

Abstract
AIM: To reviewing genetic and epigenetic make-up of metastatic colorectal cancers (mCRCs) addicted to epidermal growth factor receptor (EGFR) signalling.
METHODS: The present study summarizes the potential value of prognostic and predictive biomarkers in selecting mCRC patients treated with anti-EGFR therapy. A meta-analysis was performed using a systematic search of PubMed, Medline and Web of Science to identify eligible papers until March 21st, 2016 using these following terms: ‘‘colorectal cancer’’, “predictive biomarkers’’, “anti-EGFR therapy”, “KRAS”, “NRAS’’, “PIK3CA”, “TP53”, “PTEN”, ‘‘EGFR”, “MET”, “HER2”, “epiregulin”, “amphiregulin”, “prognostic biomarkers”, “BRAF”, “miRNA” and “antibody-dependent cell-mediated cytotoxicity (ADCC) activity”. Two investigators independently evaluated and extracted data from each identified studies based on selected criteria of inclusion and exclusion.
RESULTS: The introduction of agents targeting EGFR such as cetuximab and panitumumab increased overall survival of mCRCs. Nevertheless, it has firstly became evident that response rates to cetuximab regimens in unselected patient populations were typically lower than 30%. Clinical data confirmed the predictive value of RAS mutations for resistance to cetuximab and panitumumab leading to the license of these monoclonal antibodies exclusively for the management of patients with RAS-wild type colorectal cancers. So far the identification of predictive biomarkers have generated interesting, though preliminary and, at times, conflicting data on the importance of tumour mRNA levels of EGFR ligands, of activating mutations in other genes such as NRAS and PIK3CA. The prognostic value of selected microRNAs level and ADCC activity is under investigation, while the prognostic impact of BRAF status remains controversial.
CONCLUSION: This review focuses on the personalized treatment of mCRC and discusses the potential of new prognostic and predictive biomarkers in selecting patients treated with anti-EGFR therapy.
Keywords: Metastatic colorectal cancer, Anti-epidermal growth factor receptor therapy, KRAS, Biomarkers, Antibody-dependent cell-mediated cytotoxicity
Core tip: This review focuses on progress in the metastatic colorectal cancer (mCRC) personalized treatment and on the role of prognostic and predictive biomarkers available for selecting patients treated with anti-epidermal growth factor receptor (EGFR) therapy. Not only the KRAS mutational status but also BRAF, NRAS, PIK3CA, TP53 and PTEN alterations might be useful in selecting patients who likely will respond to anti-EGFR treatments. In particular, we focused on the following points: (1) predictive biomarkers of response to anti-EGFR therapy; (2) prognostic biomarkers; and (3) new prognostic value of antibody-dependent cell-mediated cytotoxicity activity induced by cetuximab in mCRC.
INTRODUCTION
Colorectal cancer represents the third most frequent neoplastic disorder worldwide and one of the main causes of tumour-related mortality[1].
Treatments of metastatic colorectal cancer (mCRC) in the last 20 years have been improved and median overall survival (OS) increased approximately from 10 to 30 mo.
This significant increase of OS is due to the introduction, in systemic treatments, of biologic drugs targeting either angiogenesis such as bevacizumab, aflibercept and regorafenib, or epidermal growth factor receptor (EGFR) such as cetuximab and panitumumab[2].
EGFR on the cancer cell surface allows to transmit signals of proliferation, angiogenesis, metastasis. Cetuximab, a chimeric IgG1 monoclonal antibody (mAb) and panitumumab, a humanised IgG2 mAb, are now approved for patients with mCRC. They are used in combination with chemotherapy, either in first or in second line, or alone in refractory disease. Identification of tumors addicted to EGFR signalling and so susceptible to anti-EGFR therapy became mandatory, since, at first, response rates to cetuximab in unselected patients were less than 30%[3].
KRAS is a cytoplasmic GTP-binding protein with low GTPase activity. When GTP binds KRAS, signals of cellular proliferation and inhibition of apoptosis are released, thus KRAS acts as a classical oncogene. KRAS mutations were found mainly in exon 2, causing the abrogation of the GTPase activity and the lock of KRAS protein in the active form.
Those mutations, activating the RAS/RAF/MAPK pathway, make the targeting of EGFR therapeutically unuseful[4]. The value of KRAS exon 2 mutations in predicting resistance to cetuximab and panitumumab were confirmed by clinical data; thus these mAbs were licenced exclusively for in KRAS-wild type (WT) CRC patients[5,6].
KRAS and NRAS are closely related RAS oncogene family members. Alterations in exons 2, 3 and 4 of either gene constitutively activate RAS and are mutually exclusive, which suggests functional redundancy. So far, several retrospective, non-prespecified analyses of randomized clinical trials validated the pan-RAS mutations as negative predictive factors for anti-EGFR therapy[7,8].
On this base, the European regulatory authority (EMA) restricted the use of cetuximab and panitumumab to patients not having any mutation in KRAS or in NRAS codon 12, 13, 59, 61, 117 and 146 hotspots, defined as RAS-WT patients.
Interesting and, sometimes, conflicting preliminary data indicated new potential predictive biomarkers as the tumour mRNA levels of EGFR ligands and activating mutations in BRAF and PIK3CA[9].
BRAF activating mutations, mainly V600E, identify molecularly a subgroup (8%-10%) of CRCs. BRAF mutant (BRAF-mut) tumours have been thus defined to present specific clinical and histopathological characteristics, as typically are associated with female sex, old age, right-sided CRC, high-grade mucinous histotype, MSI, methylator phenotype and peritoneal and lymph node metastases[10].
This review focuses on progress in the mCRC personalized treatment and on the role of prognostic and predictive biomarkers available for selecting patients treated with anti-EGFR therapy.
MATERIALS AND METHODS
The selected literatures were determined via an electronic search of Medline, PubMed and Web of Science using these following terms: “colorectal cancer”, “predictive biomarkers”, “anti-EGFR therapy”, “KRAS”, “NRAS’’, “PIK3CA”, “TP53”, “PTEN”, ‘‘EGFR”, “MET”, “HER2”, “epiregulin”, “amphiregulin”, “prognostic biomarkers”, “BRAF”, “miRNA” and “antibody-dependent cell-mediated cytotoxicity (ADCC) activity”.
