

Abstract
Background
Leishmaniasis is a neglected tropical parasitic diseases affecting millions of people around the globe. Quinazolines are a group of compounds with diverse pharmacological activities. Owing to their promising antileishmanial activities, some 3-aryl-2-(substitutedstyryl)-4(3H)-quinazolinones were synthesized in good yields (65.2% to 86.4%).
Results
The target compounds were synthesized by using cyclization, condensation, and hydrolysis reactions. The structures of the synthesized compounds were determined using elemental microanalysis, infrared (IR), and proton nuclear magnetic resonance (1H NMR). The in vitro antileishmanial activities of the synthesized compounds were evaluated using Leishmania donovani strain. All the synthesized compounds displayed appreciable antileishmanial activities (IC50 values, 0.0128 to 3.1085 μg/ml) as compared to the standard drug miltefosine (IC50 = 3.1911 μg/ml). (E)-2-(4-chlorostyryl)-3-p-tolyl-4(3H)-quinazolinone (7) is the compound with the most promising antileishmanial activities (IC50 = 0.0128 μg/ml) which is approximately 4 and 250 times more active than the standard drugs amphotericin B deoxycholate (IC50 = 0.0460 μg/ml) and miltefosine (IC50 = 3.1911 μg/ml), respectively.
Conclusions
The results obtained from this investigation indicate that the synthesized and biologically evaluated quinazoline compounds showed promising antileishmanial activities and are good scaffolds for the synthesis of different antileishmanial agents.
Electronic supplementary material
The online version of this article (doi:10.1186/s13588-014-0010-1) contains supplementary material, which is available to authorized users.
Keywords: Quinazolinones, Leishmania, Antileishmanial activities
Background
Leishmanisis is a neglected tropical disease resulting from infection of macrophages by obligate intracellular parasites of the genus Leishmania[1]-[3]. It is a public health problem in at least 88 countries with an estimated 350 million people at risk. The estimated global prevalence of all forms of the disease is 12 million. Every year, 1.5 to 2 million new cases and 70,000 deaths occur due to cutaneous leishmaniasis (CL). In addition, 500,000 new cases and 59,000 deaths from visceral leishmaniasis (VL) occur annually [4]. The number of cases of leishmaniasis is increasing globally due to Leishmania/HIV co-infection [5],[6], international travel, and migration of immigrants and refugees from endemic regions [7],[8].
The prophylactic treatment of leishmaniasis mainly rely on vector and reservoir control [9]-[11]. Control of reservoir host and vector is difficult due to high coast, operational difficulties, and frequent relapses in the host [12]. Although considerable effort has been made to produce vaccine candidates for the treatment of leishmaniasis, there is no vaccine against any form of human leishmaniasis yet [13]-[17].
Pentavalent antimonials (SbV) have been used for the treatment of leishmania infections. Unfortunately, in many parts of the world, the parasite has become resistant to SbV[18]. Treatment failure to sodium stibogluconate (SSG) is observed in Eastern Sudan [19] and in Tigray, Northern Ethiopia [20]. Recent reports showed that pentamidine also developed resistance as well as difficulties in treating patients with Leishmania/HIV co-infection [21].
Combination chemotherapy has improved prospects for decreasing the emergence of drug resistance, increasing activity, and reducing required doses and thereby toxic side effects. In the previous study, WR 279,396 (a topical formulation containing 15% paromomycin and 0.5% gentamicin) was found to be safe and effective against CL caused by Leishmania major[22]. In addition, AmBisome-paromomycin is the most cost-effective combination among miltefosine-paromomycin and AmBisome-miltefosine [23]. So far, no combination chemotherapy has been used in treatment programs, except paromomycin/SSG [24].
Tremendous quinazoline derivatives are synthesized in the past two decades, using different synthetic pathways [25]-[30], due to their diverse pharmacological activities [31]-[36] including antileishmanial activities [37]-[40]. These reports indicate that several quinazolines were synthesized and tested for their antileishmanial activities, with the aim of discovering alternative chemotherapeutic agents for the development of new antileishmanials. Promising antileishmanial activities were observed in some 4-aminoquinazoline [37], indolo[2,1-b]quinazoline-6,12-dione [38], and 2,3-disubstituted-4(3H)-quinazolinone derivatives [39],[40]. As part of the efforts to discover less toxic and more effective drug analogues for the treatment of leishmaniasis, we synthesized some 2,3-disubstituted-4(3H)-quinazolinones and tested their in vitro antileishmanial activities.
Methods
Chemicals and reagents
Anthranilic acid, acetic anhydride, aniline, p-toluidine, o-toluidine, acetone, dimethylsulfoxide, anhydrous zinc chloride, p-chlorobenzaldehyde, p-nitrobenzaldehyde, p-hydroxybenzaldehyde, chloroform, absolute ethanol, resazurin sodium salt, anhydrous petroleum ether, distilled water, iodine, HCl, and KOH were used in the study.
Instruments and apparatuses
Melting points were determined in open capillaries using electro-thermal 9100 melting point apparatus and were uncorrected. Infrared (IR) spectra in nujol were recorded with the SHIMADZU 8400SP FT-IR spectrophotometer (Shimadzu Corporation, Nakagyo-ku, Kyoto, Japan), and proton nuclear magnetic resonance (1H NMR) spectral data were performed on Bruker Avance DMX400 FT-NMR spectrometer (Bruker, Billerica, MA, USA) using tetramethyl silane (TMS) as internal standard. Silica gel TLC plates of 0.25-mm thickness were used in the study.
