

Abstract
One major aim in quantitative and translational neuroscience is to achieve a precise and fast neuronal counting method to work on high throughput scale to obtain reliable results. Here, we tested the isotropic fractionator (IF) method for evaluating neuronal and non-neuronal cell loss in different models of central nervous system (CNS) pathologies. Sprague-Dawley rats underwent: (i) ischemic brain damage; (ii) intraperitoneal injection with kainic acid (KA) to induce epileptic seizures; and (iii) monolateral striatal injection with quinolinic acid (QA) mimicking human Huntington’s disease. All specimens were processed for IF method and cell loss assessed. Hippocampus from KA-treated rats and striatum from QA-treated rats were carefully dissected using a dissection microscope and a rat brain matrix. Ischemic rat brains slices were first processed for TTC staining and then for IF. In the ischemic group the cell loss corresponded to the neuronal loss suggesting that hypoxia primarily affects neurons. Combining IF with TTC staining we could correlate the volume of lesion to the neuronal loss; by IF, we could assess that neuronal loss also occurs contralaterally to the ischemic side. In the epileptic group we observed a reduction of neuronal cells in treated rats, but also evaluated the changes in the number of non-neuronal cells in response to the hippocampal damage. In the QA model, there was a robust reduction of neuronal cells on ipsilateral striatum. This neuronal cell loss was not related to a drastic change in the total number of cells, being overcome by the increase in non-neuronal cells, thus suggesting that excitotoxic damage in the striatum strongly activates inflammation and glial proliferation. We concluded that the IF method could represent a simple and reliable quantitative technique to evaluate the effects of experimental lesions mimicking human diseases, and to consider the neuroprotective/anti-inflammatory effects of different treatments in the whole brain and also in discrete regions of interest, with the potential to investigate non-neuronal alterations. Moreover, IF could be used in addition or in substitution to classical stereological techniques or TTC staining used so far, since it is fast, precise and easily combined with complex molecular analysis.
Keywords: isotropic fractionator, cerebral ischemia, epilepsy, striatal lesion, neuroprotection
Introduction
Since its introduction (Herculano-Houzel and Lent, 2005), the isotropic fractionator (IF) method represented a significant innovation in the field of quantitative neuroscience. The issue of counting neurons and other cell types in the nervous system has been fundamental in neuroscience since the past century. Generating accurate and reproducible quantitative estimates of both neuronal and non-neuronal populations is a crucial requirement in a number of experimental setups aiming at tracking developmental or neurodegenerative events in the nervous system, yet available protocols are hampered by a number of technical and procedural limitations.
A series of methods was developed in order to overcome the problem of overestimating cell number due to double counting of profiles in histological sections, because not all of the counted objects are contained exclusively in one section (Abercrombie, 1946). The same nucleus, in this case, could lie partly within one section, partly within the adjacent section(s). Therefore, the bigger the nuclear dimension is, in relation to the section thickness, the bigger the error could be.
A first attempt to overcome double counting was the Abercrombie’s correction factor (Abercrombie, 1946; Clarke, 1992), by which the new concept of nuclear-point came to light. A nuclear-point was for Abercrombie any geometrical point of the same relative position in all nuclei. Through this equation: P = A∗(M/L + M); where P is the average number of nuclear points, A is the gross number of nuclei seen in section, M the thickness of the section, and L the average length of nuclei, the authors assumed that the error would be negligible and estimated cell numbers more accurate. In the last decade of the previous century, stereology emerged as the method for cell counting. Stereology is a mathematical method that derives global quantities (i.e., volume, surface area and length, and also object number) from measurements obtainable on sections of a given structure (Weibel, 1981; Cruz-Orive, 1987). In this field, for many years, the Optical Fractionator method was considered the gold standard for counting neurons and even synapses. The Optical Fractionator method involves counting cells/neurons through optical dissectors in a uniform and systematic sample that constitutes a fraction of the region to be analyzed (Gundersen, 1986; West et al., 1991; West, 1999). Several software tools (e.g., StereoInvestigator, Neurolucida) were developed according to this method in order to assist researchers in quantitative analysis (Tomasi et al., 2011; Apolloni et al., 2014; Boido et al., 2014; Papageorgiou et al., 2014; Deidda et al., 2015; Dudok et al., 2015; Kawagishi et al., 2015). Nevertheless, the method remains complex and time-consuming, and some authors (Clarke, 1992) still recommend the Abercrombie’s correction factor when the profile is small compared to the section thickness.
Stereological methods such as the optical dissector and fractionator can estimate the number of cells and neurons in discrete brain regions (Korbo et al., 1990; Andersen et al., 1992). However, because these estimates are obtained from cell densities, the estimates precision is related to the homogeneity of neurons in the samples (West, 1999). Thus, these methods result inaccurate to quantify for example the total cell numbers in the brain. This could be done, however, but require the burden of dividing the brain into numerous regions of homogeneous cell density. Additionally, because stereological estimates are necessarily achieved by multiplying cell density by volume, the numbers obtained are not independent variables and therefore cannot be used in statistical comparisons against volume (Harrison et al., 2002).
The possibility to overcome methodological and time limitations of standard stereological cell counting techniques led neuroscientists to get new insights into evolutionary and developmental issues in different species (Collins et al., 2010; Gabi et al., 2010; Lent et al., 2012; Herculano-Houzel et al., 2015). Developed first by Herculano-Houzel et al. (2006) to work on high-scale measurements and comparative studies (Santiago et al., 2007; Azevedo et al., 2009, 2013), the IF method briefly consists in transforming highly anisotropic brain structures into homogeneous, isotropic suspensions of cell nuclei, which can be identified immunocytochemically as neuronal or non-neuronal nuclei, then counted. The method was recently applied also in pathological contexts, such as: age-related neuronal deficit in rats (Morterá and Herculano-Houzel, 2012); mouse model of autism (Chen et al., 2015); mouse model of Alzheimer’s disease (AD; Brautigam et al., 2012) and even on human specimens from AD patients (Andrade-Moraes et al., 2013). The IF method has been recently substantially improved by an automated machine for large-scale fractionation (Azevedo et al., 2013).
This potentially ground-breaking method has not yet been widely adopted mainly due to the lack in calibrating and/or validating works that compare the IF with standard counting techniques, except for the works of Bahney and von Bartheld (2014) on human and macaque monkey samples and Miller et al. (2014) in chimpanzee primary visual cortex. Because of its fast, precise and reliable cell/neuron counts, the IF could be a good strategy to investigate for example the neuroprotective effects of different compounds in different central nervous system (CNS) pathologies.
Here, we test the use of IF method in experimental models of CNS diseases, showing that it is as precise as stereological counts, but significantly faster. To our knowledge there are few papers attempting to apply this method to investigate the neuronal loss following (i) cerebral ischemia; (ii) epileptic seizures; and (iii) striatal lesion in rat models (i.e., Lopim et al., 2016 for epilepsy in rats).
Moreover, this procedure, in comparison to stereological counts, could be implemented and optimized to work down stream of a Fluorescence-Activated Cell Sorting (FACS) technique allowing further molecular investigations such as mic roarray RNA quantification or real time quantitative PCR. This possibility has been recently explored by other groups (Guez-Barber et al., 2012) by performing these molecular analyses on NeuN-sorted cells from a cell suspension of adult rat striata.
Materials and Methods
Animals
Two- to four-month-old Sprague-Dawley (SD) male rats (Harlan-Italy, San Pietro al Natisone, Italy) were used for producing the cerebral ischemia, epilepsy and striatal lesion models. All the experimental procedures involving live animals were performed in strict accordance with European Community Council guidelines 86/609/EEC (November 24, 1986), and with the Italian Ministry of Health and University of Turin institutional guidelines on animal welfare (law 116/92 on Care and Protection of living animals undergoing experimental or other scientific procedures; authorization number 17/2010-B, June 30, 2010). Additionally, an ad hoc Ethical Committee of the University of Turin approved this study. Animals were maintained with a 12:12 light/dark cycle. Food and water were provided ad libitum (standard mouse chow 4RF25-GLP, Mucedola srl, Settimo Milanese, Italy) Particular care was taken to minimize the number of animals, their discomfort and pain.
Cerebral Ischemia
Permanent middle cerebral artery occlusion (MCAo) was performed according to Renolleau’s method (Renolleau et al., 1998). Briefly, SD rats (N = 3) were anesthetized with 5% isoflurane (Isoflurane-Vet 100%, Liquid, Merial Italy, Milan, Italy) vaporized in O2/N2O 30:70, and maintained at 1.5–2.5% isoflurane during surgery. The left MCA was exposed and electrocoagulated with a bipolar forceps (Jeweler 30665, GIMA, Milan, Italy). Then the ipsilateral common carotid artery (CCA) was clamped during 90 min to avoid collateral perfusion.
After 24 h the animals were sacrificed with an overdose of anesthetic, the brains were isolated and cut with a rat brain matrix in coronal slices at a thickness of 2 mm. Then the samples were processed for Triphenyl-Tetrazolium Chloride (TTC) staining. The TTC reaction was stopped substituting the TTC solution with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB, pH 7.4) and the brain slices post-fixed for 2 weeks. In sequence, the two hemispheres of each slice were separated and processed with the IF method singly.
Control rats (N = 3) were sacrificed with an overdose of anesthetic, the brains were isolated, cut, and processed for TTC and IF method as above mentioned.
Epileptic Seizures
Sprague-Dawley rats (N = 3) were intraperitoneally (i.p.) injected with 11 mg/kg kainic acid (KA; Tocris Bioscience, Bristol, UK) to induce epileptic seizures. One hour after the injection, rats showed the first symptoms (immobility, facial myoclonus, and head nodding), and only animals that reached the fourth and fifth stages of the Racine scale (Racine et al., 1972) were included in the study: stage 0, no seizures; stage 2, head nodding; stage 3, forelimb clonus; stage 4, rearing in addition to severe forelimb clonus; and stage 5, rearing and falling in addition to severe forelimb clonus. To minimize suffering and prevent mortality, 2 h following the symptoms onset a single i.p. injection of 4 mg/kg diazepam (Valium; Roche, Monza, Italy) blocked epileptic seizures within 30 min of administration. Control rats (N = 3) were i.p. injected with PBS.