The last search was updated in March 21st, 2016. The search strategy used both MeSH terms and free-text words to increase the sensitivity of the search. The present study was performed in accordance with the standard guidelines for systematic reviews[11].
Inclusion and exclusion criteria
The two investigators (RV, LNC) independently assessed all the eligible studies and extracted the data. Studies were considered eligible if they met the following criteria: (1) response to anti-EGFR therapy; (2) primary and acquired resistance mechanisms to anti-EGFR treatment; (3) mutations and therapeutic modulation of EGFR; and (4) published as a full paper in English. Exclusion criteria are the following: (1) abstracts; (2) studies investigating the RAS/RAF/MEK/ERK pathway not involved in the response to anti-EGFR therapy; (3) studies without usable data; (4) studies published in language other than English; and (5) duplicate publications.
Data extraction
Two investigators (RV, LNC) independently evaluated and extracted data from each identified studies based on criteria of inclusion and exclusion.
RESULTS
Predictive biomarkers of response to anti-EGFR therapy
KRAS mutations: The Erb family includes cell membrane receptors such as HER1/erbB1 (EGFR), HER2/c-neu (ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4)[12]. EGFR gene is one of the major target of biologic drugs, and favoured the use of anti-EGFR mAbs and tyrosine kinase inhibitors (TKIs). Cetuximab (anti-IgG1 mAb) and panitumumab (anti-IgG2 mAb) bind to the extracellular ligand site, while erlotinib and gefitinib, as EGFR TKIs, compete with ATP to the TK binding domain and inhibit EGFR autophosphorylation. Both mAbs and TKIs interrupt the downstream intracellular signalling pathway. First clinical trial with anti-EGFR mAb enrolled patients with high tumoural EGFR expression; however overall response rates (ORRs) were low[13], suggesting that other unknown factors might affect response to these drugs[14].
Lièvre et al[15] identified a correlation between lack of response to anti-EGFR therapy and KRAS mutations. They analysed 30 patients receiving second- or third-line treatment with cetuximab plus irinotecan. The OS was significantly higher in KRAS-WT patients than in those having a KRAS mutation (median OS: 16.3 mo vs 6.9 mo, respectively, P = 0.016)[15].
KRAS protein is a GTPase bound to the intracellular part of the cell membrane, acting in the EGFR/RAS/RAF/MEK/ERK kinase signalling. It transfers extracellular signals from the EGFR to the nucleus and regulates proliferation, cell growth and apoptosis. The KRAS oncogene belongs to the Erb family and lies in the short arm of chromosome 12. Point mutations in the KRAS gene are usually in codon 12 (82%-87%) and 13 (13%-18%) (exon 2), in codon 61 (exon 3) and in codon 146 (exon 4)[16]. In KRAS-WT patients, the binding of anti-EGFR antibodies to the receptor induces conformational changes affecting its internalization and sequentially causes the direct inhibition of TK activity and the blockage of RAS/RAF/MEK/ERK downstream pathway. In fact, KRAS mutations abrogate the mAb-induced inhibition of EGFR and constitutively activate the KRAS intracellular domain. In CRC the incidence of KRAS mutations is 30%-45%[17]. KRAS mutational analysis may be done either in the primary tumour or in the metastatic sites since mutations are usually concordant (around 95%) in those two samples[18].
Two studies demonstrated a survival benefit over best supportive care only in KRAS-WT patients treated with cetuximab (median OS: 9.5 mo vs 4.8 mo) or panitumumab (median PFS: 12.3 wk vs 7.3 wk), respectively[5,19]. In patients with KRAS mutated tumours, mAbs did not improve PFS or OS as compared to the best supportive care. Moreover, the effectiveness of first- and second-line chemotherapy increases when combined with anti-EGFR mAbs in mCRC.
KRAS exon 2 mutations are extremely specific negative biomarkers of response to anti-EGFR mAbs in mCRC, although not all KRAS mutations are equivalent in the effect on cell proliferation and drug resistance[20].
KRAS p.G13D mutation: Some preclinical evidence showed that neoplastic cells with KRAS codon 13 glycine (G) to aspartate (D) mutations (p.G13D), having an incidence of 10%-15%[21], respond to cetuximab similarly to WT clones[22]. Furthermore, about 10% of response to anti-EGFR mAbs in patients carrying a KRAS mutation in tumour tissue and a further 15% of patients obtained a long-term disease stabilization[23,24] have been reported. In responding patients, codon 13 mutation is more frequent than in the whole KRAS-mut tumour population.
As previously stated, not all KRAS mutations are equivalent in the effect on cell proliferation and drug resistance. A large retrospective analysis of 579 chemorefractory mCRC patients treated with cetuximab reported that OS and PFS were significantly longer in patients with p.G13D mutation (n = 32) than in patients with other KRAS-mutations (median OS = 7.6 mo vs 5.7 mo, P = 0.005; median PFS = 4 mo vs 1.9 mo, P = 0.004). No significant difference in terms of ORR was observed between patients with p.G13D mutations and other mutations (6.3% vs 1.6%, respectively, P = 0.15). Authors did not specify the number of patients achieving the clinical benefit in the two groups. However, the significant longer PFS observed in patients with p.G13D mutation might suggest a difference in clinical benefit. A considerable correlation between type of KRAS mutation (p.G13D vs other KRAS mutations) and OS was observed in cetuximab treatment (HR = 0.30, 95%CI: 0.14-0.67, P = 0.003)[20].
In a pooled analysis of CRYSTAL and OPUS studies, where mCRC patients were randomized to receive FOLFIRI (CRYSTAL) or FOLFOX (OPUS) with or without cetuximab as first-line treatment, the addition of cetuximab to chemotherapy in patients with KRAS p.G13D mutation was found advantageous[21]. Precisely, among 83 patients with p.G13D mutation, those receiving chemotherapy plus cetuximab performed better in PFS and OS than patients treated with chemotherapy alone; on the contrary, patients with any other KRAS mutation, did not benefit from combined treatment[21].
The role of KRAS mutations in codons 12 or 13 has been studied in a pooled analysis of clinical trials in which panitumumab was added to FOLFOX4 in first-line[25], to FOLFIRI in second-line[26], or compared with best supportive care in heavily pre-treated mCRC patients[13]. In conclusion it was found that codon 13 KRAS-mut tumours are unlikely to benefit from panitumumab in the same way as tumours mutated in codon 12.