Experimental animals and strains
Swiss albino male mice of weight 20 to 32 g and age 6 to 8 weeks (for acute toxicity test) were obtained from Biomedical Laboratory, Department of Biology, Faculty of Science, AAU. Leishmania donovani isolate used in this study was obtained from Leishmania Diagnosis and Research Laboratory (LDRL) culture bank, School of Medicine, AAU.
Culture medium and conditions
RPMI-1640, 10% heat-inactivated fetal calf serum (HIFCS), 1% penicillin-streptomycin, and 1% l-glutamine were supplied to make a complete culture medium. The L. donovani isolate was grown first on Novy-MacNeal-Nicolle (NNN) medium and then in tissue culture flasks containing RPMI-1640 medium supplemented with 10% HIFCS and 1% 100 IU penicillin/ml-100 μg/ml streptomycin solution at 22°C for promastigotes.
Reference drugs
Miltefosine/hexadecylphosphocholine (AG Scientific, San Diego, CA, USA) and amphotericin B deoxyhcholate (Fungizone®, ER Squibb, Middlesex, UK) were employed as reference drugs in the in vitro antileishmanial activity testing of the synthesized compounds.
Preparation of stock and working solutions
Stock solutions of 10 mg/ml of the synthesized compounds were prepared by dissolving each compound in DMSO. Stock solutions were diluted using complete RPMI to obtain aliquots of 10 μg/ml. Then, threefold serial dilution with complete RPMI gave the final six working concentrations (10, 3.33, 1.11, 0.37, 0.12, and 0.04 μg/ml) of each of the synthesized compounds. Amphotericin B deoxycholate and miltefosine, which were used as a positive control for comparison of the antileishmanial activities of the test compounds, were also made in threefold serial dilutions. All the prepared drugs were stored at −20°C and retrieved only during use [41].
In vitro antileishmanial activity
In a 96-well microtiter plate, 100 μl of each of the seven threefold serial dilutions of synthesized compounds were added in triplicate wells. Then, 100 μl of suspension of parasites (3.0 × 106 promastigotes/ml of L. donovani) was added in duplicate. Some of the wells contained only the parasites which served as a positive control. The media and DMSO alone acted as a negative control. The contents of the plates were then maintained in humidified atmosphere at 22°C under 5% CO2.
After 68 h of incubation, 10 μl of fluorochrome resazurin solution (12.5 mg dissolved in 100 ml of distilled water) was added into each well. The fluorescence intensity was measured after a total incubation period of 72 h using Victor3 Multilabel Counter (PerkinElmer, Waltham, MA, USA), at an excitation wavelength of 530 nm and emission wavelength of 590 nm [42]. The IC50 values were evaluated from sigmoidal dose-response curves using GraphPad Prism 5.0 software (GraphPad Software, Inc., San Diego, CA, USA).
In vivo acute toxicity test
The oral acute toxicity of compound 7 that exhibited promising antileishmanial activity was investigated using male Swiss albino mice (approximately 20 g each) following reported methods [43]. The experimental animals were divided into six groups (containing six mice per group) and fasted overnight. Groups 1-5 received compound 7 suspended in a vehicle containing 1% gum acacia, in doses of 10, 50, 100, 200, and 300 mg/kg, respectively. The sixth group received vehicle containing 1% gum acacia (served as a control group) at a maximum dose of 1 ml/100 g of body weight by oral route. The mice were observed closely for 24 h with special attention to the first 4 h. Acute toxicity signs were checked in the test mice.
Statistical analysis
The IC50 values for in vitro promastigotes assay of synthesized compounds were evaluated from sigmoidal dose-response curves using computer software GraphPad Prism 5.0.
Results and discussion
Chemistry of the synthesized compounds
Synthesis of the target compounds involved the formation of 2-5 and 10 as intermediates. It was accomplished using nucleophilic reaction, nucleophilic with ring opening and closing, condensation reaction, and hydrolysis reactions. The target compounds are synthesized in a good yield, which ranged from 65.2% to 86.4% (Table 1). All the synthesized compounds were readily soluble in DMSO and chloroform except compound 12 which is readily soluble in acetone. Spectral data (IR and 1H NMR) of the synthesized compounds were in full agreement with the proposed structures.
Table 1.
Physical constants and percent yield of the synthesized compounds
| Test compound | Molecular formula | Molecular weight (g/mol) | % yield | Melting point (°C) | Rfvalues [CHCl3/C6H6(9:1)] |
|---|---|---|---|---|---|
| 6 | C22H17ClN2O | 360.85 | 68.3 | 201 to 203 | 0.520 |
| 7 | C23H19ClN2O | 374.87 | 65.2 | 189 to 191 | 0.577 |
| 8 | C23H18N3O3 | 384.41 | 74.8 | 214 to 216 | 0.422 |
| 9 | C23H18N3O3 | 384.41 | 76.2 | 235 to 237 | 0.642 |
| 11 | C26H24N2O4 | 428.49 | 86.4 | 151 to 153 | 0.781 |
| 12 | C22H18N2O2 | 342.40 | 80.3 | 298 to 300 | 0.524 |
| 13 | C24H22N2O3 | 386.45 | 82.2 | 196 to 198 | 0.711 |
Biological activity testing results
In vitro antileishmanial activity of the synthesized compounds
The antipromastigote activities of the synthesized compounds and the standard antileishmanial drugs (amphotericin B deoxycholate and miltefosine) were evaluated using the clinical isolate of L. donovani strain. The IC50 of the synthesized and reference drugs were evaluated from fluorescence characteristic of AlamarBlue® (resazurin) (Trek Diagnostic Systems, Inc., Cleveland, OH, USA) which is soluble, stable in culture medium, non-toxic to cells, and does not affect the secretary abilities of cells [44]. The test works as a cell viability and proliferation indicator through the conversion of resazurin to resorufin via reduction. The amount of fluorescence produced is proportional to the number of living cells [45],[46].