Twenty-four hours after KA administration, the animals were sacrificed with an overdose of ketamine and perfused transcardially with 4% buffered PFA. Rat brains were isolated and post-fixed in 4% PFA for 2 weeks and the hippocampus from control and treated rats was carefully dissected and processed by the IF method.
Striatal Lesion
The surgical protocol of quinolinic acid (QA) injection was performed according to the procedure described by Figueredo-Cardenas et al. (1994). SD rats (N = 3) were anesthetized with 5% isoflurane vaporized in O2/N2O 30:70, placed in a rat stereotaxic apparatus (Stoelting, Wood Dale, IL, USA) and maintained under 1.5–2.5% isoflurane during surgery. QA (210 nmol/2 μl; Sigma–Aldrich, St. Louis, MO, USA) was injected into the right striatum at the following coordinates: AP +0.6, MD +2.8, DV -5. The hemisphere contralateral to the QA injection served as control.
The animals were killed 30 days later with an overdose of ketamine and perfused transcardially with 4% buffered PFA. Dissected rat brains were post-fixed in 4% PFA for 2 weeks. The fixed brains were sliced in coronal sections with a rat brain matrix and the single striatum portions carefully dissected under a stereomicroscope with the help of a rat brain atlas (Paxinos and Watson, 2016). Striata from both sides were processed by the IF method.
Isotropic Fractionator Method
The IF was performed as in Herculano-Houzel and Lent (2005). Briefly, after the neural tissue was properly fixed, small fragments were collected and placed into a glass tissue grinder, a saline-detergent solution (40 mM sodium citrate; 1% TritonTM X-100, Sigma–Aldrich, St. Louis, MO, USA) was added, and through careful and constant translation and rotation movements of a tightly coupled pestle, the tissue was disrupted chemomechanically. This homogenization broke the cell but not the nuclear membrane. The nuclear suspension obtained after centrifugation and resuspension was stained with the fluorescent DNA dye 4′-6-diamino-2-phenylindole dihydrochloride (DAPI; DAPI, dilactate, D9564, Sigma–Aldrich, St. Louis, MO, USA). Aliquots from the isotropic suspension were charged into a hemocytometer (Neubauer chamber) and observed under fluorescence microscopy (Nikon Eclipse 80i). The average nuclei density was determined by counting the number of nuclei within sectors of the coverslipped hemocytometer (1 mm2 area; 0.1 mm depth) for four aliquots. The total number of cells originally present in the analyzed region was then obtained by multiplying density by the total volume of the suspension. To identify the fraction of neuronal nuclei among the total number of DAPI-stained nuclei, another aliquot of the isotropic suspension was collected and immunostained with mouse primary antibody against neuronal nuclear protein (NeuN, MAB377, Chemicon, Single Oak Drive, Temecula, CA, USA, 1:200 in PBS, overnight incubation at room temperature). Then, the samples were washed in saline and incubated at room temperature for at least 2 h with the secondary antibody (Cy3 conjugated anti-mouse donkey IgG, Chemicon, Single Oak Drive, Temecula, CA, USA; 1:200 in PBS) and normal donkey serum (1:10; D9663, Sigma–Aldrich, St. Louis, MO, USA). The percentage of neurons was obtained by counting the number of NeuN-labeled nuclei among at least 500 DAPI-stained nuclei. The non-neuronal cells were quantified as the difference between the total number of cells and the total number of neurons.
Data Analysis
The values from each specimen were averaged and compared to achieve the p-value as follows: ischemia and striatal lesion specimens by paired Student’s t-test, two tails; epileptic seizure specimens by unpaired Student’s t-test, two tails. Data for Student’s t-test significance were performed in Microsoft Excel. Data were expressed as mean ± SEM (standard error of the mean) and differences were considered significant when p ≤ 0.05.
Results
Cerebral Ischemia
We first assessed the ischemic volume of the lesioned rats through TTC staining. Taking into account the formula for edema correction (Dohare et al., 2008), the ischemic lesion was 15.65% ± 0.44% of the whole brain, and the edema volume estimated to have an average value of 86.97 ± 20.43 mm3 affecting the 7.34 ± 1.58% of the whole brain (Figure 1).
FIGURE 1.
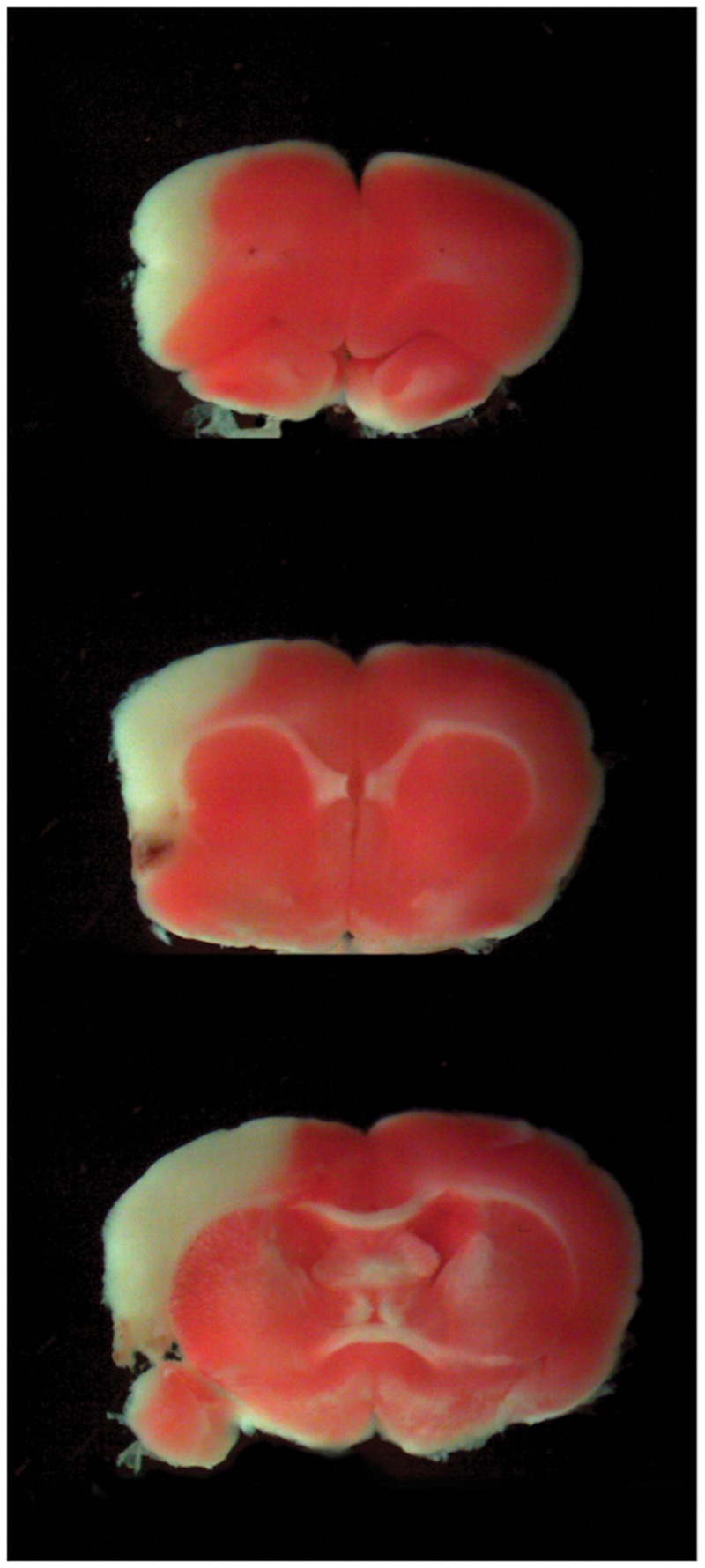
Cerebral Ischemia. TTC representative slices at different brain levels of the lesioned group. The percentage of ischemic volume was measured with Neurolucida software as described in the “Materials and Methods” section.
We found a significant difference in the number of 4′-6-diamino-2-phenylindole dihydrochloride (DAPI) positive cells in ischemic brains compared to the controls (1.06∗108± 3.95∗106 vs. 1.33∗108± 7.42∗106, p = 0.04). Our cell counts in control brains are similar to cell/neuronal numbers observed in rats (Herculano-Houzel and Lent, 2005).
The percentage of NeuN positive cells was 27.44% ± 2.06 in lesioned rats as compared to 39.73% ± 1.94 in control rats (p = 0.01). That difference results in a 2.34∗107 neuronal cell loss in ischemic brains as compared to the controls (2.91∗107± 9.89∗105 vs. 5.25∗107± 8.75∗105, p = 0.00006) and furthermore results in a significant reduction in neuronal density (2.22∗104± 8.46∗102 neurons/mg vs. 4.31∗104± 3.59∗103 neurons/mg, p = 0.02). There were no statistically significant differences in total number or density of non-neuronal cells between groups (7.70∗107 ± 4.93∗106 vs. 8.04∗107 ± 6.95∗106 p = 0.71; 5.88∗104 ± 4.42∗103 vs. 6.52∗104 ± 3.75∗103 non-neurons/mg p = 0.33; Figure 2).
FIGURE 2.