NRAS mutations: Neuroblastoma-ras (NRAS) gene belongs to the RAS oncogene family and is located on chromosome 1. It encodes for a GTPase membrane protein that shuttles between the cell membrane and the Golgi system. NRAS mutations, with a 3%-5% rate in CRC[9], are associated with anti-EGFR treatment failure. Moreover KRAS, BRAF and NRAS mutations are mutually exclusive[27].
A retrospective analysis suggested a potential negative prognostic role of RAS mutations in patients with mCRC, reporting a worse median OS in NRAS and KRAS mutated patients (25.6 mo vs 30.2 mo, respectively) in comparison to all WT (42.7 mo)[28].
PIK3CA mutations: Phosphatidylinositol 3-kinase (PI3K) activates AKT, triggering downstream pathways and promoting proliferation and cell survival. The PIK3CA gene encodes the PI3K catalytic subunit; mutations in this gene result in aberrant AKT activation and cancer growth[29,30]. PIK3CA mutations are reported in 10%-20% of CRC and their effect on clinical outcome is not yet well know[30].
Ogino et al[31] showed that, in patients with KRAS-WT, PIK3CA mutation increases tumor-specific mortality (HR = 3.80, 95%CI: 1.56-9.27), but did not observe any significant effect on mortality in KRAS-mut patients (HR = 1.25, 95%CI: 0.585-2.96)[31]. Conversely, in mCRC, a PIK3CA mutation was found in 17.7% of cetuximab-treated patients, but ORR, time-to-progression and OS did not differ between mutated and WT patients[32]. In the CAIRO2 study, PIK3CA mutation was not linked to outcome in KRAS-WT tumours treated with cetuximab: 5-year survival was 90% in PIK3CA-WT and 82% in PIK3CA-mut (log-rank P = 0.075)[33]. Sartore-Bianchi et al[29] examined PIK3CA mutational status in exons 9 and 20 in 110 mCRC patients treated with cetuximab or panitumumab. They identified 15 patients (13.6%) with PIK3CA mutations, and none of them responded to anti-EGFR mAbs (P = 0.038). The correlation between lack of response and PIK3CA mutations was even more evident in KRAS-WT patients (P = 0.016), reinforcing the study by Ogino et al[31]. Differently Prenen et al[30] analyzed 200 chemorefractory mCRCs and did not find a correlation between PIK3CA and anti-EGFR mAb resistance: 13% of PIK3CA-mutated patients responded to cetuximab and 11% did not (P = 0.78)[30].
These conflicting results keep a high degree of uncertainty about the predictive role of PIK3CA mutations.
TP53 mutations: p53 protein is activated when DNA damage occurs but also when oncogenes are inappropriately activated, in order to induce cell apoptosis. p53 pathway alteration has been systematically observed in non-small cell lung cancer with activating EGFR mutations, suggesting that p53 inactivation is necessary to allow expansion of a cell with EGFR activation. Moreover, it has been proposed that p53 acts as a brake for the activated PI3K transduction cascade since PI3K signalling activates p53 mediated growth suppression. These data confirm the hypothesis that EGFR activation is oncogenic, and therefore anti-EGFR mAbs might be efficient in tumours only if p53 is inactivated[34].
PTEN: PTEN is a key tumour suppressor gene involved in the homeostatic maintenance of PI3K/AKT pathway. Loss of PTEN expression, evaluated by IHC, is reported in 20%-40% of CRCs and increases phosphatidylinositol-3,4,5-triphosphate, the major substrate of PTEN; this induces a persistent activation of PI3K effectors[35,36]. Differently from KRAS status, only 60% of primary tumour are concordant in PTEN expression with metastases, since PTEN loss is more frequent in distant metastases[37,38].
Low PTEN expression in primary tumour of mCRC patients treated with cetuximab plus irinotecan did not affect outcome, but, if PTEN was measured in the metastatic tissue, ORR and PFS were significantly longer in patients with high PTEN expression than in those with low: 26% vs 5% (P = 0.007) and 4.7 mo vs 3.3 mo (P = 0.005), respectively[18].
Sartore-Bianchi et al[29] reported, in 81 mCRC patients, that PTEN loss correlted with lack of response to cetuximab and panitumumab (P = 0.001), with a shorter PFS and OS.
EGFR p.S492R mutation: The EGFR p.S492R mutation, caused either by 1476C>A or 1474A>C substitution, alters binding to cetuximab but not to panitumumab and it has been described in mCRCs with acquired resistance to cetuximab. In a retrospective analysis of patients with available samples from ASPECCT, 16% of patients in the cetuximab arm and 1% of patients in the panitumumab developed EGFR p.S492R mutation[39]. Patients with EGFR p.S492R mutation in the cetuximab arm had longer treatment duration before progressive disease and appeared to have worse OS than patients with WT p.S492 in the cetuximab arm[39] (Figure 1).
Figure 1.
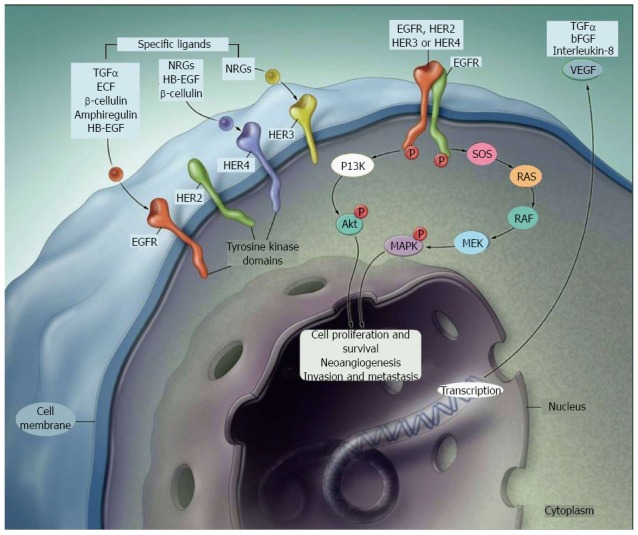
Signal transduction pathways controlled by the activation of epidermal growth factor receptor. Adapted from Ref. [64]. TGFα: Transforming growth factor alpha; ECF: Eosinophil chemotactic factor; HB-EGF: Heparin-binding EGF-like growth factor; NGR: Neuregulin; EGFR: Epidermal growth factor receptor; bFGF: Basic fibroblast growth factor; VEGF: Vascular endothelial growth factor; PI3K: Phosphoinositide 3-kinase; SOS: Son of sevenless homolog protein.
