were not different by post hoc analysis. Because ktr is a measure of total crossbridge cycling, a decreased or shallower slope would indicate increased cycling (entering and exiting) of crossbridges at a given force.
Rate of force-dependent ATP hydrolysis and tension cost.
Given the apparent differences in crossbridge cycling rates for HCM males, it was necessary to measure the rate of force-dependent ATP hydrolysis. To do this, we simultaneously measure force and ATP utilization as described previously (13). The slope between ATP hydrolyzed (in pmol·s−1·mm−3) and tension generated (in mN/mm) results in the tension cost of the cardiac tissue. All ATP hydrolysis data were collected at a SL of 2.2 μm. Figure 3 illustrates that there is a strong (statistically significant) interaction between sex and the presence of the mutation with regard to tension cost. These data are summarized in Table 2. This would indicate a higher “off” rate and a more inefficient use of ATP at a given force in HCM males with the opposite in HCM females.
Fig. 3.

ATP hydrolysis and tension cost. Tension cost is the slope relationship between the amount of ATP hydrolyzed and tension generated. A: ATP hydrolysis as a function of normalized tension in demembranated cardiac trabeculas from WT females (n = 4) and HCM females (n = 6). B: ATP hydrolysis as a function of normalized tension in demembranated cardiac trabeculas from WT males (n = 4) and HCM males (n = 6). C: bar graph representation of mean tension cost for each group. Values are presented as means ± SE. There was a significant interaction between sex and HCM transgene, with M-HCM significantly less than M-WT (P < 0.05). Data plots represent binned values. All hydrolysis data were collected at SL 2.2 μm.
Table 2.
Mean values of tension cost
| F-WT | F-HCM | M-WT | M-HCM | |
|---|---|---|---|---|
| N | 11 | 9 | 10 | 6 |
| Tension cost, (pmol·s−1·mn−1·mm−1) | 6.0 ± 0.4 | 4.3 ± 0.4 | 3.8 ± 0.5 | 6.0 ± 0.8* |
| Maximum hydrolysis, pmol·s−1·mm−3 | 318 ± 42 | 210 ± 27 | 258 ± 38 | 301 ± 35 |
Data are presented as means ± SE; n, no. of subjects. Tension cost was determined by the relationship between ATP hydrolysis and relative tension. Two-way ANOVA analysis revealed a strong interaction between sex and the HCM transgene with regard to tension cost. Bonferroni post hoc analysis found HCM males had a significantly higher tension cost compared with WT males. ATP hydrolysis at maximum tension was not different for any group.
P < 0.05 from WT counterpart.
Functional and Morphometric Cardiac Remodeling
Cardiac hypertrophy and high-resolution ultrasound.
Figure 4 displays longitudinal sections of H&E-stained hearts from each experimental group. We determined collagen content by PSR staining as previously described (10, 28). There were no differences in collagen content between all groups studied (Fig. 4A). We compared heart weight normalized to tibial length and found that there was a significant impact of sex and disease (HCM) on normalized heart weight, including a significant interaction between sex and disease (Fig. 4B). Post hoc analysis indicated significant hypertrophy in male HCM mice over male WT counterparts.
Fig. 4.

Summary of morphometric data from WT and HCM female and male mice. Top, representative hematoxylin and eosin (H&E)-stained longitudinal heart sections from each experimental group. A: bar graph summary of %collagen deposition in each experimental group. Birefringence of collagen fibers was quantified using a semiautomated imaging analysis program. B: HW/TL was determined by dividing cardiac mass (in mg) by tibial length (in mm). Echocardiographic parameters of ejection fraction (EF%; C) and relative wall thickness (RWT; D) calculated from the M-mode measurements. Values are presented as means ± SE. F-WT, n = 6; F-HCM, n = 8; M-WT, n = 8; M-HCM, n = 6. *P < 0.05 from WT counterpart.
To assess the impact of HCM on cardiac function and in situ ventricular morphometry, high-resolution two-dimensional echocardiography was performed on WT and HCM mice in each sex. A significant interaction between sex and disease was determined in ejection fraction (EF%) (Fig. 4C). Moreover, EF% was significantly elevated in female HCM compared with female WT hearts. However, there were no differences among the factors of sex or disease when we determined relative wall thickness during diastole (Fig. 4D).
MyHC isoform content.
Given that previous work has shown the importance of MyHC isoform content in the context of cardiac disease progression, as well as myosin ATPase efficiency (20), we quantified MyHC isoform expression in 2-mo HCM and WT mice (Fig. 5). In line with previous work (55), β-MyHC content was minimal (<2%) in either right or left ventricle samples for HCM mice of either sex. Therefore, the mechanical and kinetic differences seen cannot be attributed to a detrimental isoform shift occurring.