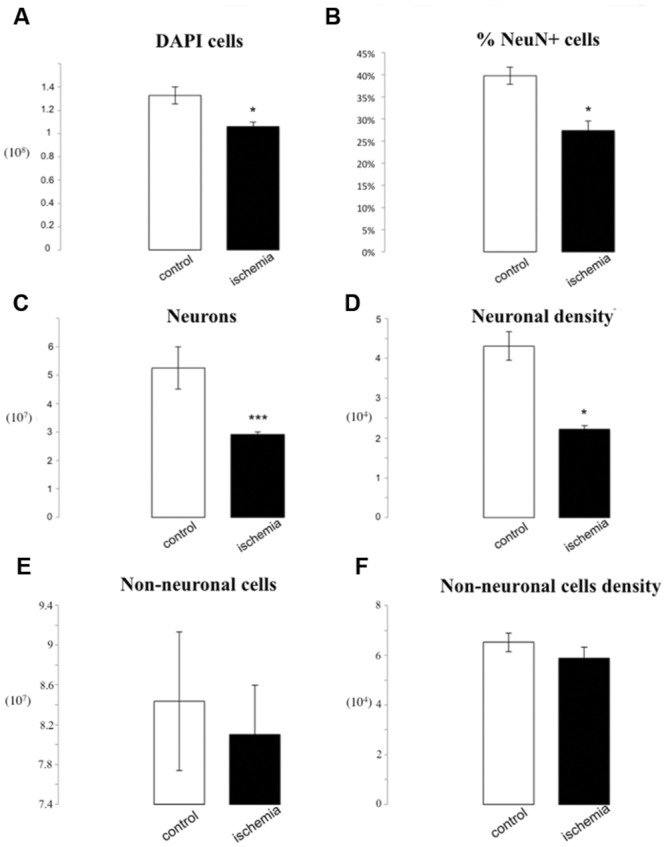
IF quantification after cerebral ischemia. Histograms representing the total number of DAPI positive cells (A), the percentage of NeuN positive nuclei upon DAPI+ cells (B), the total number of neurons (C), and the neuron density, as cells/mg (D), as well as the total number of non-neuronal cells (E) and non-neuronal density (F). Data are presented as mean ± SEM and control vs. ischemic group (T-test, ∗p ≤ 0.5, ∗∗∗p ≤ 0.001, n = 3).
We also analyzed in more detail the difference between ipsilateral and contralateral sides of lesioned brains. We observed a tendency for a decrease in total cell number loss on the side ipsilateral to the medial cerebral artery obstruction (MCAo), reaching 5.92∗106 DAPI positive profiles reduction as compared to the contralateral side (5.01∗107± 2.82∗106 vs. 5.6∗107± 1.14∗106, p = 0.07). When we considered the percentage of NeuN positive nuclei, we found no differences between groups (24.74% ± 3.04 vs. 30.14% ± 1.51, p = 0.16). This corresponded to a depletion in the number of neuronal profiles after ischemia on the ipsilateral side (1.23∗107± 8.45∗105 vs. 1.69∗107± 6.35∗105, p = 0.05). Moreover, we found a statistically significant reduction in neuronal density (8.52∗103 neurons/mg) on the ipsilateral side (1.81∗104± 1.32∗103 vs. 2.66∗104± 4.32∗102, p = 0.01). There were no statistically significant differences of the total number or density of non-neuronal cells between ipsilateral and contralateral sides (3.78∗107 ± 3.47∗106 vs. 3.92∗107 ± 1.54∗106 p = 0.74; 5.59∗104 ± 5.29∗103 vs. 6.20∗104 ± 4.34∗103 non-neurons/mg; p = 0.41; Figure 3).
FIGURE 3.
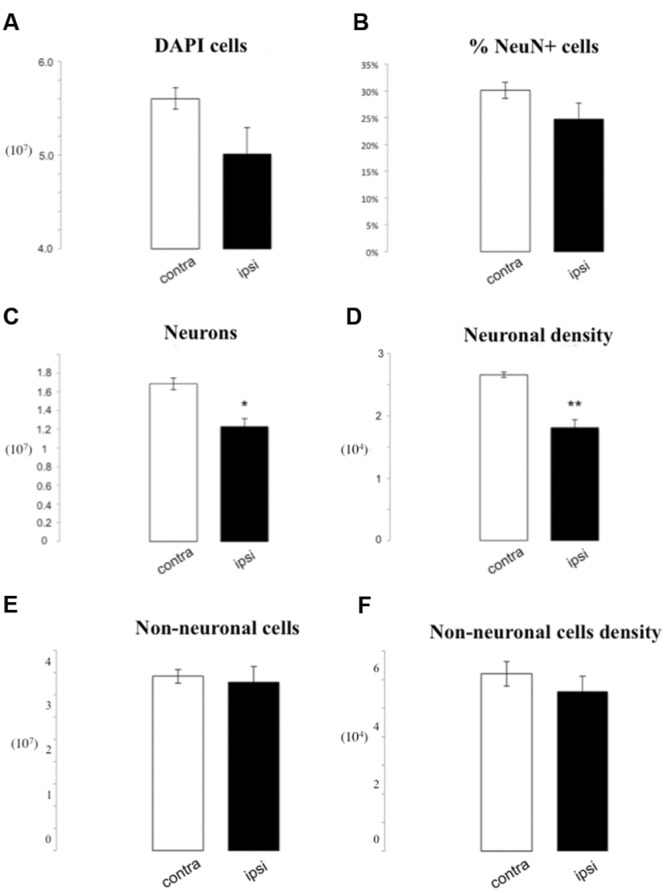
IF quantification after cerebral ischemia, differences between ipsilateral and contralateral side after lesion. Histograms representing the total number of DAPI positive cells (A), the percentage of NeuN positive nuclei upon DAPI+ cells (B), the total number of neurons (C), and the neuron density, as cells/mg (D), as well as the total number of non-neuronal cells (E) and non-neuronal cells density (F). Data are presented as mean ± SEM and contra vs. ipsilateral side (T-test, ∗p ≤ 0.5, ∗∗p ≤ 0.01, n = 3).
Epileptic Seizures
The overall number of DAPI stained nuclei was unchanged after epileptic seizures induced by KA (9.29∗106± 1.16∗105 vs. 9.79∗106± 1.03∗106, p = 0.65). If we consider instead the number of NeuN+ nuclei, we observe a significant loss in the lesioned group following KA injection (2.04∗106± 2.56∗105 vs. 3.61∗106± 1.26∗105, p = 0.005). This caused a significant decrease of 46% of the number of neurons in the lesioned hippocampus (21.01% ± 2.1% vs. 38.91% ± 1.69%, p = 0.002). Also, neuronal density was significantly reduced (9.22∗103 neurons/mg) following KA injection (1.53∗104± 2.09∗103 vs. 2.46∗104± 1.30∗103 p = 0.02). Concerning non-neuronal cells there was a tendency for an increase of the total non-neuronal cell number in lesioned rats as compared to controls (7.74∗106 ± 8.68∗105 vs. 5.68∗106 ± 2.14∗105 p = 0.08), while we observed a significant increase in non-neuronal cell density in lesioned hippocampus (5.73∗104 ± 3.67∗103 vs. 3.85∗104 ± 7.31∗102 non-neurons/mg p = 0.007; Figures 4 and 5).
FIGURE 4.
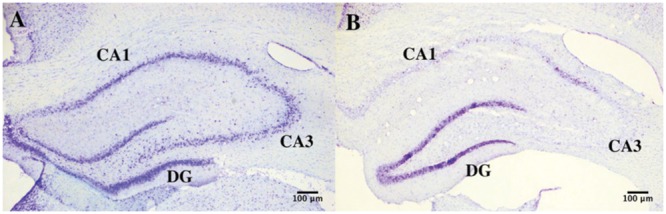
Hippocampal degeneration. Representative images of Nissl-stained sections of the hippocampus in control (A) and KA-treated (B) mice showing neuronal degeneration after KA injection.
FIGURE 5.
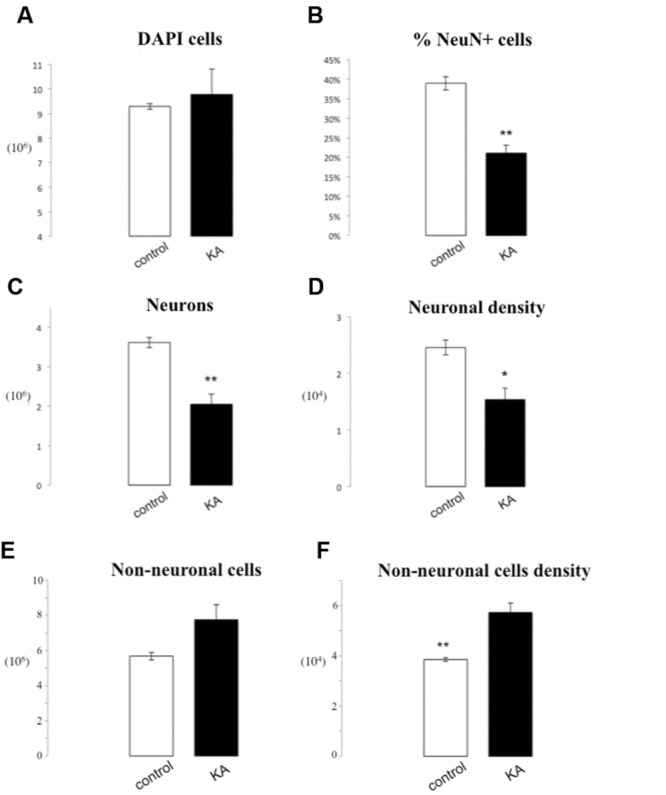
IF quantification after KA administration. Histograms representing the total number of DAPI positive cells (A) in the hippocampus, the percentage of NeuN positive nuclei upon DAPI+ cells (B), the total number of neurons (C), and the neuron density, as cells/mg (D), as well as the total number of non-neuronal cells (E) and non-neuronal cells density (F). Data are presented as mean ± SEM and control vs. ischemic group (T-test, ∗p ≤ 0.5, ∗∗p ≤ 0.01, n = 3).
Striatal Lesion
The number of DAPI positive nuclei in striata lesioned through parenchymal injection of QA was reduced by 5.81∗105 (8.00∗106± 5.67∗105 vs. 8.59∗106± 8.63∗105, p = 0.76), a non-significant difference. Neuronal nuclei were significantly depleted in the QA-lesioned striatum (1.4∗106± 1.46∗105 vs. 3.33∗106± 3.63∗105, p = 0.04). This corresponded to a 21.5% reduction in the percentage of NeuN+ cells, statistically significant (17.5% ± 2.75% vs. 38.99% ± 4.7%, p = 0.003). Moreover, we observed a significant reduction of 3.04∗104 neurons per mg in neuronal density of the QA lesioned striatum by (2.16∗104± 1.54∗103 vs. 5.2∗104± 4.91∗103, p = 0.02). There were no statistically significant differences for the number or density of non-neuronal cells in lesioned rats as compared to controls (6.61∗106 ± 5.03∗105 vs. 5.25∗106 ± 6.25∗105 p = 0.16; 1.02∗105 ± 5.88∗103 vs. 8.40∗104 ± 1.66∗104 non-neurons/mg, p = 0.35; Figures 6 and 7).