Acquired resistance mechanisms
KRAS mutant clones: A number of studies have identified KRAS somatic mutations as a biomarker of intrinsic resistance to anti-EGFR drugs in CRC patients, but the molecular basis for acquired resistance are still obscure. Alterations in KRAS gene, causing drug resistance, can be due either to the selection of pre-existent KRAS mutant and to the amplified clones or to new mutations, since the pressure of cetuximab might induce de novo KRAS mutation. KRAS mutant alleles are detected in the 0.4% to 17% of resistant tumors[40].
MET: The MET gene encodes the tyrosine kinase receptor for hepatocyte growth factor (HGF) and has an oncogenic role in several solid tumors, where it is activated by gene amplification, overexpression, activating mutations or autocrine stimulation. MET and its ligand HGF are involved in acquired resistance to targeted therapies[41].
MET inhibitors (including the clinically approved drug crizotinib for patients with mutant ALK or ROS-1) are effective. Preclinical data in which CRC xenopatients carrying MET amplification are treated is encouraging. MET gene amplification is a novel mechanism of both primary and acquired resistance to cetuximab or panitumumab. The rate of MET amplification in untreated mCRC is around 1%; it correlated with resistance to anti-EGFR therapy and might be overcome by MET kinase inhibitors[41].
HER2 amplification: HER2 gene amplification and protein overexpression were identified in about 3%-6% of CRC patients[42].
HER2 mutations activate intracellular signaling pathways, increase anchorage-independent growth in soft agar and produce resistance to the EGFR monoclonal antibodies (cetuximab and panitumumab) in colon cell lines. Therefore HER2 activating mutations may themselves be a drug target for the treatment of colorectal cancer. It was demonstrate that these HER2 mutations cause oncogenic transformation of colon epithelial cells and produce resistance to cetuximab and panitumumab in two colorectal cancer cell lines[43].
Epiregulin and amphiregulin: The EGFR ligands epiregulin (EREG) and amphiregulin (AREG) are commonly overexpressed in CRC. In a prospectively planned retrospective biomarker study from the PICCOLO trial, high ligand expression AREG/EREG is a predictive marker for panitumumab therapy benefit on PFS in RAS-WT patients; conversely, patients with low ligand expression gained no benefit[44].
Furthermore, patients with EREG and AREG mRNA expression had longer survival than those with low-expression tumors[45].
Prognostic biomarkers
BRAF mutations: The cytoplasmic serine-threonine kinase BRAF is immediately downstream of KRAS, acting as one of its main effectors, and needs be phosphorylated by KRAS for its activation.
BRAF activating mutations, mainly V600E, define a molecularly specific subset (8%-10%) of CRCs. The V600E is caused by a CTG to CAG point mutation at codon 600 and results in the RAS/RAF/MEK/ERK pathway constitutive activation, as KRAS mutations do. In CRC mutations in KRAS and BRAF are mutually exclusive[46].
BRAF V600E mutation correlated with a very aggressive phenotype and poor prognosis which usually led to a median OS < 1 year[46].
In addition to this prognostic value in CRC, retrospective studies also suggested that BRAF mutations might predict primary resistance to anti-EGFR mAbs[9,46].
The BRAF V600E mutation was retrospectively analysed in a 113 patients treated with cetuximab or panitumumab, with or without chemotherapy, and was detected in 11 (13.9%) of 79 KRAS-WT patients, None of them reported an objective tumour response[46].
Nine percent of patients in the CRYSTAL study had BRAF mutations and experienced a shorter median OS in both the FOLFIRI (10.3 mo) and FOLFIRI/cetuximab (14.1 mo) arms in respect to KRAS-WT/BRAF-WT patients, where survival was 21.6 and 25.1 mo, respectively. On the other hand, BRAF mutations were not related to cetuximab efficacy[6]. Even in the pooled analysis of the CRYSTAL and OPUS data, BRAF mutation was identified as a negative prognostic marker, with lower PFS and OS in the BRAF-mut patients independently of the treatment[47]. De Roock et al[9] reported a mutation rate of 4.7% in 761 chemorefractory patients treated with cetuximab plus chemotherapy. In comparison to BRAF-WT, BRAF-mut patients had a significantly lower ORR and a worse PFS and OS[9].
The negative prognostic value of BRAF mutation emerged also from the PRIME trial[7], where patients with RAS-WT but BRAF-mut tumours had a shorter PFS and OS compared to the RAS-WT and BRAF-WT ones. In the RAS-WT/BRAF-mut patients, panitumumab added to chemotherapy a small benefit in DFS and OS (P = 0.12 and 0.76, respectively), that was not significant[7]. The PICCOLO phase III prospective trial investigated in 1198 KRAS-WT mCRCs the addition of panitumumab to single-agent irinotecan, in second or subsequent-line[48]. BRAF-mut patients (13.6%) showed a shorter OS compared to BRAF-WT, and panitumumab produced a detrimental effect on survival (HR = 1.84, 95%CI: 1.10-3.08, P = 0.029)[48].
BRAF mutations at codon 594 and 596, identify mCRCs with different pathological, clinical, and prognostic characteristics when compared to BRAF V600E mutated ones. In BRAF V600E mutated tumors, the frequency of microsatellite instability is relatively high, even in the metastatic stage (about 20%); on the contrary, all the codons 594 or 596 BRAF mutated tumors were considered as microsatellite stable.
Cremolini et al[49] found that patients mutated in BRAF codons 594 or 596 showed a trend in longer OS if compared to BRAF-WT [(62.0 mo vs 35.9 mo) HR = 0.55 (95%CI: 0.29-1.05), P = 0.081] and a significant longer OS than BRAF V600E mutated [62.0 mo vs 12.6 mo; HR = 0.36 (95%CI: 0.20-0.64), P = 0.002].