The quinazolinone derivatives synthesized were shown to have good antileishmanial activity which was in line with the previous reports [37]-[40]. All the tested compounds exhibited better antileishmanial activity than the standard drug miltefosine as shown in Table 2. Among them, compound 7 was found to have a very promising antileishmanial activity with an IC50 value of 0.0128 μg/ml which was 250 times superior than miltefosine (3.1911 μg/ml). Compounds 8 and 11 were 30 times more active than miltefosine. Compounds 6 and 12 were 10 times and twice more active than miltefosine, respectively. Compounds 9 and 13 were as active as miltefosine.
Table 2.
Antipromastigote activity (IC 50 ) of the synthesized compounds
| Test compounds | IC50values (μg/ml) | IC50values (ng/ml) |
|---|---|---|
| 6 | 0.3014 | 301.40 |
| 7 | 0.0128 | 12.80 |
| 8 | 0.1085 | 108.50 |
| 9 | 2.7017 | 2,701.70 |
| 11 | 0.1086 | 108.60 |
| 12 | 1.6472 | 1,647.20 |
| 13 | 3.1085 | 3,108.50 |
| Miltefosine | 3.1911 | 3,191.10 |
| Amphotericin B | 0.0460 | 46.00 |
IC50: effective concentration required to achieve 50% growth inhibition (in μg/ml).
All the synthesized compounds except compound 7 displayed weak antileishmanial activities as compared to amphotericin B deoxycholate. Better antipromastigote activity was observed for (E)-2-(4-chlorostyryl)-3-p-tolyl-4(3H)-quinazolinone (7) with an IC50 value of 0.0128 μg/ml which is four times higher than the standard drug amphotericin B deoxycholate with an IC50 value of 0.0128 μg/ml. (E)-2-(4-chlorostyryl)-3-p-tolyl-4(3H)-quinazolinone (7) was found to be 4 times more active than amphotericin B deoxycholate and 250 times more active than miltefosine.
Oral acute toxicity study
Compound (E)-2-(4-chlorostyryl)-3-p-tolyl-4(3H)-quinazolinone (7) was observed to be devoid of any inherent acute toxicities at a maximum dose of 300 mg/kg.
Experimental
Synthesis of target compounds
The synthesis of target compounds, 3-aryl-2-(substitutedstyryl)-4(3H)-quinazolinones (6-9 and 11-13), was achieved using cyclization, condensation, and hydrolysis reactions. It involved the synthesis of acetanthranil (2-methyl-3,1-benzoxazin-4-one (2)) and 3-aryl-2-methyl-4(3H)-quinazolinones (3-5) as intermediates (Scheme 1). The details of each reactions and reaction conditions, the summarized characteristic stretching and bending IR vibration frequencies, the elemental microanalysis, and the 1H NMR chemical shift data for each of the synthesized target compounds are given below.
Scheme 1.
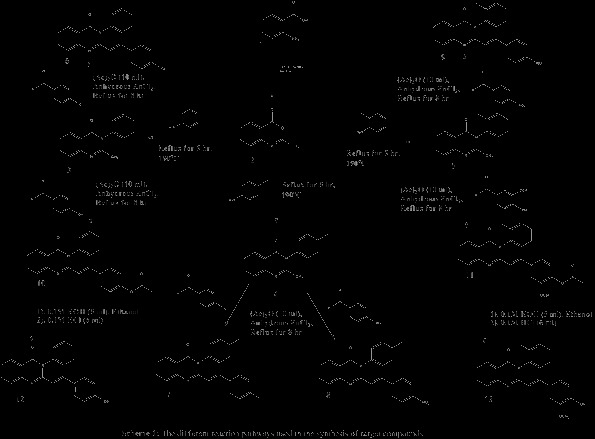
Synthesis of 3-aryl-2-(substitutedstyryl)-4(3 H )-quinazolinones using acetanthranil (2-methyl-3,1-benzoxazin-4-one (2)) and 3-aryl-2-methyl-4(3 H )-quinazolinones (3-5) as intermediates.
General procedure for the synthesis of 2-methyl-3,1-benzoxazin-4-one (2)
A solution of anthranillic acid (1) (10 g, 0.073 mol) in acetic anhydride (25 ml) was heated under reflux for 1 h. The precipitate formed on cooling was filtered and the excess acetic anhydride was washed with anhydrous petroleum ether, where upon a solid mass is obtained. This solid mass (2), without purification, was used for subsequent reaction [47].
General procedure for the synthesis of 3-aryl-2-methyl-4(3H)-quinazolinones (3-5)
A mixture of 2-methyl-3,1-benzoxazin-4-one (2) (3 g, 0.017 mol) and equimolar amounts of aromatic amines (aniline, p-toluidine, and o-toluidine, respectively) was heated under reflux at 190°C for 5 h. The dark sticky mass formed were cooled and recrystallized from ethanol to yield compounds 3-5, respectively [48].
General procedure for the synthesis of 3-aryl-2-(4-chlorostyryl)-4(3H)-quinazolinones (6 and 7)
To a solution of 3 or 4 (0.5 g each) in acetic anhydride (10 ml), an equimolar amount of p-chlorobenzaldehyde was added in the presence of 10 mg of anhydrous zinc chloride as a catalyst. The reaction mixture was heated under reflux for 8 h, cooled, and poured into ice-cooled water. The solid products formed (6 or 7) were filtered, dried, and recrystallized from chloroform/ethanol (2:1) [49].