Fig. 5.

α- and β-Myosin heavy chain (MyHC) isoform content. Several dilutions of the same sample are used to capture both α- and β-MyHC in the linear densiometric range. Skeletal soleus muscle was used as a standard to visualize separation of α (band on top)- and β (band on bottom)-isoforms. At 2 mo, WT or HCM mice of either sex display no significant expression of β-MyHC (<2%).
PTM of Sarcomeric Proteins
Global PTM.
Sarcomeric proteins are the target for many kinases and remain a central site for integration of regulatory signaling. Considering the differential Ca2+ sensitivity, ktr, and tension cost observed, we predicted that HCM males and females would have a unique pattern of proteins that were posttranslationally modified and potentially underlying differences in myofilament function. Using SDS-PAGE followed by ProQ Diamond phosphoprotein staining, we were able to quantify global phosphorylation levels for sarcomeric proteins, including cTnI, cTnT, myosin-binding protein C (MyBPC), and desmin (Fig. 6A).
Fig. 6.

ProQ Diamond phosphoprotein stain. A: representative lanes (n = 4/group) cropped from the same gel stained first for phosphorylation (ProQ diamond) and then for total protein (Coomassie). B: bar graph summary of ProQ diamond phosphostain normalized to Coomassie staining in female (top) and male (bottom) mice. Two-way ANOVA identified significant sex and HCM transgene effects and is summarized in text. *P < 0.05 from WT counterpart. C: tryptic digest of 97-kDa band resulted in 6 exclusive peptides and unique spectra highlighted in the peptide sequence of muscle glycogen phosphorylase accounting for 92/842 amino acids (11% coverage). D: LC-MS/MS spectrum of a single fragment with its associated sequence revealed in the inset.
Normalizing band intensity from ProQ diamond staining to Coomassie blue staining (Fig. 6B), we found a significant effect of sex on cMyBP-C PTM only. In addition, we determined that HCM impacted the levels of PTM for cMyBP-C, cTnI, and a 97-kDa protein. Mass spectrometry analysis (Fig. 6, C and D) identified the 97-kDa protein to be glycogen phosphorylase, a key energy-regulating protein that acts as the rate-limiting step in glycogenolysis by breaking up glycogen into glucose subunits. Finally, there was significant interaction between sex and HCM (disease) on PTM levels for desmin, cTnT, and cTnI. Post hoc analysis revealed that the HCM mutation elevated the 97-kDa protein (glycogen phosphorylase) in both males and females. On the other hand, PTM of cTnI and cTnT was significantly lower in WT females compared with HCM females.
Site-specific phosphorylation of cTnI and cMyBP-C.
In the heart, multiple kinase systems, including cAMP-dependent protein kinases A, D, G, and C and p21-activated kinase converge on myofilament proteins to regulate myofilament function (50). The two central and well-studied targets for PTM in the heart are cTnI and cMyBP-C. We and others have previously demonstrated that PTM of cTnI impacts myofilament and sarcomere dynamics (15, 29, 48). Considering the sex- and HCM-specific effects on the kinetics and energetics myofilament function, we interrogated the level of PTM at known target sites on cTnI and cMyBP-C.
Given that HCM males appear desensitized to Ca2+ and have increased cycling kinetics, we probed for phosphorylation of cTnI at Ser23/24 (p-cTnI-Ser23/24), known to decrease Ca2+ sensitivity when phosphorylated by Western blot analysis. We observed a sex-specific effect on the levels of p-cTnI-Ser23/24, with M-HCM having lower p-cTnI-Ser23/24 content than M-WT (Fig. 7A).
Fig. 7.

Western blot analysis of phospho (p)-cardiac troponin I (cTnI)-Ser23/24 and p-myosin-binding protein (cMyBP)-C-Ser282. Left ventricular myocardial samples from male, female, WT, or HCM mice (n = 4/group) were separated by SDS-PAGE. A: Western blot analysis and bar graph summary of p-cTnI-Ser23/24; phosphoprotein was normalized to total protein. B: Western blot analysis and bar graph summary of p-cMyBP-C-Ser282; phosphoprotein was normalized to total protein. Two-way ANOVA identified significant sex effect for p-cTnI-Ser23/24 but no difference for p-cMyBP-C-Ser282. *P < 0.05 from WT counterpart.
cMyBP-C also has multiple phosphorylation sites (45). Considering a sex-specific elevation of cMyBP-C by ProQ diamond staining, we examined cMyBP-C phosphorylation at Ser282 (p-cMyBP-C-Ser282), which is a prerequisite phosphorylation site for phosphorylation at other sites (46). Again, using Western blot analysis, we measured levels of p-cMyBP-C-Ser282 in all experimental groups. We did not observe any impact of sex or HCM in the levels of p-cMyBP-C-Ser282 (Fig. 7B).