FIGURE 6.
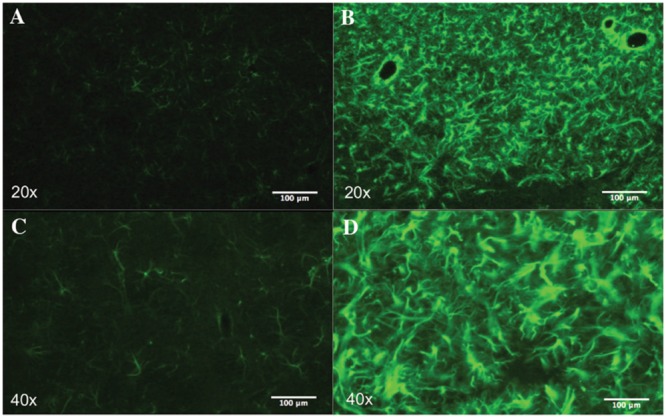
Striatal astrogliosis. Representative images of the striatum showing reactive astrogliosis after QA injection. Astrocytes were labeled with GFAP. In lesioned rats (B,D) there is a remarkable increase in GFAP immunoreactivity compared to the control group (A,C).
FIGURE 7.
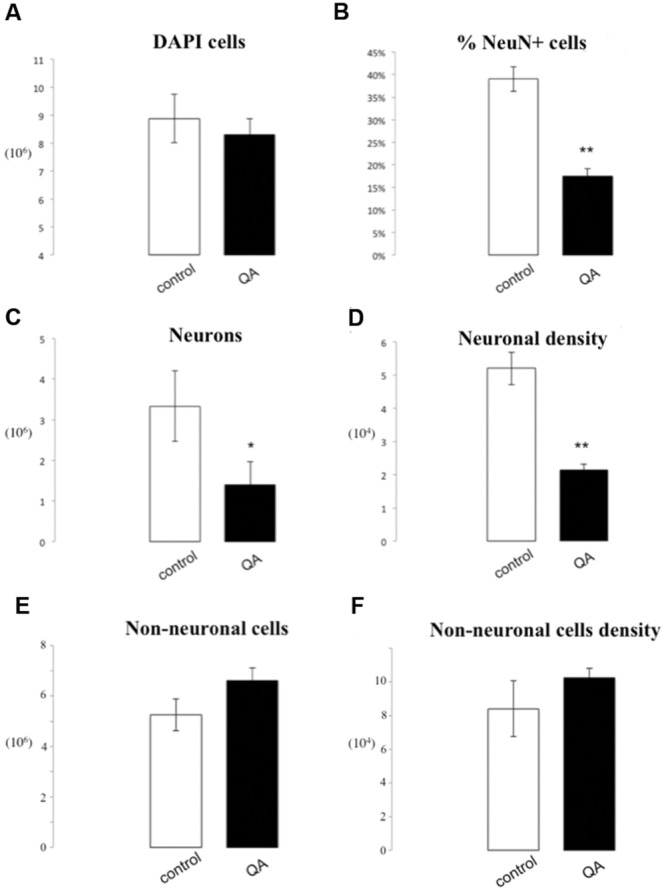
IF quantification after QA injection. Histograms representing the total number of DAPI positive cells (A); the percentage of NeuN positive nuclei upon DAPI+ cells (B), the total number of neurons (C), and the neuron density, as cells/mg (D), as well as the total number of non-neuronal cells (E) and non-neuronal cells density (F). Data are presented as mean ± SEM and control vs. QA group (T-test, ∗p ≤ 0.05, ∗∗p ≤ 0.01, n = 3).
Discussion
The aim of this study was to test the use of the IF method for the study of CNS diseases in order to provide a novel tool to assess quantitative parameters related to the effects of therapeutic strategies. The IF method was previously adopted for determining age-related neuronal loss in rats (Morterá and Herculano-Houzel, 2012), synucleinopathy (Aldrin-Kirk et al., 2014), in a mouse model of AD (Brautigam et al., 2012) and even on AD human patients (Andrade-Moraes et al., 2013). Here, we used it to detect changes in cell numbers occurring after cerebral ischemia, epileptic seizures and striatal lesion mimicking Huntington’s Disease (HD).
A major goal in the field of clinical neuroscience is to develop and characterize neuroprotective agents which could either reduce or delay brain damage, or modulate regenerative responses in the parenchyma (e.g., neurogenesis, angiogenesis, axonal sprouting, and synaptogenesis) which are directly stimulated by a lesion such as an ischemic insult (Zhang and Chopp, 2009).
In general, there are three major types of cell death induced by a pathogenic process: necrosis, apoptosis, and autophagic death. The characteristic features of apoptosis are cellular shrinkage and blebbing, nuclear fragmentation, and formation of apoptotic bodies (Kerr et al., 1972). Apoptosis is involved in neuronal cell loss in the penumbra of an ischemic brain injury (Mehta et al., 2007); in epileptic seizures (Cole-Edwards et al., 2006) and in Parkinson’s disease (PD; Ciechanover and Brundin, 2003; Olanow, 2007). Such a cellular death alters the morphology of cell nuclei and in turn the number of viable/not viable neurons that an automatic counting method for IF (Collins et al., 2010) can detect (i.e., counting a picnotic nucleus like a vial nucleus), causing a bias in particular in studies focused on neuronal cells rescue or replacement.
Noteworthy, preclinical research has a low translational success rate. Even if related to a specific pathology (e.g., stroke), the update of Stroke Therapy Academic Industry Roundtable (STAIR) recommendation pointed out (Fisher et al., 2009) that it is mandatory to improve sample size, rigor, standardization, and minimize bias within the experimental protocols. We think that especially for the first point, the IF is appealing for stroke studies because of its capability to obtain reliable results in short time over a large number of samples. Moreover, it would be useful not only for stroke research but also for the most of neuroprotection/neurorestorative studies upon different CNS diseases.
We believe that IF done by manual counting can give more consistent results than an automatic counting procedure based on FACS; nevertheless, within the flow-cytometry-based techniques for cell counting, the FAST-FIN could be the more promising especially for the capability to discriminate not only neuronal-vs. non-neuronal nuclei but also glial vs. neuronal cells (Marion-Poll et al., 2014), even if this characterization is made only on nuclear size.
Of great interest, especially for disease-related investigations, could be understanding whether a disease (such as stroke or epilepsy) could differentially affect different types of glial cells rather than neurons. There is a growing body of evidence supporting the notion that glial cells play a crucial role in pathogenesis and progression of CNS disorders (Takeuchi, 2013).
Unfortunately, even if a nuclear marker for oligodendrocytes is reliable and well characterized (the transcriptional factor SOX10, Kuhlbrodt et al., 1998) there are not so far appreciable nuclear markers for the other glial cell types that work upon this protocol. There are some unpublished data exploring the use of Olig2 in the IF method as a marker for specific type of oligodendrocytes in mouse models of psychiatric diseases, but the main issue to solve is whether it is universal (stains all oligodendrocytes in a brain region) and specific (stains only oligodendrocytes in that brain region). This issue has been solved for neuronal markers such as NeuN (Mullen et al., 1992; Wolf et al., 1996; Sarnat et al., 1998) but still not for those of glial subtypes.
Cerebral Ischemia
In cerebral ischemia induced by a permanent MCAo in adult rats, we observed a significant reduction in the total number of nuclei in ischemic brains as compared to the controls. In fact, the decrease in the number of DAPI+ nuclei on ischemic brains (2.67∗107) is very close to that of NeuN+ nuclei (2.34∗107). During our examination we found that the percentage of NeuN+ nuclei in control brains was almost 40% of the total number of nuclei. This percentage is similar to that observed by Herculano-Houzel and Lent (2005). Slight differences may derive from different post-fixation times in the two papers. In fact, it is known that a longer fixation time could mask NeuN epitope retrieval (Gill et al., 2005). Also, since we used a different rat strain (SD instead of Wistar in Herculano-Houzel and Lent, 2005), it is possible that this could be a source of some difference in numbers. On the other hand, our neuronal counts are consistent with those from Aldrin-Kirk et al. (2014) in adult SD rat forebrains, especially regarding neuronal density (neurons/mg).
In the ischemic brains, we counted separately the ipsilateral side to the MCAo and the contralateral side. Interestingly, beside a significant reduction of 17.93% of NeuN stained cells in the ipsilateral side as compared to the contralateral, we found a significant difference also in the percentage of NeuN between the control hemispheres and contralateral side (39.73% ± 1.94% vs. 30.14% ± 1.51%, p = 0.019), suggesting an impact of the ischemic procedure beyond the directly affected hemisphere. In fact, these data are consistent with the well-known influence of a focal ischemia on the whole brain, causing neuronal damage also in the contralateral hemisphere through neuroinflammation and blood brain barrier (BBB) leakage (Garbuzova-Davis et al., 2013).
Therefore, we confirmed that neurons are more sensitive to a hypoxic insult than other cell types in the cerebral cortex. It is known that astrocytes, especially in the ischemic boundary zone (IBZ), may resist to a slight reduction in glucose and oxygen delivery, with a prolonged survival compared to neurons (Swanson et al., 2004; Zhao and Rempe, 2010). In addition, astrocytes are more resistant to oxygen and glucose deprivation in vitro (Panickar and Norenberg, 2005). Microglial cells seem to be resistant to an increase of Ca2+ influx into the cell membrane due to excitotoxicity reactive expression of the GluA2 subunit of α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (Beppu et al., 2013).
An important advantage of our protocol employing the IF method in cerebral ischemia is highlighted by the percentage of NeuN positive cells between groups. Neuronal loss was found to be 44.54%, much higher than expected by an ischemic volume lesion assessed with TTC on 15.65% of the whole brain. Thus, IF was able to detect cell death out of the ischemic core, beyond the penumbra, reaching as far as the contralateral hemisphere. Even though apoptotic nuclei can be detected by stereological analysis even outside the ischemic core, its occurrence and quantification was hardly explored before probably due to the complex and time-consuming protocols.