E)-2-(4-chlorostyryl)-3-phenylquinazolin-4(3H)-one (6)
IR (Nujol) (cm−1): 1,682 (C = O), 1,597 (C = N), and 1,224 (C-Cl). 1H NMR (CDCl3) δ (ppm): 6.33 (d, 1H, J = 15.49 Hz, vinyl-C2 H), 7.23 (d, 2H, J = 8.53 Hz, 4-chlorophenyl C3,5 H), 7.28 (d, 2H, J = 8.47 Hz, 4-chlorophenyl C2,6 H), 7.34 (d, 2H, J = 6.82 Hz, phenyl C2,6 H), 7.45 to 7.49 (m, 1H, quina-C6 H), 7.58 to 7.63 (m, 3H, phenyl C3,4,5 H), 7.75 to 7.79 (m, 2H, quina-C7,8 H), 7.91 (d, 1H, J = 15.47 Hz, vinyl-C1 H), 8.29 (d, 1H, J = 7.95 Hz, quina-C5). Anal. calcd. for C22H17ClN2O: C, 73.23; H, 4.75; Cl, 9.83; N, 7.76. Found: C, 73.64; H, 4.92; Cl, 10.22; N, 7.54.
E)-2-(4-chlorostyryl)-3-p-tolylquinazolin-4(3H)-one (7)
IR (Nujol) (cm−1): 1,682 (C = O), 1,597 (C = N), and 1,224 (C-Cl). 1H NMR (CDCl3) δ (ppm): 2.5 (s, 3H, p- tolyl CH3), 6.42 (d, 1H, J = 15.70 Hz, vinyl-C2 H), 7.21 (d, 2H, 4-chlorophenyl C3,5 H), 7.26 to 7.32 (m, 4H, p-tolyl C2,3,5,6 H), 7.40 (d, 2H, 4-chlorophenyl C2,6 H), 7.47 to 7.51 (m, 1H, quina-C6 H), 7.79 to 7.83 (m, 2H, quina-C7,8 H), 7.93 (d, 1H, vinyl-C1 H), 8.32 (d, 1H, quina-C5 H). Anal. calcd. for C23H19ClN2O: C, 73.69; H, 5.12; Cl, 9.46; N, 7.47. Found: C, 73.98; H, 5.38; Cl, 9.35; N, 7.21.
General procedure for the synthesis of 3-aryl-2-(4-nitrostyryl)-4(3H)-quinazolinones (8 and 9)
To a solution of 4 or 5 (0.5 g each) in acetic anhydride (10 ml), an equimolar amount of p-nitrobenzaldehyde was added in the presence of 10 mg of anhydrous zinc chloride as a catalyst. The reaction mixture was heated under reflux for 8 h, cooled, and poured into ice-cooled water. The solid products formed (8 or 9) were filtered, dried, and recrystallized from chloroform/ethanol (2:1) [49].
E)-2-(4-nitrostyryl)-3-p-tolylquinazolin-4(3H)-one (8)
IR (Nujol) (cm−1): 1,684 (C = O), 1,593 (C = N), 1,556 and 1,377 (NO2). 1H NMR (CDCl3) δ (ppm): 2.5 (s, 3H, p- tolyl CH3), 6.56 (d, 1H, J = 15.52 Hz, vinyl-C2 H), 7.21 (d, 2H, J = 8.19 Hz, p- tolyl C3,5 H), 7.41 (d, 2H, J = 7.97 Hz, p-tolyl C2,6 H), 7.46 to 7.53 (m, 3H, 4-nitrophenyl C2,6 and quina-C6), 7.77 to 7.81 (m, 2H, quina-C7,8), 8.00 (d, 1H, J = 15.52 Hz, vinyl-C1), 8.19 (d, 2H, J = 8.74 Hz, 4-nitrophenyl C3,5), 8.30 (d, 1H, J = 8.01 Hz, quina-C5). Anal. calcd. for C23H18N3O3: C, 71.86; H, 4.72; N, 10.93. Found: C, 72.12; H, 4.35; N, 11.10.
E)-2-(4-nitrostyryl)-3-o-tolylquinazolin-4(3H)-one (9)
IR (Nujol) (cm−1): 1,682 (C = O), 1,593 (C = N), 1,556 and 1,377 (NO2). 1H NMR (CDCl3) δ (ppm): 2.17 (s, 3H, o-tolyl CH3), 6.47 (d, 1H, J = 15.66 Hz, vinyl-C2 H), 7.25 (d, 1H, J = 7.91 Hz, o-tolyl C3 H), 7.44 to 7.46 (m, 3H, 4-nitrophenyl C2,6 and o-tolyl C6 H), 7.47 to 7.58 (m, 3H, o-tolyl C4,5 and quina-C6 H), 7.82 to 7.89 (m, 2H, quina-C7,8 H), 8.05 (d, 1H, J = 15.56 Hz, vinyl-C1 H), 8.19 (d, 2H, J = 8.73 Hz, 4-nitrophenyl C4,6 H), 8.36 (d, 1H, J = 8.25 Hz, quina-C5 H). Anal. calcd. for C23H18N3O3: C, 71.86; H, 4.72; N, 10.93. Found: C, 71.68; H, 4.93; N, 11.24.
General procedure for the synthesis of 3-aryl-2-(4-acetylatedstyryl)-4(3H)-quinazolinones (10)
To a solution of 3 (0.5 g) in acetic anhydride (10 ml), an equimolar amount of p-hydroxybenzaldehyde was added. Anhydrous zinc chloride (10 mg) is added as a catalyst. The reaction mixture is heated under reflux for 8 h, cooled, and poured into ice-cooled water. The solid product formed (10) was filtered, dried, and recrystallized from ethanol [49].