Pietrantonio et al[50], in a meta-analysis of BRAF-mut CRCs, demonstrated that the addition of anti-EGFR mAbs not increased OS, PFS nor ORR in first- and subsequent-line treatments.
In the meta-analysis of Rowland et al[51], there was no sufficient evidence to definitively state a treatment benefit of anti-EGFR mAbs in RAS-WT/BRAF-mut mCRCs compared to RAS-WT/BRAF-WT.
Ultimately BRAF mutations act as a negative prognostic factor more than a predictive marker of resistance to anti-EGFR mAb. BRAF-mut patients presented a shorter survival than the BRAF-WT ones, independently from treatment. They might have a minimal benefit from anti-EGFR therapy than BRAF-WT patients.
microRNAs high level: microRNA (miRNAs) are endogenous, short (17-25 bases), non-coding single-stranded RNAs involved in the post-transcriptional regulation of target gene expression.
Experimental and clinical data, addressing the clinical role of Let-7a level in relation to the SNP in the Let-7a KRAS mRNA binding site and the type of KRAS mutation, concluded that high Let-7 miRNA level might correlate with a relevant antitumor activity from anti-EGFR therapy in the presence of KRAS mutations[52].
Cappuzzo et al[53] showed that KRAS-WT patients with high miR-99a/Let-7c/miR-125b cluster expression showed longer OS and PFS than patients with low levels.
Antibody-dependent cell-mediated cytotoxicity activity: ADCC has been proposed as a parallel mechanism of cetuximab activity[54].
ADCC is a response of innate immune cells that exerts antitumor cytotoxicity and is activated when the Fc fragment of the antibody interacts with the Fc receptor on the immune cells. Some polymorphisms regulating Fc:FcR interactions has been reported as relevant in the level of ADCC induced by cetuximab[55]. In particular, response to therapeutic mAbs has been correlated with specific SNPs in FCGR2A (H131R) and FCGR3A (V158F) genes[56]. However, data up to now are conflicting, since came mostly from low-powered studies with small sample sizes[57].
Drugs that target Natural Killer (NK) cells, γδ T cells, macrophages and dendritic cells might augment the immune response and enhance the antitumor activity of the mAbs[58].
Invariant CD1d-restricted natural killer T (iNKT) cells are T lymphocytes with an invariant T-cell antigen receptor-α-chain rearrangement that co-express NK markers[59].
Molling et al[60] observed that a circulating iNKT cells deficit is linked to poor clinical outcome in HNSCC, suggesting a critical role in immune response against tumor. Moreover, determination of iNKT cells level might help in determining which patients can benefit from immunotherapeutic adjuvant therapies which aim to reconstitute the circulating iNKT cells reservoir[60].
The question if ADCC is associated with EGFR expression and/or RAS and BRAF mutations remains not yet clear in CRC. Seo et al[61] observed that the ADCC is significantly related to EGFR levels, but not with mutations in KRAS and/or BRAF.
Lo Nigro et al[62] investigated the prognostic role of iNKT and ADCC in 41 KRAS-WT mCRC patients treated with cetuximab in II and III lines. Authors demonstrated that patients with basal level of ADCC above the median (71%) presented a longer OS in comparison to those with ADCC below (16 mo vs 8 mo, P = 0.026). No significant correlation of iNKT cells with OS (P = 0.19) was seen, but a tendency of a better OS after 10 mo in patients with high iNKT cells basal level (above median of 0.382 cells/mL). However, patients with both high ADCC activity and high circulating iNKT cells showed a beneficial effect compared to low ADCC and low iNKT. This benefit seems superior to the role of ADCC alone supporting the hypothesis of a positive interplay between iNKT and ADCC effector cells. Correlation of key SNPs involved in ADCC ability and OS and PFS revealed not to be significant, in line with other reports[63]. Patients having both alleles with A in FCGR2A and TT in FCGR3A showed a longer, although not significant, PFS (9 mo vs 5 mo, P = 0.064) (Table 1).
Table 1.
Novel molecular biomarkers
| Biomarker | Incidence | Prognostic value | Predictive value | Ref |
| K-RAS mutations | 40% | Controversial | Predictor of resistance | [20-22,24] |
| K-RAS G13D mutation | 15%-20% | Controversial | Faint resistance | [21] |
| N-RAS mutations | 3%-5% | Controversial | Predictor of resistance | [9,27,28] |
| PI3KCA mutations | 10%-20% | Controversial | Controversial | [29-31] |
| TP53 | 15%-50% | - | Controversial | [34] |
| PTEN expression | 20%-40% | Controversial | Controversial | [18,29,36] |
| S492R mutation | 16% | - | Controversial | [39] |
| KRAS mutant clones | 0.4%-17% | - | Controversial | [40] |
| MET amplification | 1% | - | Controversial | [41] |
| HER2 amplification | 3%-6% | Controversial | - | [42,43] |
| Epiregulin and amphiregulin | - | Controversial | - | [44,45] |
| B-RAF mutations | 4%-15% | Poor prognosis | Controversial | [7,46-51] |
| miRNAs high level | - | Controversial | - | [52,53] |
| Cetuximab basal ADCC activity | - | Controversial | - | [62] |
ADCC: Antibody-dependent cell-mediated.
DISCUSSION
The anti-EGFR mAbs cetuximab and panitumumab, by blocking MAPK pathway, are important in the treatment of mCRC.
KRAS and NRAS mutations in exons 2, 3 and 4 (overall found in around 50% of mCRCs) are predictive of anti-EGFR mAbs resistance. Thus, guidelines limit the use of cetuximab and panitumumab to RAS-WT patients. However, some RAS-WT tumours do not respond to anti-EGFR therapy. Since the cost of mAbs is high and treatment-related toxicity might be severe, the need of identifying additional predictive markers in anti-EGFR therapy beside KRAS is still compelling.
Mutations in KRAS exons 3 and 4, NRAS and not functional PTEN should be searched in mCRCs to improve clinical benefit of anti-EGFR mAbs. Uncertain is the predictive role of PIK3CA mutations.
Controversial remains the prognostic impact of BRAF mutations in CRC. A recent meta-analysis, suggested that anti-EGFR mAb therapy do not provide benefit in BRAF mutated mCRCs[49]. Conversely, another meta-analysis, concluded that there is not sufficient evidence to consider BRAF-WT as a definitive negative predictive biomarker in mCRC patients treated with anti-EGFR mAbs. The gain in OS and PFS for BRAF-WT tumours may be small or less evident, but further studies are necessary to shed light to this point[50].