Additionally, we probed for phosphorylation of cTnI at Ser150 (p-cTnI-Ser150), which is known to be the preferential site for phosphorylation by p21-activated kinase and adenosine monophosphate-activated protein kinase (31, 32). We did not observe any difference between sex and disease in p-cTnI-Ser150 levels (data not shown).
PhosTag.
Because cTnI and cMyBP-C harbor multiple phosphorylation sites, the specific distribution of phosphorylation on each respective protein may act to differentially impact myofilament function (21, 45, 49, 60). We were therefore interested if the HCM transgene or sex had an impact on the overall phosphospecies distribution of cTnI and cMyBP-C. To do this, we used PhosTag accompanied by site-specific phosphoantibodies to determine the distribution of cTnI- and cMyBP-C-phosphorylated species.
With the use of an antibody for total cTnI (Fig. 8A), three distinct bands were evident that we designated as high, mid, and low based on gel-shift mobility (3, 32). Using this convention, we found a significant transgene effect in the amount of the low and mid bands (Fig. 8A) and no significant differences in the low-mobility band. Moreover, the midmobility band was less in both F-HCM and M-HCM compared with WT counterparts.
Fig. 8.

Phosphate-affinity SDS-PAGE (SDS-PAGE-PhosTag) for total cTnI or cMyBP-C and p-cTnI at p-cTnI-Ser23/24 or cMyBP-C at p-cMyBP-C-Ser282. SDS-PAGE-PhosTag of left ventricular myocardial samples from male, female, WT, or HCM mice (n = 4–6/group) followed by Western blot analysis with cTnI antibodies targeting p-cTnI-Ser23/24 or p-cMyBP-C-Ser282 only when phosphorylated. A, top: representative SDS-PAGE-PhosTag followed by Western blot with a total cTnI (left) or p-cTnI-Ser23/24 (right) antibody illustrating distinct bands corresponding to low, mid, and high gel mobility. Stacked bar graph summary indicating the relative amounts of phosphospecies for each experimental group. B, top: representative SDS-PAGE-PhosTag followed by Western blot with a total cMyBP-C or p-cMyBP-C-Ser282 antibody illustrating 2 distinct bands corresponding to low and high gel mobility. Stacked bar graph summary indicating the relative amounts of phosphospecies. Each lane was normalized to total cTnI or cMyBP-C. A 2-way ANOVA followed by a Bonferroni post hoc test determined differences between HCM and WT groups. *P < 0.05 from cTnI WT counterpart; *,*P < 0.05 when comparing low- and high-mobility bands between HCM and WT males.
PhosTag was performed again using a site-specific antibody for p-cTnI-Ser23/24 (3). Again, we defined the bands based on mobility such that we identified a low- and midmobility band, which correspond with the low- and midmobility bands of total cTnI (Fig. 8). There was a significant interaction between sexes and the HCM transgene, with HCM males demonstrating significantly less of the mid- and low-mobility bands compared with WT males. We additionally used PhosTag with a site-specific antibody for p-cTnI-Ser150 (data not shown). There was no sex or disease-specific effect in the distribution of phosphorylation.
PhosTag followed by immunoblotting for total or p-Ser282-MyBPC revealed the presence of two phosphospecies designated as either high or low mobility as previously described (1). Despite some emerging trends, statistical analysis using a two-way ANOVA determined that there were no differences in distribution between either sex or HCM disease (Fig. 8B).
DISCUSSION
Because gender/sex is recognized as a potent modifier in cardiac disease progression and severity, the goal of our study was to examine this at the level of crossbridge kinetics and energy use. Our study supports this notion with several key findings but also points to the difficulty of attributing sex dimorphisms to a central cellular and/or molecular mechanism. The major findings of this study are 1) HCM males showed decreased Ca2+ sensitivity and increased crossbridge cycling at low activation levels, 2) HCM males had increased tension cost while HCM females show reduced tension cost, 3) both HCM males and HCM females displayed a similar “stress” response with an increase in glycogen phosphorylase, and 4) both sex and disease impacted myofilament phosphorylation content and patterning.