General procedure for the synthesis of 3-aryl-2-(4-acetylatedstyryl)-4(3H)-quinazolinones (11)
To a solution of 5 (0.5 g) in acetic anhydride (10 ml), an equimolar amount of vanillin was added. Anhydrous zinc chloride (10 mg) is added as a catalyst. The reaction mixture is heated under reflux for 8 h, cooled, and poured into ice-cooled water. The solid product (11) was filtered, dried, and recrystallized from ethanol [49].
1E)-2-[-3,4-dihydro-3-(2-methylphenyl)-4-oxoquinazoline-2-yl)]vinyl}-2-methoxyphenyl acetate (11)
IR (Nujol) (cm−1): 1,761 (C = O), 1,682 (C = O), 1,634 (C = N), 1,260 and 1,149 (C-O-C). 1H NMR (CDCl3) δ (ppm): 2.15 (s, 3H, phenylacetate CH3), 2.33 (s, 3H, o-tolyl CH3), 3.80 (s, 3H, methoxy -O-CH3), 6.27 (d, 1H, J = 15.44 Hz, vinyl-C2 H), 6.88 to 6.93 (m, 2H, 2-methoxyphenyl C3,5 H), 6.98 (d, 1H, J = 8.12 Hz, 2-methoxyphenyl C6 H), 7.24 (d, 1H, J = 7.52 Hz, o-tolyl C3 H), 7.42 to 7.53 (m, 4H, o-tolyl C4,5,6 H and quina-C6 H), 7.82 to 7.83 (m, 2H, quina-C7,8 H), 7.96 (d, 1H, J = 15.48 Hz, vinyl-C1 H), 8.34 (d, 1H, J = 7.88 Hz, quina-C5 H). Anal. calcd. for C26H24N2O4: C, 72.88; H, 5.65; N, 6.54. Found: C, 73.11; H, 5.89; N, 6.42.
General procedure for the synthesis of 3-aryl-2-(4-deacetylatedstyryl)-4(3H)-quinazolinones (12 and 13)
Subsequent treatment of 10 and 11 with 0.1 M alcoholic KOH (5 ml) in the presence of ethanol followed by 0.1 M HCl (6 ml) gave the corresponding 4-hydroxyl containing compounds 12 and 13 after recrystallization from ethanol [49].
E)-2-(4-hydroxystyryl)-3-phenylquinazolin-4(3H)-one (12)
IR (Nujol) (cm−1): 3,290 (OH), 1,652 (C = O), and 1,604 (C = N). 1H NMR (acetone-d6) δ (ppm): 6.24 (d, 1H, J = 15.39 Hz, vinyl-C2 H), 6.80 (d, 2H, J = 8.64 Hz, 4-hydroxyphenyl C3,5 H), 7.24 (d, 2H, J = 8.62 Hz, 4-hydroxyphenyl C2,6 H), 7.46 to 7.50 (m, 3H, phenyl C3,4,5 H), 7.60 to 7.66 (m, 3H, quina-C6, phenyl C2,6 H), 7.73 (d, 1H, J = 8.07 Hz, quina-C8 H), 7.81 to 7.85 (m, 1H, quina-C7 H), 7.92 (d, 1H, J = 15.43 Hz, vinyl-C1 H), 8.02 (s, 1H, 4-hydroxyphenyl -OH), 8.17 (d, 1H, J = 9.18 Hz, quina-C5 H). Anal. calcd. for C22H18N2O2: C, 7.17; H, 5.23; N, 8.18. Found: C, 76.86; H, 5.02; N, 8.38.
E)-2-(4-hydroxy-3-methoxystyryl)-3-o-tolylquinazolin-4(3H)-one (13)
IR (Nujol) (cm−1): 3,400 (OH), 1,683 (C = O), 1,634 (C = N), 1,211 and 1,148 (C-O-C). 1H NMR (CDCl3) δ (ppm): 2.15 (s, 3H, o-tolyl CH3), 3.80 (s, 3H, 4-hydroxy-2-methoxyphenyl -O-CH3), 6.10 (s, 1H, 4-hydroxy-2-methoxyphenyl -OH), 6.27 (d, 1H, J = 15.44 Hz, vinyl-C2 H), 6.88 to 6.93 (m, 2H, 4-hydroxy-2-methoxyphenyl C3,5 H), 6.98 (d, 1H, J = 8.12 Hz, 4-hydroxy-2-methoxyphenyl C6 H), 7.24 (d, 1H, J = 7.52 Hz, o-tolyl C3 H), 7.42 to 7.53 (m, 4H, o-tolyl C4,5,6 H and quina-C6 H), 7.82 (m, 2H, quina-C7,8 H), 7.96 (d, 1H, J = 15.48 Hz, vinyl-C1 H), 8.34 (d, 1H, J = 7.884 Hz, quina-C5 H). Anal. calcd. for C24H22N2O3: C, 74.59; H, 5.74; N, 7.23. Found: C, 74.28; H, 5.96; N, 7.56.
Conclusions
Some 3-aryl-2-(substitutedstyryl)-4(3H)-quinazolinone derivatives were synthesized and tested for their antileishamanial activities. Most of the synthesized compounds displayed better antileishmanial activities as compared to the standard drug miltefosine and lower antileishmanial activity as compared to amphotericin B deoxycholate except (E)-2-(4-chlorostyryl)-3-p-tolyl-4(3H)-quinazolinone (7). Compound 7 showed pronounced antileishmanial activities as compared to miltefosine and amphotericin B deoxycholate. Thus, 2,3-disubstituted-4(3H)-quinazolinones containing an aromatic substitution at 3-position and substituted styryl moiety at 2-position represent a promising matrix for the development of antileishmanial agents.