Cell membrane EGFR expression does not seem to influence therapy efficacy. Researchers are now ongoing to assess the predictive value of the number of EGFR and HER2 copies, mutations in the NRAS, PI3KCA, TP53 and PTEN genes, concentration of EGFR ligands, expression of epiregulin and amphiregulin and SNPs in the EGF and EGFR, and in the FCGR2A and FCGR3A genes.
The definition of predictive and prognostic role of PIK3CA mutation, PTEN deletion and TP53 mutation is of interest, but there is still no consensus among clinicians on their use in clinical practice and in decision-making. Beyond a doubt, in the future, NRAS, PIK3CA and PTEN, in addition with KRAS and BRAF mutation analysis, will be useful in selecting mCRC patients that might benefit from anti-EGFR therapy.
NK cells are considered as major mediators of the therapeutic effect of cetuximab due to ADCC. iNKT cells number and ADCC level, exerted by NKs in the presence of cetuximab, might be useful prognostic or predictive markers of response[62].
In the next future, a robust analysis of many genes and different mutations, is likely to help in selecting patients and predicting the efficacy of anti-EGFR treatment. This approach will hopefully identify a mCRC subset with specific biological behaviour and treatment response. This will be an important step forward the “personalized medicine” of CRC patients and will inform the correct use of anti-EGFR antibodies.
COMMENTS
Background
The new therapeutic approach integrates novel molecular biomarkers with the pathologic features of a tumour to improve the prediction of prognosis and treatment efficacy. In recent years we have tried to study the molecular mechanisms underlying resistance to epidermal growth factor receptor (EGFR) inhibitors in order to obtain a better selection of patients for these treatments and improve the clinical outcome of patients treated with anti-EGFR mAbs.
Research frontiers
The metastatic colorectal cancer (mCRC) treatment has been linked to molecular progresses, which led to the discovery of prognostic and predictive biomarkers of response to anti-EGFR therapy. In the present study we summarize the potential value of prognostic and predictive biomarkers in selecting mCRC patients treated with anti-EGFR therapy.
Innovations and breakthroughs
It is clear that the evaluation of not only the KRAS mutational status but also NRAS, PIK3CA, TP53, PTEN, EGFR, MET, HER2, epiregulin and amphiregulin alterations might be beneficial to the selection of patients who are likely to respond to anti-EGFR therapies. Controversial remain the prognostic role of BRAF in addition to new potential prognostic factors such as iNKT cells and basal antibody-dependent cell-mediated cytotoxicity activity.
Applications
In summary, this review might be identify a subgroup of mCRC patients with distinct biological behaviour and response to treatments, including anti-EGFR antibodies. All of this will be a step forward in the “personalized medicine” treatment of CRC patients.
Terminology
EGFR on the cancer cell surface allows to transmit signals of proliferation, angiogenesis, metastasis. Cetuximab, a chimeric IgG1 monoclonal antibody (mAb) and panitumumab, a humanised IgG2 mAb, are currently licensed for the treatment of patients with mCRC. Markers able to select tumours addicted to EGFR signalling and so susceptible to anti-EGFR therapeutic modulation have been so far identified as KRAS (V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog), NRAS (neuroblastoma RAS viral oncogene homolog), PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha), TP53 (tumour protein p53), PTEN (phosphatise and tensin homolog), MET (proto-oncogene receptor tyrosine kinase) and HER2 (erb-b2 receptor tyrosine kinase 2).
Peer-review
This manuscript focuses on progress in the personalized treatment of mCRC and discusses the potential of new prognostic and predictive biomarkers in selecting patients treated with anti-EGFR therapy. The authors evaluated not only the KRAS mutational status but also BRAF, NRAS, PIK3CA and PTEN alterations which might be beneficial to the selection of patients who are likely to respond to anti-EGFR therapies.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C, C
Grade D (Fair): D
Grade E (Poor): 0
Conflict-of-interest statement: Merlano MC received honoraria as consultant from Merckgroup; The other authors have no financial and personal conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 25, 2016
First decision: May 12, 2016
Article in press: July 6, 2016
P- Reviewer: Kim SM, Li CF, LinY, Liu YP, Ziogas DE S- Editor: Ma YJ L- Editor: A E- Editor: Ma S
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Peeters M, Price T. Biologic therapies in the metastatic colorectal cancer treatment continuum--applying current evidence to clinical practice. Cancer Treat Rev. 2012;38:397–406. doi: 10.1016/j.ctrv.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Linardou H, Briasoulis E, Dahabreh IJ, Mountzios G, Papadimitriou C, Papadopoulos S, Bafaloukos D, Kosmidis P, Murray S. All about KRAS for clinical oncology practice: gene profile, clinical implications and laboratory recommendations for somatic mutational testing in colorectal cancer. Cancer Treat Rev. 2011;37:221–233. doi: 10.1016/j.ctrv.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F, Wittinghofer A. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 5.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 6.Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pintér T, Lim R, Bodoky G, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 7.Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 8.Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmüller C, Kahl C, Seipelt G, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1065–1075. doi: 10.1016/S1470-2045(14)70330-4. [DOI] [PubMed] [Google Scholar]
- 9.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 10.Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, Agarwal A, Maru DM, Sieber O, Desai J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–4632. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151:264–269, W64. doi: 10.7326/0003-4819-151-4-200908180-00135. [DOI] [PubMed] [Google Scholar]