Previous studies involving the R403Q mutation either from human tissue or mouse models point toward a gain-of-function at higher energy cost (5, 39, 57). The findings herein suggest that hearts from male HCM mice were consistent with previous observations indicated by a relative increase in crossbridge cycling rates and higher tension cost. We also found differences in Ca2+ sensitivity between HCM males and HCM females, where HCM male trabeculas displayed Ca2+ desensitization compared with WT counterparts, whereas HCM females did not. This is different from a previous study that found no sex-dependent differences in Ca2+ sensitivity at a similar age (10–20 wk). However, a potential cause for this discrepancy is that the prior study used a different R403Q model than the one used for the current study (39).
Kinetic parameters of the crossbridge cycle were determined through ktr. We found a significant effect of the HCM transgene on ktr represented as a shallower slope in HCM males compared with WT males and females of either group. An increase in submaximal ktr indicates that, at a given force, myosin in HCM males must cycle at a faster rate of attachment and detachment than WT males. Previous work on human heart samples carrying the R403Q mutation also demonstrates increased rates of an equivalent parameter (5).
To better interpret the underlying mechanism of this increased rate of tension redevelopment, we measured the rate of force-dependent ATP hydrolysis at varying [Ca2+] to determine the tension cost of contraction. We found a strong interaction between sex and the HCM mutation in tension cost. This indicates that HCM males have a significantly increased tension cost compared with their WT counterparts. To interpret this combinatorial data, we use a two-state crossbridge model, with ktr representing the summed rate of the crossbridges entering and exiting (fapp + gapp, respectively) a force-generating state (8). The increased ktr and increased tension cost (gapp) for HCM males indicates that gapp (crossbridge detachment) must be increasing, with fapp (crossbridge attachment) either increasing or remaining the same. The suggestion is more inefficient use of ATP to generate force and a gain-of-function, matching studies in human R403Q cardiac fibers (5, 57). For the first time, we showed that female HCM fibers demonstrate reduced cost of tension and, consequently, a lower detachment rate, or gapp. With an unchanged ktr, female fibers are more efficient at using ATP to generate force.
Of particular interest was this sex dimorphism in force-dependent ATP utilization, especially since these mice are age matched at an early time point where there is little evidence of pathological cardiac remodeling. Hallmarks of cardiac remodeling include cardiac hypertrophy, a recapitulation of the fetal gene program that includes elevated expression of β-MyHC, and cardiac fibrosis (42). While both male and female HCM hearts did not show an elevation of cardiac collagen deposition, male HCM hearts were significantly larger than WT males and females from each group. On the other hand, HCM females demonstrated an increase in EF% indicative of a hypercontractile state. To correlate this HCM phenotype with pathological markers, we examined expression of β-MyHC. Expression of β-MyHC can negatively impact cardiac performance (54) by decreasing power output (24) or increasing the Ca2+ sensitivity of tension development (20, 43). On the other hand, fibers expressing β-MyHC have an improved tension cost (44). Interestingly, both female and male HCM hearts expressed little, if any, β-MyHC despite having altered EF%, crossbridge cycling, and mechanics. This clearly illustrates that the HCM mutation manifests differently in each sex at an early time point, yet, the exact cause underlying this HCM phenotype cannot be explicitly attributed to a “pathological” phenotype. The suggestion is that sex-specific markers must be elucidated early on that will predict the HCM phenotype in each sex.
To gain a better understanding of the cellular and molecular mechanisms underlying sex-specific crossbridge mechanics, we examined PTM of key myofilament proteins known to impact sarcomere function. PTM of myofilament proteins is dynamically regulated by several kinases and phosphatases and has known effects on Ca2+ sensitivity (29), crossbridge cycling rate (27), and Ca2+-binding characteristics (6). The β-adrenergic pathway and resultant activation of PKA is central to acute and long-term cardiac adaptation under stress (31). PKA targeting of cTnI (Ser23/24) and cMyBP-C (Ser273/282/302) decreases the sensitivity of the myofilaments to Ca2+ (14, 29) and accelerates the rate of crossbridge cycling (2, 9, 27, 35, 41). Moreover, recent work attributes sex dimorphisms in cardiac adaptation to a mutant MyBP-C transgene (34). Using SDS-PAGE and phosphospecific antibodies, we found a significant sex effect in p-cTnI-Ser23/24 characterized by a decrease in males with a further decrease in HCM males. On the other hand, we found no differences in p-cMyBP-C-Ser282, which is a prerequisite phosphorylation site for phosphorylation at other sites (46). Considering the ability of p-cMyBP-C-Ser282 to mediate, in part, kinase targeting at alternative sites (21), it is not surprising that differences in phosphorylation levels may exist between sites on unique proteins (p-cTnI-Ser23/24 and p-cMyBP-C-Ser282) targeted by the same kinase. Clearly, compartmentalized regulation exists within myofilament “microdomains” to control kinase activity. Incidentally, we found no differences in p-cTnI-Ser150 at this time point differing from our previous work showing sex and disease differences in in HCM mice at a much later time point (12 mo) (32).