Acknowledgements
The authors are thankful to Prof. Wondimagegn Mammo for his considerable support in running and interpreting the 1H NMR data. The following were acknowledged: The Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Alexandria University for performing the elemental microanalysis and providing some chemicals, Debre Markos University for granting study leave to Mr. Yihenew, and Addis Ababa University for financially supporting this research work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Footnotes
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Yihenew Simegniew Birhan, Email: yihenews@gmail.com.
Adnan Ahmed Bekhit, Email: adnbekhit@yahoo.com.
Ariaya Hymete, Email: hymete@yahoo.com.
References
- 1.Renslo AR, McKerrow JH. Drug discovery and development for neglected parasitic diseases. Nat Chem Biol. 2006;2:701–710. doi: 10.1038/nchembio837. [DOI] [PubMed] [Google Scholar]
- 2.McConville MJ, Souza D, Saunders E, Likic VA, Thomas N. Living in a phagolysosome; metabolism of Leishmania amastigotes. Trends Parasitol. 2007;23:368–375. doi: 10.1016/j.pt.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Sharma U, Singh S. Insect vectors of Leishmania: distribution, physiology and their control. J Vector Borne Dis. 2008;45:255–272. [PubMed] [Google Scholar]
- 4.Manandhar KD, Yadav TP, Prajapati VK, Kumar S, Rai M, Dube A, Srivastava ON, Sundar S. Antileishmanial activity of nano-amphotericin B deoxycholate. J Antimicrob Chemother. 2008;62:376–380. doi: 10.1093/jac/dkn189. [DOI] [PubMed] [Google Scholar]
- 5.Nascimento ET, Moura MLN, Queiroz JW, Barroso AW, Araujo AF, Rego EF, Wilson ME, Pearson RD, Jeronimo SM. The emergence of concurrent HIV-1/AIDS and visceral leishmaniasis in Northeast Brazil. Trans R Soc Trop Med Hyg. 2011;105:298–300. doi: 10.1016/j.trstmh.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cenderello G, Pasa A, Dusi A, Dentone C, Toscanini F, Bobbio N, Bondi E, Bono VD, Izzo M, Riccio G, Anselmo M, Giacchino R, Marazzi MG, Pagano G, Cassola G, Viscoli C, Ferrea G, De Maria A. Varied spectrum of clinical presentation and mortality in a prospective registry of visceral leishmaniasis in a low endemicity area of Northern Italy. BMC Infect Dis. 2013;13:248. doi: 10.1186/1471-2334-13-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herremans T, Pinelli E, Casparie M, Nozari N, Roelfsema J, Kortbeek L. Increase of imported leishmaniasis in the Netherlands: a twelve-year overview (1996-2007) Int Health. 2010;2:42–46. doi: 10.1016/j.inhe.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Ignatius R, Loddenkemper C, Woitzik J, Schneider T, Harms G. Localized leishmanial lymphadenopathy: an unusual manifestation of leishmaniasis in a traveler in southern Europe. Vector-Borne Zoonotic Dis. 2011;11(8):1213–1215. doi: 10.1089/vbz.2011.0642. [DOI] [PubMed] [Google Scholar]
- 9.Bray DP, Hamilton JGC. Insecticide-impregnated netting as a potential tool for long-lasting control of the leishmaniasis vector Lutzomyia longipalpis in animal shelters. Parasites Vectors. 2013;6:133. doi: 10.1186/1756-3305-6-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mascari TM, Stout RW, Foil LD. Laboratory evaluation of oral treatment of rodents with systemic insecticides for control of blood feeding sand flies (Diptera: Psychodidae) Vector-Borne Zoonotic Dis. 2012;12(8):699–704. doi: 10.1089/vbz.2011.0833. [DOI] [PubMed] [Google Scholar]
- 11.Chaves LF, Calzada JE, Rigg C, Valderrama A, Gottdenker NL, Saldaña A. Leishmaniasis sand fly vector density reduction is less marked in destitute housing after insecticide thermal fogging. Parasites Vectors. 2013;6:164. doi: 10.1186/1756-3305-6-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thakur CP, Kumar K. Post kala-azar dermal leishmaniasis: a neglected aspect of kala-azar control programmes. Ann Trop Med Parasitol. 1992;86:355–359. doi: 10.1080/00034983.1992.11812678. [DOI] [PubMed] [Google Scholar]
- 13.Topuzogullari M, Koc RC, Isoglu SD, Bagirova M, Akdeste Z, Elcicek S, Oztel ON, Baydar SY, Ates SC, Allahverdiyev AM. Conjugation, characterization and toxicity of lipophosphoglycan-polyacrylic acid conjugate for vaccination against leishmaniasis. J Biomed Sci. 2013;20:35. doi: 10.1186/1423-0127-20-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaur J, Kaur T, Kaur S. Studies on the protective efficacy and immunogenicity of Hsp70 and Hsp83 based vaccine formulations in Leishmania donovani infected BALB/c mice. Acta Trop. 2011;119:50–56. doi: 10.1016/j.actatropica.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 15.Abdian N, Gholami E, Zahedifard F, Safaee N, Rafati S. Evaluation of DNA/DNA and prime-boost vaccination using LPG3 against Leishmania major infection in susceptible BALB/c mice and its antigenic properties in human leishmaniasis. Experimental Parasitol. 