Almost all recent works on neuroprotective/neurorestorative agents for cerebral ischemia use TTC staining (i.e., Yu et al., 2014; Zhu et al., 2014) or standard stereological techniques (i.e., Liu et al., 2014; Morris et al., 2014) in order to evaluate the ischemic damage. Even though all these analyses were followed by neurologic scores derived from different behavioral tasks, the use of IF to these methods could have brought more precise data on the outcome of treatment, with a reasonable time investment. With IF, any amelioration on behavioral tasks could be attributed to the number of neurons in extra core lesion areas, even in the contralateral hemisphere, which in turn could suggest whether an improvement is the result of physiological/induced plastic changes in the lesioned area/contralateral side (Takatsuru et al., 2013) or is a specific effect of the candidate neuroprotective agent administration.
Epileptic Seizures
Kainic acid is an analog of glutamic acid that acts on both AMPA/kainate receptors. Experimentally, an i.p. KA injection is currently used to induce excitotoxic cell death (Yang et al., 1997; Wang et al., 2005; Chihara et al., 2009). In rodents, KA leads to recurrent seizures, behavioral changes, and subsequent degeneration of selective populations of neurons in the brain (McKhann et al., 2003; Tripathi et al., 2009; Spigolon et al., 2010; Zhao et al., 2012; Grande et al., 2014). Here, we observed that, in spite of a critical loss in neuronal nuclei (1.57∗106), the overall number of nuclei is unchanged. This can be explained by the occurrence of massive reactive gliosis and astrocyte proliferation which mask neuronal loss. KA-induced neuronal death in fact activates microglia and astrocytes (Chen et al., 2005; Ravizza et al., 2005). In KA-induced hippocampal injury, microglial activation is believed to contribute to neuroinflammation and neurodegeneration, thus, a reduction in glial cells activation is followed by a reduction in neuronal cell death (Cho et al., 2008). Our findings suggest that the total number of nuclei counted by use of the IF is not changed. This could be due the increase in the number of non-neuronal nuclei (Figure 5). Astrocytes are the most prominent glial cell population in the CNS and a proliferative response of astrocytes to KA administration was already observed almost 35 years ago (Murabe et al., 1982). Recently, an increase in glial fibrillary acidic protein (GFAP) expression was shown from 1/3 days up to 1 month after KA intra-hippocampal injection (Bendotti et al., 2000). By contrast, GFAP expression in the hippocampus was not affected 1 day after KA systemic administration, while a 20% decrease was observed in the amygdala/pyriform cortex (Ding et al., 2000). However, our previous experiments showed a 20% increase of GFAP immunostaining 1 day after i.p. administration (Spigolon et al., 2010). The massive reduction of the number of hippocampal neuronal cells (43%) in KA treated group observed here with IF is similar to the one observed by our previous experiments with histological counts (40%, Spigolon et al., 2010). However, this neuronal reduction is different from the one observed by Lopim et al. (2016) with IF in Wistar rats (26% at 30 days post lesion), but it is important to point out that, rather then the rat strains, both the model (pilocarpine vs. KA) and the time points (1 day vs. 30 days) were different. From the translational point of view, the IF method applied to epileptic seizures could be used as a control for the number of glial cells that can be changed following anti-oxidant, anti-inflammatory/proliferative therapies (Chung and Han, 2003; Takemiya et al., 2006; Miyamoto et al., 2008; Gupta et al., 2009). Finally, regarding the histological type of neuronal death following epileptic seizures, sometimes the TUNEL staining for apoptotic death fails to show any significant increase (Spigolon et al., 2010) and the Fluoro-Jade B (FJB) staining used by other groups (Cole-Edwards et al., 2006) is not directly related to neuronal death. Even in this case, the IF could be helpful to rapidly control the KA-induced neuronal death, occurring by apoptosis rather than necrosis.
Striatal Lesion
Huntington’s Disease is an autosomal dominantly inherited neurodegenerative disease, in which an expansion of the cytosine–adenine–guanine (CAG) repeat in the gene encoding for the N-terminal region of the huntingtin protein (htt) leads to the formation of a polyglutamine stretch (mhtt; Bano et al., 2011). The behavioral symptoms are typically involuntary choreiform movements, cognitive impairment, and mood disorders, eventually compromising daily functional abilities (Walker, 2007; Paulsen et al., 2008). Unilateral QA induced striatal lesions are highly reminiscent of histological (selective loss of GABAergic and cholinergic neurons) and neurochemical characteristics of HD in experimental animals (Delli Carri et al., 2013; Kumar et al., 2013; Pérez-Severiano et al., 2013; Serrano Sánchez et al., 2014). Overexcitation of N-methyl-D-aspartate (NMDA) receptors following QA administration results in: profound oxidative damage; lipid peroxidation; mitochondrial dysfunction; and apoptosis (Estrada Sánchez et al., 2008; Pérez-De La Cruz et al., 2012). In fact, we observed that the neuronal loss consists of 44.87% of NeuN+ nuclei after lesion. Furthermore, 30 days after QA injection we found a difference of 6.77% of DAPI+ nuclei. Also in this case, similarly to KA experiments on the hippocampus, the reduction in the number of neuronal nuclei is higher than the difference in the number of total nuclei. We can ascribe this finding to an important reactive gliosis, as suggested by an increase in the number of non-neuronal nuclei (Figure 7). Microglial activation in the pathogenesis of HD has been addressed by clinical studies demonstrating a direct correlation between abnormal microglial activity and disease progression (Tai et al., 2007). While microglial activation is unlikely to be the initiating event in these neurodegenerative diseases, it may cause cell death via various pathways. When activated, microglia produce cytotoxic substances including pro-inflammatory cytokines (e.g., TNF-α and IL-1β) and reactive oxygen species (e.g., hydrogen peroxide and superoxide). During acute inflammatory reactions there is also a rearrangement of the extracellular matrix, and matrix metalloproteinases (MMPs) involved in this process have a prominent role in microglial genesis (Kierdorf et al., 2013).
Besides, QA can alter the BBB (Guillemin, 2012), leaving the brain parenchyma permissive to the infiltration of inflammatory responsive cells. The importance of microglial activation was underlined, moreover, by an excellent paper by Neher et al. (2012), who found that inflamed microglia could phagocyte viable neurons. QA administration also leads to an intense astrogliosis (Björklund et al., 1986; Dihné et al., 2001).
All these mechanisms could contribute to explain our finding by a proliferation of glial-cells following excitotoxicity-induced neurodegeneration. Most studies on HD treatment use complex stereological counting techniques to assess the parenchymal damage in the striatum (Mazurová et al., 2014; Southwell et al., 2015). The IF method for analyzing a discrete region such as the corpus striatum could be an additional/substitute strategy to obtain lesion specific information of induced neuronal loss, and even an indirect quantification of reactive gliosis (when using anti-inflammatory/glial genesis compounds), by observing the difference among different cell populations between experimental groups.
In another interesting context, when injected into the striatum of adult rodents to model HD, QA strongly stimulates the subventricular zone (SVZ) and striatal neurogenesis (Tattersfield et al., 2004; Collin et al., 2005). An adult reactive neurogenic process was also obtained in zebrafish with a telencephalic administration of QA (Skaggs et al., 2014). Neural progenitor cells (NPCs) in the SVZ have been proposed as an endogenous source of new neurons that could be mobilized to repair brain circuits damaged due to injury or disease (Kernie and Parent, 2010). The high percentage (80%) of newborn neurons that succeed to differentiate (NeuN expression) 6 weeks after the lesion, as found by Collin et al. (2005), rises a question to our quantification analysis. To what extent our NeuN+ percentage is influenced by newly born neurons in the striatum vs. pre-existing neurons escaped from excitotoxicity-induced neuronal death?
Recently, Deierborg et al. (2009) labeled newborn cells by i.p. injection of bromo–deoxy–uridine (BrdU), or by green fluorescent protein (GFP)-expressing lentiviral vectors injected into the SVZ. They did not detect any GFP+ cells that co-labeled with NeuN into the lesioned striatum at any time point studied (1, 2, and 3 weeks post lesion; Deierborg et al., 2009). Despite evidence that a certain amount of reactive neurogenesis occurs after an ischemic insult in particular in the acute phase (Yamashita et al., 2006; Liu et al., 2009; Wei et al., 2011), the potential of the newborn cells to replace dying medium spiny neurons is controversial (Arvidsson et al., 2002; Luzzati et al., 2006). In fact, in the mouse model of slow progressive degeneration (Creb1Camkcre4Crem-/- double mutant mice) newborn neuronal cells show a transient existence and they do not express any specific marker of striatal projection neurons (Luzzati et al., 2011).
Conclusion
Our results (summarized in Supplementary Table S1) support the use of IF as a simple and reliable method to evaluate the effects of experimental lesions mimicking human diseases and the outcome of therapeutic measures. Moreover, we have shown that IF can provide additional information about neuronal death and glial proliferation: the finding of new specific markers for detecting astroglial and microglial nuclei could further improve the method. The IF method in fact can miss some details when the cell loss is type- or even subtype-specific. Nevertheless, the IF allows to count quickly the amount of cell loss, and it can easily discriminate neurons and glia by NeuN IHC. In cerebral ischemia, this is a valid complement to the evaluation of the volume of the infarct, and also allows to detect cell loss in the surrounding penumbra and in the contralateral hemisphere avoiding time consuming stereological counts. This holds true also for more discrete structures, such as the hippocampus and the striatum, where the inhomogeneity of the areas and the tissue (with myelin fascicles intermingled to neurons), respectively, make stereological counts complicated.
Author Contributions
IR contributed to the design of the work, performed acquisition, analysis and interpretation of all the experiments; drafted and revised the work, RM performed acquisition and analysis of most of the experiment and revised the work, MT performed part of the experiments (epileptic seizures) and revised the work, ST contributed to the conception and design of the work and revised it, AA performed part of the experiments and revised the work, C-HA-M contributed to the interpretation of the data and revised the work, RL contributed to the conception of the work and revised it AV contributed to the conception and design of the work and revised it.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Neuroscience Institute Cavalieri Ottolenghi (NICO), University of Turin and Department of Neuroscience “Rita Levi Montalcini” in Turin, that aided the efforts of the authors.