Similarly, SDS-PAGE PhosTag of cTnI and cMyBP-C revealed a unique patterning of PTM that was dependent on sex, the HCM transgene, and the interaction between sex and HCM. Moreover, the site-specific distribution of PTM proteins paralleled our other methods of PTM interrogation but cannot directly underlie crossbridge kinetics and energetics. Inconsistencies in PTM of cTnI, cMyBP-C, and crossbridge myofilament Ca2+ sensitivity are not surprising given that multiple kinase pathways converge on cTnI and cMyBP-C, resulting in at least 10 (cTnI) and 4 (cMyBP-C) possible phosphorylation sites (45, 49), and that the myofilaments act as a nodal point for multiple cellular kinases. Therefore, it is important to note that, while SDS-PAGE PhosTag is highly useful for providing information on phosphorylation distribution, it is likely that this technique is not able to distinguish all possible phosphorylation states and only has the resolution for the most prevalent states in the densiometric range. It is also likely that discrepancies and inconsistencies may arise between the techniques used in this study (ProQ-Diamond phosphostain, immunoblot of 1D SDS-PAGE and SDS-PAGE PhosTag) due to the nature of the phosphoprotein. Nevertheless, SDS-PAGE-PhosTag remains a useful tool for highlighting unique and distinct patterns of phosphorylation.
Of particular interest, ProQ Diamond showed increased phosphorylation of glycogen phosphorylase in both HCM males and females. Glycogen phosphorylase functions to catalyze the cleavage of glycogen into glucose units and can be activated either allosterically or by phosphorylation. This increase in activity indicates that both HCM males and females are most likely under energy stress, forcing an increase in glucose metabolism. The suggestion is that HCM females have compensatory mechanisms in place that alter crossbridge kinetics differently from HCM males. The disease- and sex-specific effects are summarized in Table 3.
Table 3.
Summary of disease- or sex-specific effects
| Factor | Sex | HCM | Sex × HCM | Post Hoc |
|---|---|---|---|---|
| Ca2+ sensitivity | * | − | − | ↓M-HCM |
| ktr | − | * | − | ↑M-HCM |
| Tension cost | − | − | * | ↑M-HCM |
| Cardiac remodeling | ||||
| Collagen, % | − | − | − | − |
| HW/TL | * | * | * | ↑M-HCM |
| EF, % | − | − | * | ↑F-HCM |
| RWTd | − | − | − | − |
| ProQ Diamond | ||||
| cMyBP-C | * | * | − | − |
| 97 kDa | − | * | − | ↑F/M-HCM |
| Desmin | − | − | * | − |
| cTnT | − | − | * | ↑F-HCM |
| cTnI | − | * | * | ↑F-HCM |
| Western blot | ||||
| p-cTnI-Ser23/24 | * | − | − | ↓M-HCM |
| p-cMyBP-C-Ser282 | − | − | − | − |
Values are means ± SE; n, no. of subjects. EF, ejection fraction; RWTd, relative wall thickness during diastole; cMyBP-C, cardiac myosin-binding protein C; cTnT, cardiac troponin T; cTnI, cardiac troponin I, p, phospho. This summarizes the observed effects of sex and the HCM transgene in WT, HCM, male, and female mice. Using a 2-way ANOVA followed by Bonferroni's post hoc test to compare differences between groups, we designate a significant
(P < 0.05) effect or no change (−) with each factor. In addition, post hoc analysis determined whether HCM increased (↑) or decreased (↓) the parameter with respect to sex.
Physiological Implications and Future Directions
In this study, we illustrate sex-dependent differences in crossbridge kinetics in cardiac trabeculas from HCM hearts. HCM males showed an increase in tension cost while HCM females showed a decrease in tension cost. This difference occurs early on in the absence of a severe pathological phenotype. Yet, at the level of the myofilament, there are indications that both HCM males and females are under energy stress presumably initiated by the HCM transgene. What is evident from studies using this HCM model is that, although the primary defect may reside in the sarcomere, the development of the HCM phenotype depends on the interaction of the initiated signaling pathways, environmental stressors, and individual genotype (including sex/gender). The suggestion is, however, that HCM females recruit a unique cellular pathway to improve tension cost by altering crossbridge kinetics. Future studies may be targeted at identifying these pathways, which appear to be activated (or deactivated) very early on.