2011;127:627–636. doi: 10.1016/j.exppara.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 16.Salazara MBM, Domíngueza JD, Estradaa JS, Bonillab CG, Becker I. Vaccination with Leishmania mexicana LPG induces PD-1 in CD8+ and PD-L2 in macrophages thereby suppressing the immune response: a model to assess vaccine efficacy. Vaccine. 2014;32:1259–1265. doi: 10.1016/j.vaccine.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 17.Carrión J, Folgueira C, Soto M, Fresno M, Requena JM. Leishmania infantum HSP70-II null mutant as candidate vaccine against leishmaniasis: a preliminary evaluation. Parasites Vectors. 2011;4:150. doi: 10.1186/1756-3305-4-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ait-Oudhia K, Gazanion E, Oury B, Vergnes B, Sereno D. The fitness of antimony-resistant Leishmania parasites: lessons from the field. Trends Parasitol. 2011;27:141–142. doi: 10.1016/j.pt.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Adam GK, Abdulla MA, Ahmed AA, Adam I. Maternal and perinatal outcomes of visceral leishmaniasis (kala-azar) treated with sodium stibogluconate in eastern Sudan. Int J Gynecol Obstet. 2009;107:208–210. doi: 10.1016/j.ijgo.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Dorlo TPC, Kager PA. Comment on: cutaneous and mucocutaneous leishmaniasis in Tigray, northern Ethiopia: clinical aspects and therapeutic concerns. Trans R Soc Trop Med Hyg. 2010;104:84–85. doi: 10.1016/j.trstmh.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 21.Bray PG, Barrett MP, Ward SA, Koning HP. Pentamidine uptake and resistance in pathogenic protozoa: past, present and future. Trends Parasitol. 2003;19:232–239. doi: 10.1016/S1471-4922(03)00069-2. [DOI] [PubMed] [Google Scholar]
- 22.Ben Salah A, Buffet PA, Morizot G, Ben Massoud N, Zaatour A, Ben Alaya N, Hamida NBH, El Ahmadi Z, Downs MT, Smith PL, Dellagi K, Grogl M. WR279,396, a third generation aminoglycoside ointment for the treatment of Leishmania major cutaneous leishmaniasis: a phase 2, randomized, double blind, placebo-controlled study. PLoS Negl Trop Dis. 2009;3:e432. doi: 10.1371/journal.pntd.0000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sundar S, Sinha PK, Rai M, Verma DK, Nawin K, Alam S, Chakravarty J, Vaillant M, Verma N, Pandey K, Kumari P, Lal CS, Arora R, Sharma B, Ellis S, Strub-Wourgaft N, Balasegaram M, Olliaro P, Das P, Modabber F. Comparison of short-course multidrug treatment with standard therapy for visceral leishmaniasis in India: an open-label, non-inferiority, randomised controlled trial. Lancet. 2011;377:477–486. doi: 10.1016/S0140-6736(10)62050-8. [DOI] [PubMed] [Google Scholar]
- 24.Melaku Y, Collin SM, Keus K, Gatluak F, Ritmeijer K, Davidson RN. Treatment of kala-azar in southern Sudan using a 17-day regimen of sodium stibogluconate combined with paromomycin: a retrospective comparison with 30-day sodium stibogluconate monotherapy. Am J Trop Med Hyg. 2007;77:89–94. [PubMed] [Google Scholar]
- 25.Adib M, Ansari S, Mohammadi A, Bijanzadeh HR. A novel, one-pot, solvent and catalyst-free synthesis of 2-aryl/alkyl-4(3H)-quinazolinones. Tetrahedron Lett. 2010;51:30–32. doi: 10.1016/j.tetlet.2009.06.034. [DOI] [Google Scholar]
- 26.Kumar M, Sharma K, Sharma DK. Diversity oriented one-pot three-component sequential synthesis of annulated benzothiazoloquinazolines. Org Med Chem Lett. 2012;2:10. doi: 10.1186/2191-2858-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nouira I, Kostakis IK, Dubouilh C, Chosson E, Iannelli M, Besson T. Decomposition of formamide assisted by microwaves, a tool for synthesis of nitrogen-containing heterocycles. Tetrahedron Lett. 2008;49:7033–7036. doi: 10.1016/j.tetlet.2008.09.135. [DOI] [Google Scholar]
- 28.Ye C, You J, Li XF, You R, Weng Y, Li J, Wang Y. Design, synthesis and anticoccidial activity of a series of 3-(2-(2-methoxyphenyl)-2-oxoethyl) quinazolinone derivatives. Pestic Biochem Physiol. 2010;97:194–198. doi: 10.1016/j.pestbp.2010.02.001. [DOI] [Google Scholar]
- 29.Omar MA, Conrad J, Beifuss U. Copper-catalyzed domino reaction between 1-(2-halophenyl)methanamines and amidines or imidates for the synthesis of 2-substituted quinazolinones. Tetrahedron. 2014;70:3061–3072. doi: 10.1016/j.tet.2014.02.066. [DOI] [Google Scholar]
- 30.Safaei HR, Shekouhy M, Shafiee V, Davoodi M. Glycerol based ionic liquid with a borone core: a new highly efficient and reusable promoting medium for the synthesis of quinazolinones. J Mol Liq. 2013;180:139–144. doi: 10.1016/j.molliq.2013.01.013. [DOI] [Google Scholar]
- 31.Fischer C, Shah S, Hughes BL, Nikov GN, Crispino JL, Middleton RE, Szewczak AA, Munoz B, Shearman MS. Quinazolinones as γ-secretase modulators. Bioorg Med Chem Lett. 2011;21:773–776. doi: 10.1016/j.bmcl.2010.11.111. [DOI] [PubMed] [Google Scholar]