Footnotes
Funding. We want to thank SMArathon Onlus for providing fellowship grant for RI and Department of Neuroscience “Rita Levi Montalcini” in Turin for funding the research.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fncel.2016.00190
References
- Abercrombie M. (1946). Estimation of nuclear population from microtome sections. Anat. Rec. 94 239–247. 10.1002/ar.1090940210 [DOI] [PubMed] [Google Scholar]
- Aldrin-Kirk P., Davidsson M., Holmqvist S., Li J.-Y., Björklund T. (2014). Novel AAV-Based Rat model of forebrain synucleinopathy shows extensive pathologies and progressive loss of cholinergic interneurons. PLoS ONE 9:e100869 10.1371/journal.pone.0100869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen B. B., Korbo L., Pakkenberg B. (1992). A quantitative study of the human cerebellum with unbiased stereological techniques. J. Comp. Neurol. 326 549–560. 10.1002/cne.903260405 [DOI] [PubMed] [Google Scholar]
- Andrade-Moraes C. H., Oliveira-Pinto A. V., Castro-Fonseca E., da Silva C. G., Guimarães D. M., Szczupak D., et al. (2013). Cell number changes in Alzheimer’s disease relate to dementia, not to plaques and tangles. Brain 136 3738–3752. 10.1093/brain/awt273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apolloni S., Amadio S., Parisi C., Matteucci A., Potenza R. L., Armida M., et al. (2014). Spinal cord pathology is ameliorated by p2x7 antagonism in a sod1-mutant mouse model of amyotrophic lateral sclerosis. Dis. Model Mech. 7 1101–1109. 10.1242/dmm.017038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson A., Collin T., Kirik D., Kokaia Z., Lindvall O. (2002). Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 8 963–970. 10.1038/nm747 [DOI] [PubMed] [Google Scholar]
- Azevedo F. A., Andrade-Moraes C. H., Curado M. R., Oliveira-Pinto A. V., Guimarães D. M., Szczupak D., et al. (2013). Automatic isotropic fractionation for large-scale quantitative cell analysis of nervous tissue. J. Neurosci. Methods 212 72–78. 10.1016/j.jneumeth.2012.09.015 [DOI] [PubMed] [Google Scholar]
- Azevedo F. A., Carvalho L. R., Grinberg L. T., Farfel J. M., Ferretti R. E., Leite R. E., et al. (2009). Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 513 532–541. 10.1002/cne.21974 [DOI] [PubMed] [Google Scholar]
- Bahney J., von Bartheld C. S. (2014). Validation of the isotropic fractionator: comparison with unbiased stereology and DNA extraction for quantification of glial Cells. J. Neurosci. Methods 222 165–174. 10.1016/j.jneumeth.2013.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bano D., Zanetti F., Mende Y., Nicotera P. (2011). Neurodegenerative processes in huntington’s disease. Cell Death Dis. 2:e228 10.1038/cddis.2011.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendotti C., Guglielmetti F., Tortarolo M., Samanin R., Hirst W. D. (2000). Differential expression of s100β and glial fibrillary acidic protein in the hippocampus after kainic acid-induced lesions and mossy fiber sprouting in adult rat. Exp. Neurol. 161 317–329. 10.1006/exnr.1999.7262 [DOI] [PubMed] [Google Scholar]
- Beppu K., Kosai Y., Kido M. A., Akimoto N., Mori Y., Kojima Y., et al. (2013). Expression, subunit composition, and function of ampa-type glutamate receptors are changed in activated microglia; possible contribution of glua2 (glur-b)-deficiency under pathological conditions. Glia 61 881–891. 10.1002/glia.22481 [DOI] [PubMed] [Google Scholar]
- Björklund H., Olson L., Dahl D., Schwarcz R. (1986). Short- and long-term consequences of intracranial injections of the excitotoxin, quinolinic acid, as evidenced by gfa immunohistochemistry of astrocytes. Brain Res. 371 267–277. 10.1016/0006-8993(86)90362-8 [DOI] [PubMed] [Google Scholar]
- Boido M., Piras A., Valsecchi V., Spigolon G., Mareschi K., Ferrero I., et al. (2014). Human mesenchymal stromal cell transplantation modulates neuroinflammatory milieu in a mouse model of amyotrophic lateral sclerosis. Cytotherapy 16 1059–1072. 10.1016/j.jcyt.2014.02.003 [DOI] [PubMed] [Google Scholar]
- Brautigam H., Steele J. W., Westaway D., Fraser P. E., St George-Hyslop P. H., Gandy S., et al. (2012). The isotropic fractionator provides evidence for differential loss of hippocampal neurons in two mouse models of Alzheimer’s disease. Mol. Neurodegener. 7:58 10.1186/1750-1326-7-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Wen-Chin H., Séjourné J., Clipperton-Allen A. E., Page D. T. (2015). Pten mutations alter brain growth trajectory and allocation of cell types through elevated β-catenin signaling. J. Neurosci. 35 10252–10267. 10.1523/JNEUROSCI.5272-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Duan R. S., Quezada H. C., Mix E., Nennesmo I., Adem A., et al. (2005). Increased microglial activation and astrogliosis after intranasal administration of kainic acid in c57bl/6 mice. J. Neurobiol. 62 207–218. 10.1002/neu.20099 [DOI] [PubMed] [Google Scholar]
- Chihara K., Saito A., Murakami T., Hino S., Aoki Y., Sekiya H., et al. (2009). Increased vulnerability of hippocampal pyramidal neurons to the toxicity of kainic acid in oasis-deficient mice. J. Neurochem. 110 956–965. 10.1111/j.1471-4159.2009.06188.x [DOI] [PubMed] [Google Scholar]
- Cho I. H., Hong J., Suh E. C., Kim J. H., Lee H., Lee J. E., et al. (2008). Role of microglial ikkβ in kainic acid-induced hippocampal neuronal cell death. Brain 131 3019–3033. 10.1093/brain/awn230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S. Y., Han S. H. (2003). Melatonin attenuates kainic acid-induced hippocampal neurodegeneration and oxidative stress through microglial inhibition. J. Pineal Res. 34 95–102. 10.1034/j.1600-079X.2003.00010.x [DOI] [PubMed] [Google Scholar]
- Ciechanover A., Brundin P. (2003). The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40 427–446. 10.1016/S0896-6273(03)00606-8 [DOI] [PubMed] [Google Scholar]
- Clarke P. G. (1992). How inaccurate is the abercrombie correction factor for cell counts? Trends Neurosci. 15 211–212. 10.1016/0166-2236(92)90036-8 [DOI] [PubMed] [Google Scholar]
- Cole-Edwards K. K., Musto A. E., Bazan N. G. (2006). C-Jun n-terminal kinase activation responses induced by hippocampal kindling are mediated by reactive astrocytes. J. Neurosci. 26 8295–8304. 10.1523/JNEUROSCI.1986-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin T., Arvidsson A., Kokaia Z., Lindvall O. (2005). Quantitative analysis of the generation of different striatal neuronal subtypes in the adult brain following excitotoxic injury. Exp. Neurol. 195 71–80. 10.1016/j.expneurol.2005.03.017 [DOI] [PubMed] [Google Scholar]
- Collins C. E., Airey D. C., Young N. A., Leitch D. B., Kaas J. H. (2010). Neuron densities vary across and within cortical areas in primates. Proc. Natl. Acad. Sci. 107 15927–15932. 10.1073/pnas.1010356107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Orive L. M. (1987). Particle number can be estimated using a disector of unknown thickness: the selector. J. Microsc. 145 121–142. [PubMed] [Google Scholar]
- Deidda G., Allegra M., Cerri C., Naskar S., Bony G., Zunino G., et al. (2015). Early depolarizing GABA controls critical-period plasticity in the rat visual cortex. Nat. Neurosci. 18 87–96. 10.1038/nn.3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deierborg T., Staflin K., Pesic J., Roybon L., Brundin P., Lundberg C. (2009). Absence of striatal newborn neurons with mature phenotype following defined striatal and cortical excitotoxic brain injuries. Exp. Neurol. 219 363–367. 10.1016/j.expneurol.2009.05.002 [DOI] [PubMed] [Google Scholar]
- Delli Carri A., Onorati M., Lelos M. J., Castiglioni V., Faedo A., Menon R., et al. (2013). Developmentally coordinated extrinsic signals drive human pluripotent stem cell differentiation toward authentic DARPP-32+ medium-sized spiny neurons. Development 140 301–312. 10.1242/dev.084608 [DOI] [PubMed] [Google Scholar]
- Dihné M., Block F., Korr H., Töpper R. (2001). Time course of glial proliferation and glial apoptosis following excitotoxic CNS injury. Brain Res. 902 178–189. 10.1016/S0006-8993(01)02378-2 [DOI] [PubMed] [Google Scholar]
- Ding M., Haglid K. G., Hamberger A. (2000). Quantitative immunochemistry on neuronal loss, reactive gliosis and BBB damage in cortex/striatum and hippocampus/amygdala after systemic kainic acid administration. Neurochem. Int. 36 313–318. 10.1016/S0197-0186(99)00139-4 [DOI] [PubMed] [Google Scholar]
- Dohare P., Garg P., Jain V., Nath C., Ray M. (2008). Dose dependence and therapeutic window for the neuroprotective effects of curcumin in thromboembolic model of rat. Behav. Brain Res. 193 289–297. 10.1016/j.bbr.2008.06.012 [DOI] [PubMed] [Google Scholar]
- Dudok B., Barna L., Ledri M., Szabó SI., Szabadits E., Pintér B., et al. (2015). Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nat. Neurosci. 18 75–86. 10.1038/nn.3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada Sánchez A. M., Mejía-Toiber J., Massieu L. (2008). Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch. Med. Res. 39 265–276. 10.1016/j.arcmed.2007.11.011 [DOI] [PubMed] [Google Scholar]