GRANTS
This work was supported by National Institutes of Health (NIH) Grant HL-098256, by a National Mentored Research Science Development Award (K01-AR-052840), and an Independent Scientist Award (K02-HL-105799) from the NIH awarded to J. P. Konhilas, the Interdisciplinary Training in Cardiovascular Research (HL-007249), and the Cardiovascular Biomedical Engineering Training Grant (HL-007955). Support was received from the Sarver Heart Center at the University of Arizona and the Steven M. Gootter Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.B. and J.P.K. conception and design of research; C.B., S.B., M.L.-P., and C.S. performed experiments; C.B., S.B., M.L.-P., C.S., and H.G. analyzed data; C.B., S.B., C.D., Y.L., H.G., and J.P.K. interpreted results of experiments; C.B. and J.P.K. prepared figures; C.B. and J.P.K. drafted manuscript; C.B., H.G., and J.P.K. edited and revised manuscript; H.G. and J.P.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Eleni Constantopoulos for help with animal maintenance.
REFERENCES
- 1.Ackermann MA, Kerr JP, King B, CWW, Kontrogianni-Konstantopoulos A. The phosphorylation profile of myosin binding protein-C slow is dynamically regulated in slow-twitch muscles in health and disease. Sci Rep 5: 12637, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bardswell SC, Cuello F, Rowland AJ, Sadayappan S, Robbins J, Gautel M, Walker JW, Kentish JC, Avkiran M. Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J Biol Chem 285: 5674–5682, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behunin SM, Lopez-Pier MA, Birch CL, McKee LA, Danilo C, Khalpey Z, Konhilas JP. LKB1/Mo25/STRAD uniquely impacts sarcomeric contractile function and posttranslational modification. Biophys J 108: 1484–1494, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belin RJ, Sumandea MP, Kobayashi T, Walker LA, Rundell VL, Urboniene D, Yuzhakova M, Ruch SH, Geenen DL, Solaro RJ, de Tombe PP. Left ventricular myofilament dysfunction in rat experimental hypertrophy and congestive heart failure. Am J Physiol Heart Circ Physiol 291: H2344–H2353, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Belus A, Piroddi N, Scellini B, Tesi C, D'Amati G, Girolami F, Yacoub M, Cecchi F, Olivotto I, Poggesi C. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J Physiol 586: 3639–3644, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biesiadecki BJ, Kobayashi T, Walker JS, John Solaro R, de Tombe PP. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ Res 100: 1486–1493, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PA. Troponin I phosphorylation in the normal and failing adult human heart. Circulation 96: 1495–1500, 1997. [DOI] [PubMed] [Google Scholar]
- 8.Brenner B, Eisenberg E. Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc Natl Acad Sci USA 83: 3542–3546, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 573: 188–197, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Hwang H, McKee LA, Perez JN, Regan JA, Constantopoulos E, Lafleur B, Konhilas JP. Temporal and morphological impact of pressure overload in transgenic FHC mice. Frontiers Physiol 4: 205, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuda G, Fananapazir L, Epstein ND, Sellers JR. The in vitro motility activity of beta-cardiac myosin depends on the nature of the beta-myosin heavy chain gene mutation in hypertrophic cardiomyopathy. J Muscle Res Cell Motil 18: 275–283, 1997. [DOI] [PubMed] [Google Scholar]
- 12.de Tombe PP, Solaro RJ. Integration of cardiac myofilament activity and regulation with pathways signaling hypertrophy and failure. Ann Biomed Eng 28: 991–1001, 2000. [DOI] [PubMed] [Google Scholar]
- 13.de Tombe PP, Stienen GJ. Impact of temperature on cross-bridge cycling kinetics in rat myocardium. J Physiol 584: 591–600, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Tombe PP, Stienen GJ. Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circ Res 76: 734–741, 1995. [DOI] [PubMed] [Google Scholar]
- 15.de Tombe PP, Stienen GJM. Protein Kinase A does not alter economy of force maintenance in skinned cardiac trabeculae. Circ Res 76: 734–741, 1995. [DOI] [PubMed] [Google Scholar]
- 16.de Windt LJ, Willems J, Reneman RS, Van der Vusse GJ, Arts T, Van Bilsen M. An improved isolated, left ventricular ejecting, murine heart model. Functional and metabolic evaluation. Pflugers Arch 437: 182–190, 1999. [DOI] [PubMed] [Google Scholar]