- 12.Heinemann V, Stintzing S, Kirchner T, Boeck S, Jung A. Clinical relevance of EGFR- and KRAS-status in colorectal cancer patients treated with monoclonal antibodies directed against the EGFR. Cancer Treat Rev. 2009;35:262–271. doi: 10.1016/j.ctrv.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–1664. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 14.Siena S, Sartore-Bianchi A, Di Nicolantonio F, Balfour J, Bardelli A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101:1308–1324. doi: 10.1093/jnci/djp280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 16.Puerta-García E, Cañadas-Garre M, Calleja-Hernández MÁ. Molecular biomarkers in colorectal carcinoma. Pharmacogenomics. 2015;16:1189–1222. doi: 10.2217/PGS.15.63. [DOI] [PubMed] [Google Scholar]
- 17.Wong R, Cunningham D. Using predictive biomarkers to select patients with advanced colorectal cancer for treatment with epidermal growth factor receptor antibodies. J Clin Oncol. 2008;26:5668–5670. doi: 10.1200/JCO.2008.19.5024. [DOI] [PubMed] [Google Scholar]
- 18.Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, Masi G, Graziano F, Cremolini C, Rulli E, et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol. 2009;27:2622–2629. doi: 10.1200/JCO.2008.20.2796. [DOI] [PubMed] [Google Scholar]
- 19.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 20.De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M, Piessevaux H, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812–1820. doi: 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 21.Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. 2012;30:3570–3577. doi: 10.1200/JCO.2012.42.2592. [DOI] [PubMed] [Google Scholar]
- 22.Molinari F, Felicioni L, Buscarino M, De Dosso S, Buttitta F, Malatesta S, Movilia A, Luoni M, Boldorini R, Alabiso O, et al. Increased detection sensitivity for KRAS mutations enhances the prediction of anti-EGFR monoclonal antibody resistance in metastatic colorectal cancer. Clin Cancer Res. 2011;17:4901–4914. doi: 10.1158/1078-0432.CCR-10-3137. [DOI] [PubMed] [Google Scholar]
- 23.Lee CN, Chen HY, Liu HE. Favorable response to erlotinib in a lung adenocarcinoma with both epidermal growth factor receptor exon 19 deletion and K-ras G13D mutations. J Clin Oncol. 2010;28:e111–e112. doi: 10.1200/JCO.2009.24.0747. [DOI] [PubMed] [Google Scholar]
- 24.De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–515. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 25.Peeters M, Douillard JY, Van Cutsem E, Siena S, Zhang K, Williams R, Wiezorek J. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J Clin Oncol. 2013;31:759–765. doi: 10.1200/JCO.2012.45.1492. [DOI] [PubMed] [Google Scholar]
- 26.Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJ, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28:4706–4713. doi: 10.1200/JCO.2009.27.6055. [DOI] [PubMed] [Google Scholar]
- 27.Hawkes E, Cunningham D. Relationship between colorectal cancer biomarkers and response to epidermal growth factor receptor monoclonal antibodies. J Clin Oncol. 2010;28:e529–e331; author reply e532-e533. doi: 10.1200/JCO.2010.29.5626. [DOI] [PubMed] [Google Scholar]
- 28.Schirripa M, Cremolini C, Loupakis F, Morvillo M, Bergamo F, Zoratto F, Salvatore L, Antoniotti C, Marmorino F, Sensi E, et al. Role of NRAS mutations as prognostic and predictive markers in metastatic colorectal cancer. Int J Cancer. 2015;136:83–90. doi: 10.1002/ijc.28955. [DOI] [PubMed] [Google Scholar]
- 29.Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1851–1857. doi: 10.1158/0008-5472.CAN-08-2466. [DOI] [PubMed] [Google Scholar]
- 30.Prenen H, De Schutter J, Jacobs B, De Roock W, Biesmans B, Claes B, Lambrechts D, Van Cutsem E, Tejpar S. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res. 2009;15:3184–3188. doi: 10.1158/1078-0432.CCR-08-2961. [DOI] [PubMed] [Google Scholar]
- 31.Ogino S, Nosho K, Kirkner GJ, Shima K, Irahara N, Kure S, Chan AT, Engelman JA, Kraft P, Cantley LC, et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol. 2009;27:1477–1484. doi: 10.1200/JCO.2008.18.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cappuzzo F, Varella-Garcia M, Finocchiaro G, Skokan M, Gajapathy S, Carnaghi C, Rimassa L, Rossi E, Ligorio C, Di Tommaso L, et al. Primary resistance to cetuximab therapy in EGFR FISH-positive colorectal cancer patients. Br J Cancer. 2008;99:83–89. doi: 10.1038/sj.bjc.6604439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tol J, Dijkstra JR, Klomp M, Teerenstra S, Dommerholt M, Vink-Börger ME, van Cleef PH, van Krieken JH, Punt CJ, Nagtegaal ID. Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. Eur J Cancer. 2010;46:1997–2009. doi: 10.1016/j.ejca.2010.03.036. [DOI] [PubMed] [Google Scholar]
- 34.Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 2007;27:662–677. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 36.Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 37.Molinari F, Martin V, Saletti P, De Dosso S, Spitale A, Camponovo A, Bordoni A, Crippa S, Mazzucchelli L, Frattini M. Differing deregulation of EGFR and downstream proteins in primary colorectal cancer and related metastatic sites may be clinically relevant. Br J Cancer. 2009;100:1087–1094. doi: 10.1038/sj.bjc.6604848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santini D, Loupakis F, Vincenzi B, Floriani I, Stasi I, Canestrari E, Rulli E, Maltese PE, Andreoni F, Masi G, et al. High concordance of KRAS status between primary colorectal tumors and related metastatic sites: implications for clinical practice. Oncologist. 2008;13:1270–1275. doi: 10.1634/theoncologist.2008-0181. [DOI] [PubMed] [Google Scholar]
- 39.Price TJ, Newhall K, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, Venkatasatya SA, Thomas A, Tjulandin S, et al. Prevalence and outcomes of patients (pts) with EGFR S492R ectodomain mutations in ASPECCT: Panitumumab (pmab) vs. cetuximab (cmab) in pts with chemorefractory wild-type KRAS exon 2 metastatic colorectal cancer (mCRC) J Clin Oncol. 2015;33(suppl 3):abstr 740. [Google Scholar]