- Figueredo-Cardenas G., Anderson K. D., Chen Q., Veenman C. L., Reiner A. (1994). Relative survival of striatal projection neurons and interneurons after intrastriatal injection of quinolinic acid in rats. Exp. Neurol. 129 37–56. 10.1006/exnr.1994.1145. [DOI] [PubMed] [Google Scholar]
- Fisher M., Feuerstein G., Howells D. W., Hurn P. D., Kent T. A., Savitz S. I., et al. (2009). Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 40 2244–2250. 10.1161/STROKEAHA.108.541128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabi M., Collins C. E., Wong P., Torres L. B., Kaas J. H., Herculano-Houzel S. (2010). Cellular scaling rules for the brains of an extended number of primate species. Brain Behav. Evol. 76 32–44. 10.1159/000319872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbuzova-Davis S., Rodrigues M. C., Hernandez-Ontiveros D. G., Tajiri N., Frisina-Deyo A., Boffeli S. M., et al. (2013). Blood-brain barrier alterations provide evidence of subacute diaschisis in an ischemic stroke rat model. PLoS ONE 8:e63553 10.1371/journal.pone.0063553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S. K., Ishak M., Rylett R. J. (2005). Exposure of nuclear antigens in formalin-fixed, paraffin-embedded necropsy human spinal cord tissue: detection of neun. J. Neurosci. Methods 148 26–35. 10.1016/j.jneumeth.2005.03.008 [DOI] [PubMed] [Google Scholar]
- Grande V., Manassero G., Vercelli A. (2014). Neuroprotective and anti-inflammatory roles of the phosphatase and tensin homolog deleted on chromosome ten (PTEN) inhibition in a mouse model of temporal lobe epilepsy. PLoS ONE 9:e114554 10.1371/journal.pone.0114554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guez-Barber D., Fanous S., Harvey B. K., Zhang Y., Lehrmann E., Becker K. G., et al. (2012). FACS purification of immunolabeled cell types from adult rat brain. J. Neurosci. Methods 203 10–18. 10.1016/j.jneumeth.2011.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemin G. J. (2012). Quinolinic acid, the inescapable neurotoxin. FEBS J. 279 1356–1365. 10.1111/j.1742-4658.2012.08485.x [DOI] [PubMed] [Google Scholar]
- Gundersen H. J. (1986). Stereology of arbitrary particles. a review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J. Microsc. 143 3–45. 10.1111/j.1365-2818.1986.tb02764.x [DOI] [PubMed] [Google Scholar]
- Gupta Y. K., Briyal S., Sharma M. (2009). Protective effect of curcumin against kainic acid induced seizures and oxidative stress in rats. Indian J. Physiol. Pharmacol. 53 39–46. [PubMed] [Google Scholar]
- Harrison K. H., Hof P. R., Wang S. S. H. (2002). Scaling laws in the mammalian neocortex: does form provide clues to function? J. Neurocytol. 31 289–298. 10.1023/A:1024178127195 [DOI] [PubMed] [Google Scholar]
- Herculano-Houzel S., Catania K., Manger P. R., Kaas J. H. (2015). Mammalian brains are made of these: a dataset of the numbers and densities of neuronal and nonneuronal cells in the brain of glires, primates, scandentia, eulipotyphlans, afrotherians and artiodactyls, and their relationship with body mass. Brain Behav. Evol. 86 145–163. 10.1159/000437413 [DOI] [PubMed] [Google Scholar]
- Herculano-Houzel S., Lent R. (2005). Isotropic fractionator: a simple, rapid method for the quantification of total cell and neuron numbers in the brain. J. Neurosci. 25 2518–2521. 10.1523/JNEUROSCI.4526-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herculano-Houzel S., Mota B., Lent R. (2006). Cellular scaling rules for rodent brains. Proc. Natl. Acad. Sci. 103 12138–12143. 10.1073/pnas.0604911103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagishi K., Ando M., Yokouchi K., Sumitomo N., Karasawa M., Fukushima N., et al. (2015). Stereological estimation of olfactory receptor neurons in rats. Chem. Senses 40 89–95. 10.1093/chemse/bju062 [DOI] [PubMed] [Google Scholar]
- Kernie S. G., Parent J. M. (2010). Forebrain neurogenesis after focal ischemic and traumatic brain injury. Neurobiol. Dis. 37 267–274. 10.1016/j.nbd.2009.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. F., Wyllie A. H., Currie A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26 239–257. 10.1038/bjc.1972.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K., Erny D., Goldmann T., Sander V., Schulz C., Perdiguero E. G., et al. (2013). Microglia emerge from erythromyeloid precursors via pu.1- and irf8-dependent pathways. Nat. Neurosci. 16 273–280. 10.1038/nn.3318 [DOI] [PubMed] [Google Scholar]
- Korbo L., Pakkenberg B., Ladefoged O., Gundersen H. J., Arlien-Søborg P., Pakkenberg H. (1990). An efficient method for estimating the total number of neurons in rat brain cortex. J. Neurosci. Methods 31 93–100. 10.1016/0165-0270(90)90153-7 [DOI] [PubMed] [Google Scholar]
- Kuhlbrodt K., Herbarth B., Sock E., Hermans-Borgmeyer I., Wegner M. (1998). Sox10 a novel transcriptional modulator in glial cells. J. Neurosci. 18 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Chaudhary T., Mishra J. (2013). Minocycline modulates neuroprotective effect of hesperidin against quinolinic acid induced huntington’s disease like symptoms in rats: behavioral, biochemical, cellular and histological evidences. Eur. J. Pharmacol. 720 16–28. 10.1016/j.ejphar.2013.10.057 [DOI] [PubMed] [Google Scholar]
- Lent R., Azevedo F. A., Andrade-Moraes C. H., Pinto A. V. (2012). How many neurons do you have? some dogmas of quantitative neuroscience under revision. Eur. J. Neurosci. 35 1–9. 10.1111/j.1460-9568.2011.07923.x [DOI] [PubMed] [Google Scholar]
- Liu F., You Y., Li X., Ma T., Nie Y., Wei B., et al. (2009). Brain injury does not alter the intrinsic differentiation potential of adult neuroblasts. J. Neurosci. 29 5075–5087. 10.1523/JNEUROSCI.0201-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P., Liu X., Liou A. K.-F., Xing J., Jing Z., Ji X., et al. (2014). The neuroprotective mechanism of erythropoietin-TAT fusion protein against neurodegeneration from ischemic brain injury. CNS Neurol. Disord. Drug Targets 13 1465–1474. 10.2174/1871527313666140806155259 [DOI] [PubMed] [Google Scholar]
- Lopim G. M., Vannucci Campos D., Gomes da Silva S., de Almeida A. A., Lent R., Cavalheiro E. A., et al. (2016). Relationship between seizure frequency and number of neuronal and non-neuronal cells in the hippocampus throughout the life of rats with epilepsy. Brain Res. 1634 179–186. 10.1016/j.brainres.2015.12.055 [DOI] [PubMed] [Google Scholar]
- Luzzati F., De Marchis S., Fasolo A., Peretto P. (2006). Neurogenesis in the caudate nucleus of the adult rabbit. J. Neurosci. 26 609–621. 10.1523/JNEUROSCI.4371-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzati F., De Marchis S., Parlato R., Gribaudo S., Schütz G., Fasolo A., et al. (2011). New striatal neurons in a mouse model of progressive striatal degeneration are generated in both the subventricular zone and the striatal parenchyma. PLoS ONE 6:e25088 10.1371/journal.pone.0025088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion-Poll L., Montalban E., Munier A., Hervé D., Girault J. A. (2014). Fluorescence-activated sorting of fixed nuclei: a general method for studying nuclei from specific cell populations that preserves post-translational modifications. Eur. J. Neurosci. 39 1234–1244. 10.1111/ejn.12506 [DOI] [PubMed] [Google Scholar]
- Mazurová Y., Anderova M., Němečková I., Bezrouk A. (2014). Transgenic rat model of Huntington’s disease: a histopathological study and correlations with neurodegenerative process in the brain of HD patients. Biomed. Res. Int. 2014:291531 10.1155/2014/291531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G. M., II, Wenzel H. J., Robbins C. A., Sosunov A. A., Schwartzkroin P. A. (2003). Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience 122 551–561. 10.1016/S0306-4522(03)00562-1 [DOI] [PubMed] [Google Scholar]
- Mehta S. L., Manhas N., Raghubir R. (2007). Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. Rev. 54 34–66. 10.1016/j.brainresrev.2006.11.003 [DOI] [PubMed] [Google Scholar]
- Miller D. J., Balaram P., Young N. A., Kaas J. H. (2014). Three counting methods agree on cell and neuron number in chimpanzee primary visual cortex. Front. Neuroanat. 8:36 10.3389/fnana.2014.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto R., Shimakawa S., Suzuki S., Ogihara T., Tamai H. (2008). Edaravone prevents kainic acid-induced neuronal death. Brain Res. 1209 85–91. 10.1016/j.brainres.2008.02.064 [DOI] [PubMed] [Google Scholar]
- Morris D. C., Cui Y., Cheung W. L., Lu M., Zhang L., Zhang Z. G., et al. (2014). A Dose–response study of thymosin β4 for the treatment of acute stroke. J. Neurol. Sci. 345 61–67. 10.1016/j.jns.2014.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morterá P., Herculano-Houzel S. (2012). Age-related neuronal loss in the rat brain starts at the end of adolescence. Front. Neuroanat. 6:45 10.3389/fnana.2012.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen R. J., Buck C. R., Smith A. M. (1992). NeuN, a neuronal specific nuclear protein in vertebrates. Development 116 201–211. [DOI] [PubMed] [Google Scholar]
- Murabe Y., Ibata Y., Sano Y. (1982). Morphological studies on neuroglia. IV. proliferative response of non-neuronal elements in the hippocampus of the rat to kainic acid-induced lesions. Cell Tissue Res. 222 223–226. [DOI] [PubMed] [Google Scholar]