- 17.Dimitrow PP, Czarnecka D, Kawecka-Jaszcz K, Dubiel JS. The influence of age on gender-specific differences in the left ventricular cavity size and contractility in patients with hypertrophic cardiomyopathy. Int J Cardiol 88: 11–17, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res 113: 709–724, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Yeh YH, Nattel S, Dobrev D, Eschenhagen T, Carrier L. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J Mol Cell Cardiol 43: 223–229, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Ford SJ, Mamidi R, Jimenez J, Tardiff JC, Chandra M. Effects of R92 mutations in mouse cardiac troponin T are influenced by changes in myosin heavy chain isoform. J Mol Cell Cardiol 53: 542–551, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J 14: 1952–1960, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science 272: 731–734, 1996. [DOI] [PubMed] [Google Scholar]
- 23.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 62: 999–1006, 1990. [DOI] [PubMed] [Google Scholar]
- 24.Herron TJ, Korte FS, McDonald KS. Loaded shortening and power output in cardiac myocytes are dependent on myosin heavy chain isoform expression. Am J Physiol Heart Circ Physiol 281: H1217–H1222, 2001. [DOI] [PubMed] [Google Scholar]
- 25.Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res 81: 412–419, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res 81: 412–419, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Phosphorylation of troponin i by protein kinase a accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res 88: 1059–1065, 2001. [DOI] [PubMed] [Google Scholar]
- 28.Konhilas JP, Watson PA, Maass A, Boucek DM, Horn T, Stauffer BL, Luckey SW, Rosenberg P, Leinwand LA. Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circ Res 98: 540–548, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Konhilas JP, Wolska B, Martin AF, Solaro RJ, de Tombe PP. PKA modulates length-dependent activation in murine myocardium. Biophys J 78: 108A, 2000. [Google Scholar]
- 30.Kooij V, Saes M, Jaquet K, Zaremba R, Foster DB, Murphy AM, Dos Remedios C, van der Velden J, Stienen GJ. Effect of troponin I Ser23/24 phosphorylation on Ca2+-sensitivity in human myocardium depends on the phosphorylation background. J Mol Cell Cardiol 48: 954–963, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res 66: 12–21, 2005. [DOI] [PubMed] [Google Scholar]
- 32.McKee LA, Chen H, Regan JA, Behunin SM, Walker JW, Walker JS, Konhilas JP. Sexually dimorphic myofilament function and cardiac troponin I phosphospecies distribution in hypertrophic cardiomyopathy mice. Arch Biochem Biophys 535: 39–48, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNally EM, Barefield DY, Puckelwartz MJ. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab 21: 174–182, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Najafi A, Schlossarek S, van Deel ED, van den Heuvel N, Guclu A, Goebel M, Kuster DW, Carrier L, van der Velden J. Sexual dimorphic response to exercise in hypertrophic cardiomyopathy-associated MYBPC3-targeted knock-in mice. Pflugers Arch 467: 1303–1317, 2015. [DOI] [PubMed] [Google Scholar]
- 35.Najafi A, Sequeira V, Helmes M, Bollen IA, Goebel M, Regan JA, Carrier L, Kuster DW, Van Der Velden J. Selective phosphorylation of PKA targets after beta-adrenergic receptor stimulation impairs myofilament function in Mybpc3-targeted HCM mouse model. Cardiovasc Res 110: 200–214, 2016. [DOI] [PubMed] [Google Scholar]
- 36.Neubauer S. The failing heart–an engine out of fuel. N Engl J Med 356: 1140–1151, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Olsson MC, Palmer BM, Leinwand LA, Moore RL. Gender and aging in a transgenic mouse model of hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 280: H1136–H1144, 2001. [DOI] [PubMed] [Google Scholar]
- 38.Palmer BM, Fishbaugher DE, Schmitt JP, Wang Y, Alpert NR, Seidman CE, Seidman JG, VanBuren P, Maughan DW. Differential cross-bridge kinetics of FHC myosin mutations R403Q and R453C in heterozygous mouse myocardium. Am J Physiol Heart Circ Physiol 287: H91–H99, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Palmer BM, Wang Y, Teekakirikul P, Hinson JT, Fatkin D, Strouse S, Vanburen P, Seidman CE, Seidman JG, Maughan DW. Myofilament mechanical performance is enhanced by R403Q myosin in mouse myocardium independent of sex. Am J Physiol Heart Circ Physiol 294: H1939–H1947, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Palmiter KA, Tyska MJ, Haeberle JR, Alpert NR, Fananapazir L, Warshaw DM. R403Q and L908V mutant beta-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J Muscle Res Cell Motil 21: 609–620, 2000. [DOI] [PubMed] [Google Scholar]