- 40.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–673. doi: 10.1158/2159-8290.CD-12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seo AN, Kwak Y, Kim DW, Kang SB, Choe G, Kim WH, Lee HS. HER2 status in colorectal cancer: its clinical significance and the relationship between HER2 gene amplification and expression. PLoS One. 2014;9:e98528. doi: 10.1371/journal.pone.0098528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, Searleman AC, Shen W, Monsey J, Trusolino L, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5:832–841. doi: 10.1158/2159-8290.CD-14-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seligmann JF, Elliott F, Richman SD, Jacobs B, Hemmings G, Brown S, Barrett JH, Tejpar S, Quirke P, Seymour MT. Combined Epiregulin and Amphiregulin Expression Levels as a Predictive Biomarker for Panitumumab Therapy Benefit or Lack of Benefit in Patients With RAS Wild-Type Advanced Colorectal Cancer. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2015.6065. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 45.Llovet P, Sastre J, Ortega JS, Bando I, Ferrer M, García-Alfonso P, Donnay O, Carrato A, Jiménez A, Aranda E, et al. Prognostic Value of BRAF, PI3K, PTEN, EGFR Copy Number, Amphiregulin and Epiregulin Status in Patients with KRAS Codon 12 Wild-Type Metastatic Colorectal Cancer Receiving First-Line Chemotherapy with Anti-EGFR Therapy. Mol Diagn Ther. 2015;19:397–408. doi: 10.1007/s40291-015-0165-0. [DOI] [PubMed] [Google Scholar]
- 46.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 47.Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, Celik I, Köhne CH. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48:1466–1475. doi: 10.1016/j.ejca.2012.02.057. [DOI] [PubMed] [Google Scholar]
- 48.Seymour MT, Brown SR, Middleton G, Maughan T, Richman S, Gwyther S, Lowe C, Seligmann JF, Wadsley J, Maisey N, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14:749–759. doi: 10.1016/S1470-2045(13)70163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cremolini C, Di Bartolomeo M, Amatu A, Antoniotti C, Moretto R, Berenato R, Perrone F, Tamborini E, Aprile G, Lonardi S, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol. 2015;26:2092–2097. doi: 10.1093/annonc/mdv290. [DOI] [PubMed] [Google Scholar]
- 50.Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, Cabiddu M, Iacovelli R, Bossi I, Lonati V, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51:587–594. doi: 10.1016/j.ejca.2015.01.054. [DOI] [PubMed] [Google Scholar]
- 51.Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, Sorich MJ. Meta-analysis comparing the efficacy of anti-EGFR monoclonal antibody therapy between KRAS G13D and other KRAS mutant metastatic colorectal cancer tumours. Eur J Cancer. 2016;55:122–130. doi: 10.1016/j.ejca.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 52.Ruzzo A, Graziano F, Vincenzi B, Canestrari E, Perrone G, Galluccio N, Catalano V, Loupakis F, Rabitti C, Santini D, et al. High let-7a microRNA levels in KRAS-mutated colorectal carcinomas may rescue anti-EGFR therapy effects in patients with chemotherapy-refractory metastatic disease. Oncologist. 2012;17:823–829. doi: 10.1634/theoncologist.2012-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cappuzzo F, Sacconi A, Landi L, Ludovini V, Biagioni F, D’Incecco A, Capodanno A, Salvini J, Corgna E, Cupini S, et al. MicroRNA signature in metastatic colorectal cancer patients treated with anti-EGFR monoclonal antibodies. Clin Colorectal Cancer. 2014;13:37–45.e4. doi: 10.1016/j.clcc.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 54.Iannello A, Ahmad A. Role of antibody-dependent cell-mediated cytotoxicity in the efficacy of therapeutic anti-cancer monoclonal antibodies. Cancer Metastasis Rev. 2005;24:487–499. doi: 10.1007/s10555-005-6192-2. [DOI] [PubMed] [Google Scholar]
- 55.Taylor RJ, Chan SL, Wood A, Voskens CJ, Wolf JS, Lin W, Chapoval A, Schulze DH, Tian G, Strome SE. FcgammaRIIIa polymorphisms and cetuximab induced cytotoxicity in squamous cell carcinoma of the head and neck. Cancer Immunol Immunother. 2009;58:997–1006. doi: 10.1007/s00262-008-0613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol. 2013;6:1. doi: 10.1186/1756-8722-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kjersem JB, Skovlund E, Ikdahl T, Guren T, Kersten C, Dalsgaard AM, Yilmaz MK, Fokstuen T, Tveit KM, Kure EH. FCGR2A and FCGR3A polymorphisms and clinical outcome in metastatic colorectal cancer patients treated with first-line 5-fluorouracil/folinic acid and oxaliplatin +/- cetuximab. BMC Cancer. 2014;14:340. doi: 10.1186/1471-2407-14-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kohrt HE, Houot R, Marabelle A, Cho HJ, Osman K, Goldstein M, Levy R, Brody J. Combination strategies to enhance antitumor ADCC. Immunotherapy. 2012;4:511–527. doi: 10.2217/imt.12.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Porcelli S, Yockey CE, Brenner MB, Balk SP. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J Exp Med. 1993;178:1–16. doi: 10.1084/jem.178.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Molling JW, Langius JA, Langendijk JA, Leemans CR, Bontkes HJ, van der Vliet HJ, von Blomberg BM, Scheper RJ, van den Eertwegh AJ. Low levels of circulating invariant natural killer T cells predict poor clinical outcome in patients with head and neck squamous cell carcinoma. J Clin Oncol. 2007;25:862–868. doi: 10.1200/JCO.2006.08.5787. [DOI] [PubMed] [Google Scholar]
- 61.Seo Y, Ishii Y, Ochiai H, Fukuda K, Akimoto S, Hayashida T, Okabayashi K, Tsuruta M, Hasegawa H, Kitagawa Y. Cetuximab-mediated ADCC activity is correlated with the cell surface expression level of EGFR but not with the KRAS/BRAF mutational status in colorectal cancer. Oncol Rep. 2014;31:2115–2122. doi: 10.3892/or.2014.3077. [DOI] [PubMed] [Google Scholar]
- 62.Lo Nigro C, Ricci V, Vivenza D, Monteverde M, Strola G, Lucio F, Tonissi F, Miraglio E, Granetto C, Fortunato M, et al. Evaluation of antibody-dependent cell-mediated cytotoxicity activity and cetuximab response in KRAS wild-type metastatic colorectal cancer patients. World J Gastrointest Oncol. 2016;8:222–230. doi: 10.4251/wjgo.v8.i2.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Srivastava RM, Lee SC, Andrade Filho PA, Lord CA, Jie HB, Davidson HC, López-Albaitero A, Gibson SP, Gooding WE, Ferrone S, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19:1858–1872. doi: 10.1158/1078-0432.CCR-12-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]