- 32.Manivannan E, Chaturvedi SC. Analogue-based design, synthesis and molecular docking analysis of 2,3-diarylquinazolinones as non-ulcerogenic anti-inflammatory agents. Bioorg Med Chem. 2011;19:4520–4528. doi: 10.1016/j.bmc.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 33.Rhee H-K, Yoo JH, Lee E, Kwon YJ, Seo H-R, Lee Y-S, Choo H-YP. Synthesis and cytotoxicity of 2-phenylquinazolin-4(3H)-one derivatives. Eur J Med Chem. 2011;46:3900–3909. doi: 10.1016/j.ejmech.2011.05.061. [DOI] [PubMed] [Google Scholar]
- 34.Jagani CL, Sojitra NA, Vanparia SF, Patel TS, Dixit RB, Dixit BC: Microwave promoted synthesis and antimicrobial activity of 3-thiazole substituted 2-styryl-4(3H)-quinazolinone derivatives.J Saudi Chem Soc. doi:10.1016/j.jscs.2011.02.001
- 35.Shivananda MK, Holla BS. Antifungal activity studies of some quinazolinone derivatives. J Chem Pharm Res. 2011;3:83–86. [Google Scholar]
- 36.Špulák M, Pourová J, Vopršálová M, Mikušek J, Kuneš J, Vacek J, Ghavre M, Gathergood N, Pour M. Novel bronchodilatory quinazolines and quinoxalines: synthesis and biological evaluation. Eur J Med Chem. 2014;74:65–72. doi: 10.1016/j.ejmech.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 37.Berman JD, King M, Edwards N. Antileishmanial activities of 2,4-diaminoquinazoline putative dihydrofolate reductase inhibitors. Antimicrobial Agents Chemother. 1989;33:1860–1863. doi: 10.1128/AAC.33.11.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhattacharjee AK, Skanchy DJ, Jennings B, Hudson TH, Brendle JJ, Werbovetz KA. Analysis of stereoelectronic properties, mechanism of action and pharmacophore of synthetic indolo [2,1-b]quinazoline-6,12-dione derivatives in relation to antileishmanial activity using quantum chemical, cyclic voltammetry and 3-D-QSAR catalyst procedures. Bioorg Med Chem. 2002;10:1979–1989. doi: 10.1016/S0968-0896(02)00013-5. [DOI] [PubMed] [Google Scholar]
- 39.Arfan M, Khan R, Khan MA, Anjum S, Choudhary MI, Ahmad M. Synthesis and antileishmanial and antimicrobial activities of some 2,3-disubstituted 3H-quinazolin-4-ones. J Enzyme Inhib Med Chem. 2010;25:451–558. doi: 10.3109/14756360903309412. [DOI] [PubMed] [Google Scholar]
- 40.Fleita DH, Mohareb RM, Sakka OK. Antitumor and antileishmanial evaluation of novel heterocycles derived from quinazoline scaffold: a molecular modeling approach. Med Chem Res. 2013;22:2207–2221. doi: 10.1007/s00044-012-0213-9. [DOI] [Google Scholar]
- 41.Lara O, Raquel E, María A, Juan J, Francisco B, José M. In vitro effect of new formulations of amphotericin B on amastigote and promastigote forms of Leishmania infantum. Int J Antimicrob Agents. 2007;30:325–329. doi: 10.1016/j.ijantimicag.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 42.Tariku Y, Hymete A, Hailu A, Rohloff J. Essential-oil composition, antileishmanial and toxicity study of Artemisia abyssinica and Satureja punctata ssp. punctata from Ethiopia. Chem Biodivers. 2010;7:1013–1016. doi: 10.1002/cbdv.200900375. [DOI] [PubMed] [Google Scholar]
- 43.Lorke D. A new approach to practical acute toxicity test. Arch Toxicol. 1983;54:275–286. doi: 10.1007/BF01234480. [DOI] [PubMed] [Google Scholar]
- 44.Nakayama GR, Caton MC, Nova MP, Parandoosh Z. Assessment of the Alamar Blue assay for cellular growth and viability in vitro. J Immunol Methods. 1997;204:205–208. doi: 10.1016/S0022-1759(97)00043-4. [DOI] [PubMed] [Google Scholar]
- 45.Al-Nasiry S, Geusens N, Hanssens M, Luyten C, Pijnenborg R. The use of Alamar Blue assay for quantitative analysis of viability, migration and invasion of choriocarcinoma cells. Hum Reprod. 2007;25:1–6. doi: 10.1093/humrep/dem011. [DOI] [PubMed] [Google Scholar]
- 46.Shimony O, Jaffe CL. Rapid fluorescent assay for screening drugs on Leishmania amastigotes. J Microbiol Methods. 2006;75:196–200. doi: 10.1016/j.mimet.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 47.Kumar A, Sharma S, Bajaj K, Sharma S, Panwar H, Singh T, Srivastava VK. Some new 2,3,6-trisubstituted quinazolinones as potent anti-inflammatory, analgesic and COX-II inhibitors. Bioorg Med Chem. 2003;11:5293–5299. doi: 10.1016/S0968-0896(03)00501-7. [DOI] [PubMed] [Google Scholar]
- 48.Errede LA. Acylanthranils 1. The pathway of quinazolone formation in the reaction of acylanthranils with anilines. J Org Chem. 1976;41:1763–1765. doi: 10.1021/jo00872a021. [DOI] [Google Scholar]
- 49.Raffa D, Edler MC, Daidone G, Maggio B, Merickech M, Plescia S, Schillaci D, Bai R, Hamel E. Synthesis, cytotoxicity, and inhibitory effects on tubulin polymerization of a new 3-heterocyclo substituted 2-styrylquinazolinones. Eur J Med Chem. 2004;39:299–304. doi: 10.1016/j.ejmech.2003.12.009. [DOI] [PubMed] [Google Scholar]