- 41.Patel JR, Fitzsimons DP, Buck SH, Muthuchamy M, Wieczorek DF, Moss RL. PKA accelerates rate of force development in murine skinned myocardium expressing α- or β-tropomyosin. Am J Physiol Heart Circ Physiol 280: H2732–H2739, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim HS, Smithies O, Rockman HA. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest 116: 1547–1560, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rice R, Guinto P, Dowell-Martino C, He H, Hoyer K, Krenz M, Robbins J, Ingwall JS, Tardiff JC. Cardiac myosin heavy chain isoform exchange alters the phenotype of cTnT-related cardiomyopathies in mouse hearts. J Mol Cell Cardiol 48: 979–988, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rundell VL, Manaves V, de Tombe PP. Cardiac myofilament tension cost in rats: relation to MHC isoform content. Biophys J 82: 397a, 2002. [Google Scholar]
- 45.Sadayappan S, de Tombe PP. Cardiac myosin binding protein-C: redefining its structure and function. Biophys Rev 4: 93–106, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko VM, Lorenz JN, Maillet M, Martin JL, Brown JH, Bers DM, Molkentin JD, James J, Robbins J. A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ Res 109: 141–150, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen W, Asai K, Uechi M, Mathier MA, Shannon RP, Vatner SF, Ingwall JS. Progressive loss of myocardial ATP due to a loss of total purines during the development of heart failure in dogs: a compensatory role for the parallel loss of creatine. Circulation 100: 2113–2118, 1999. [DOI] [PubMed] [Google Scholar]
- 48.Solaro RJ. Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. J Biol Chem 283: 26829–26833, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Solaro RJ, Henze M, Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res 112: 355–366, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solaro RJ, Kobayashi T. Protein phosphorylation and signal transduction in cardiac thin filaments. J Biol Chem 286: 9935–9940, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, Ingwall JS. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest 101: 1775–1783, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stehle R, Iorga B. Kinetics of cardiac sarcomeric processes and rate-limiting steps in contraction and relaxation. J Mol Cell Cardiol 48: 843–850, 2010. [DOI] [PubMed] [Google Scholar]
- 53.Sweeney HL, Straceski AJ, Leinwand LA, Tikunov BA, Faust L. Heterologous expression of a cardiomyopathic myosin that is defective in its actin interaction. J Biol Chem 269: 1603–1605, 1994. [PubMed] [Google Scholar]
- 54.Tardiff JC, Hewett TE, Factor SM, Vikstrom KL, Robbins J, Leinwand LA. Expression of the beta (slow)-isoform of MHC in the adult mouse heart causes dominant-negative functional effects. Am J Physiol Heart Circ Physiol 278: H412–H419, 2000. [DOI] [PubMed] [Google Scholar]
- 55.Vikstrom KL, Factor SM, Leinwand LA. Mice expressing mutant myosin heavy chains are a model for familial hypertrophic cardiomyopathy. Mol Med 2: 556–567, 1996. [PMC free article] [PubMed] [Google Scholar]
- 56.Villar AV, Llano M, Cobo M, Exposito V, Merino R, Martin-Duran R, Hurle MA, Nistal JF. Gender differences of echocardiographic and gene expression patterns in human pressure overload left ventricular hypertrophy. J Mol Cell Cardiol 46: 526–535, 2009. [DOI] [PubMed] [Google Scholar]
- 57.Witjas-Paalberends ER, Ferrara C, Scellini B, Piroddi N, Montag J, Tesi C, Stienen GJ, Michels M, Ho CY, Kraft T, Poggesi C, van der Velden J. Faster cross-bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. J Physiol 592: 3257–3272, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamashita H, Tyska MJ, Warshaw DM, Lowey S, Trybus KM. Functional consequences of mutations in the smooth muscle myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. J Biol Chem 275: 28045–28052, 2000. [DOI] [PubMed] [Google Scholar]
- 59.Yar S, Monasky MM, Solaro RJ. Maladaptive modifications in myofilament proteins and triggers in the progression to heart failure and sudden death. Pflugers Arch 466: 1189–1197, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang P, Kirk JA, Ji W, dos Remedios CG, Kass DA, Van Eyk JE, Murphy AM. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation 126: 1828–1837, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]