- Neher J. J., Neniskyte U., Brown G. C. (2012). Primary phagocytosis of neurons by inflamed microglia: potential roles in neurodegeneration. Front. Pharmacol. 3:27 10.3389/fphar.2012.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow C. W. (2007). The Pathogenesis of cell death in Parkinson’s disease – 2007. Mov. Disord. 22(Suppl. 17) S335–S342. 10.1002/mds.21675 [DOI] [PubMed] [Google Scholar]
- Panickar K. S., Norenberg M. D. (2005). Astrocytes in cerebral ischemic injury: morphological and general considerations. Glia 50 287–298. 10.1002/glia.20181 [DOI] [PubMed] [Google Scholar]
- Papageorgiou I. E., Fetani A. F., Lewen A., Heinemann U., Kann O. (2014). Widespread activation of microglial cells in the hippocampus of chronic epileptic rats correlates only partially with neurodegeneration. Brain Struct. Funct. 220 2423–2439. 10.1007/s00429-014-0802-0 [DOI] [PubMed] [Google Scholar]
- Paulsen J. S., Langbehn D. R., Stout J. C., Aylward E., Ross C. A., Nance M., et al. (2008). Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J. Neurol. Neurosurg. Psychiatry 79 874–80. 10.1136/jnnp.2007.128728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G., Watson C. (2016). The Rat Brain in Stereotaxic Coordinates. Sydney: Trove; Available at: http://trove.nla.gov.au/work/8025602 (accessed April 11, 2016). [DOI] [PubMed] [Google Scholar]
- Pérez-De La Cruz V., Carrillo-Mora P., Santamaría A. (2012). Quinolinic acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int. J. Tryptophan Res. 5 1–8. 10.4137/IJTR.S8158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Severiano F., Montes S., Gerónimo-Olvera C., Segovia J. (2013). Study of oxidative damage and antioxidant systems in two Huntington’s disease rodent models. Methods Mol. Biol. 1010 177–200. 10.1007/978-1-62703-411-1_12 [DOI] [PubMed] [Google Scholar]
- Racine R. J., Gartner J. G., Burnham W. M. (1972). Epileptiform activity and neural plasticity in limbic structures. Brain Res. 47 262–268. 10.1016/0006-8993(72)90268-5 [DOI] [PubMed] [Google Scholar]
- Ravizza T., Rizzi M., Perego C., Richichi C., Velísková J., Moshé S. L., et al. (2005). Inflammatory response and glia activation in developing rat hippocampus after status epilepticus. Epilepsia 46 113–117. 10.1111/j.1528-1167.2005.01006.x [DOI] [PubMed] [Google Scholar]
- Renolleau S., Aggoun-Zouaoui D., Ben-Ari Y., Charriaut-Marlangue C. (1998). A model of transient unilateral focal ischemia with reperfusion in the P7 neonatal rat morphological changes indicative of apoptosis. Stroke 29 1454–1461. 10.1161/01.STR.29.7.1454 [DOI] [PubMed] [Google Scholar]
- Santiago L. F., Rocha E. G., Freire M. A. M., Dias I. A., Lent R., Houzel J. C., et al. (2007). The organizational variability of the rodent somatosensory cortex. Rev. Neurosci. 18 283–294. 10.1515/REVNEURO.2007.18.3-4.283 [DOI] [PubMed] [Google Scholar]
- Sarnat H. B., Nochlin D., Born D. E. (1998). Neuronal Nuclear Antigen (NeuN): a marker of neuronal maturation in the early human fetal nervous system1. Brain Dev. 20 88–94. 10.1016/S0387-7604(97)00111-3 [DOI] [PubMed] [Google Scholar]
- Serrano Sánchez T., Alberti Amador E., Lorigados Pedre L., Blanco Lezcano L., Diaz Armesto I., Bergado J. A. (2014). BDNF in quinolinic acid lesioned rats after bone marrow cells transplant. Neurosci. Lett. 559 147–151. 10.1016/j.neulet.2013.11.060 [DOI] [PubMed] [Google Scholar]
- Skaggs K., Goldman D., Parent J. M. (2014). Excitotoxic brain injury in adult zebrafish stimulates neurogenesis and long-distance neuronal integration. Glia 62 2061–2079. 10.1002/glia.22726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell A. L., Franciosi S., Villanueva E. B., Xie Y., Winter L. A., Veeraraghavan J., et al. (2015). Anti-semaphorin 4D immunotherapy ameliorates neuropathology and some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 76 46–56. 10.1016/j.nbd.2015.01.002 [DOI] [PubMed] [Google Scholar]
- Spigolon G., Veronesi C., Bonny C., Vercelli A. (2010). C-Jun N-terminal kinase signaling pathway in excitotoxic cell death following kainic acid-induced status epilepticus. Eur. J. Neurosci. 31 1261–1272. 10.1111/j.1460-9568.2010.07158.x [DOI] [PubMed] [Google Scholar]
- Swanson R. A., Ying W., Kauppinen T. M. (2004). Astrocyte influences on ischemic neuronal death. Curr. Mol. Med. 4 193–205. 10.2174/1566524043479185 [DOI] [PubMed] [Google Scholar]
- Tai Y. F., Pavese N., Gerhard A., Tabrizi S. J., Barker R. A., Brooks D. J., Piccini P. (2007). Imaging microglial activation in Huntington’s disease. Brain Res. Bull. 72 148–151. 10.1016/j.brainresbull.2006.10.029 [DOI] [PubMed] [Google Scholar]
- Takatsuru Y., Nakamura K., Nabekura J. (2013). Compensatory contribution of the contralateral pyramidal tract after experimental cerebral ischemia. Front. Neurol. Neurosci. 32 36–44. 10.1159/000346409 [DOI] [PubMed] [Google Scholar]
- Takemiya T., Maehara M., Matsumura K., Yasuda S., Sugiura H., Yamagata K. (2006). Prostaglandin E2 produced by late induced COX-2 stimulates hippocampal neuron loss after seizure in the CA3 region. Neurosci. Res. 56 103–110. 10.1016/j.neures.2006.06.003 [DOI] [PubMed] [Google Scholar]
- Takeuchi H. (2013). “Glial communication via gap junction in neuroinflammation,” in Neuron-Glia Interaction in Neuroinflammation Vol. 7 eds Suzumura A., Ikenaka K. (New York, NY: Springer; ) 119–33. 10.1007/978-1-4614-8313-7_8 [DOI] [Google Scholar]
- Tattersfield A. S., Croon R. J., Liu Y. W., Kells A. P., Faull R. L., Connor B. (2004). Neurogenesis in the striatum of the quinolinic acid lesion model of Huntington’s disease. Neuroscience 127 319–332. 10.1016/j.neuroscience.2004.04.061 [DOI] [PubMed] [Google Scholar]
- Tomasi S., Sarmientos P., Giorda G., Gurewich V., Vercelli A. (2011). Mutant prourokinase with adjunctive C1-inhibitor is an effective and safer alternative to tPA in rat stroke. PLoS ONE 6:e21999 10.1371/journal.pone.0021999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi P. P., Sgadò P., Scali M., Viaggi C., Casarosa S., Simon H. H., et al. (2009). Increased susceptibility to kainic acid–induced seizures in Engrailed-2 knockout mice. Neuroscience 159 842–849. 10.1016/j.neuroscience.2009.01.007 [DOI] [PubMed] [Google Scholar]
- Walker F. O. (2007). Huntington’s disease. Lancet 369 218–228. 10.1016/S0140-6736(07)60111-1 [DOI] [PubMed] [Google Scholar]
- Wang Q., Yu S., Simonyi A., Sun G. Y., Sun A. Y. (2005). Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol. Neurobiol. 31 3–16. 10.1385/MN:31:1-3:003 [DOI] [PubMed] [Google Scholar]
- Wei B., Nie Y., Li X., Wang C., Ma T., Huang Z., et al. (2011). Emx1-expressing neural stem cells in the subventricular zone give rise to new interneurons in the ischemic injured striatum. Eur. J. Neurosci. 33 819–830. 10.1111/j.1460-9568.2010.07570.x [DOI] [PubMed] [Google Scholar]
- Weibel E. R. (1981). Stereological Methods: Theoretical Foundations Vol. 2 London: Academic Press. [Google Scholar]
- West M. J. (1999). Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci. 22 51–61. 10.1016/S0166-2236(98)01362-9 [DOI] [PubMed] [Google Scholar]
- West M. J., Slomianka L., Gundersen H. J. (1991). Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 231 482–497. 10.1002/ar.1092310411 [DOI] [PubMed] [Google Scholar]
- Wolf H. K., Buslei R., Schmidt-Kastner R., Schmidt-Kastner P. K., Pietsch T., Wiestler O. D., et al. (1996). NeuN: a useful neuronal marker for diagnostic histopathology. J. Histochem. Cytochem. 44 1167–1171. 10.1177/44.10.8813082 [DOI] [PubMed] [Google Scholar]
- Yamashita T., Ninomiya M., Hernández Acosta P., García-Verdugo J. M., Sunabori T., Sakaguchi M., et al. (2006). Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J. Neurosci. 26 6627–6636. 10.1523/JNEUROSCI.0149-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D. D., Kuan C. Y., Whitmarsh A. J., Rincón M., Zheng T. S., Davis R. J., et al. (1997). Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389 865–870. 10.1038/39899 [DOI] [PubMed] [Google Scholar]
- Yu S., Cheng Q., Li L., Liu M., Yang Y., Ding F. (2014). 2-(4-methoxyphenyl)ethyl-2-acetamido-2-deoxy-B-D-pyranoside confers neuro-protection in cell and animal models of ischemic stroke through calpain1/PKA/CREB-mediated induction of neuronal glucose transporter 3. Toxicol. Appl. Pharmacol. 277 259–269. 10.1016/j.taap.2014.03.025 [DOI] [PubMed] [Google Scholar]
- Zhang Z. G., Chopp M. (2009). Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol. 8 491–500. 10.1016/S1474-4422(09)70061-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Rempe D. A. (2010). Targeting astrocytes for stroke therapy. Neurotherapeutics 7 439–451. 10.1016/j.nurt.2010.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Spigolon G., Bonny C., Culman J., Vercelli A., Herdegen T. (2012). The JNK inhibitor D-JNKI-1 blocks apoptotic JNK signaling in brain mitochondria. Mol. Cell. Neurosci. 49 300–310. 10.1016/j.mcn.2011.12.005 [DOI] [PubMed] [Google Scholar]
- Zhu Y., Bu Q., Liu X., Hu W., Wang Y. (2014). Neuroprotective effect of TAT-14-3-3𝜀 fusion protein against cerebral ischemia/reperfusion injury in rats. PLoS ONE 9:e93334 10.1371/journal.pone.0093334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.