

Abstract
Reprogramming of somatic cells has become a versatile tool for biomedical research and for regenerative medicine. In the current study, we show that manipulating alternative splicing (AS) is a highly potent strategy to produce cells for therapeutic applications. We demonstrate that silencing of hTAF4-TAFH activity of TAF4 converts human facial dermal fibroblasts to melanocyte-like (iMel) cells. iMel cells produce melanin and express microphthalmia-associated transcription factor (MITF) and its target genes at levels comparable to normal melanocytes. Reprogramming of melanoma cells by manipulation with hTAF4-TAFH activity upon TAFH RNAi enforces cell differentiation towards chondrogenic pathway, whereas ectoptic expression of TAF4 results in enhanced multipotency and neural crest-like features in melanoma cells. In both cell states, iMels and cancer cells, hTAF4-TAFH activity controls migration by supporting E- to N-cadherin switches. From our data, we conclude that targeted splicing of hTAF4-TAFH coordinates AS of other TFIID subunits, underscoring the role of TAF4 in synchronised changes of Pol II complex composition essential for efficient cellular reprogramming. Taken together, targeted AS of TAF4 provides a unique strategy for generation of iMels and recapitulating stages of melanoma progression.
Alternative splicing (AS) is a key process regulating gene expression and underlying proteome diversity. By changing the activity of transcription factors, AS affects cell growth, differentiation1,2, survival3,4 and tumourigenesis5,6,7. Changes in the splicing patterns accompany frequently with reprogramming of somatic cells into induced pluripotent stem cells (iPSCs)8,9,10. The discovery of methods for generation of iPSCs by use of specific transcription factors, chromatin-modifying compounds, non-coding RNAs and low molecular weight substances has provided different promising strategies for development of tools for different disease modelling and cell therapy applications11. The pioneering study of in vitro somatic cell reprogramming used forced expression of MyoD to convert mouse fibroblasts into muscle cells12. Use of various combinations of lineage-specific transcription factors has become by now a widely acknowledged approach for direct conversion of fibroblasts into functional neurons, hepatocytes, cardiomyocytes and melanocytes13,14,15,16. Research on cellular reprogramming is growing at high speed by applying it to numerous target cells and miscellany of reprogramming factors17. However, regulated AS as a tool for effective cell reprogramming has not been actively pursued.
It is commonly known that mechanisms of cellular reprogramming share similar features with cancer initiation18. For example, pluripotency transcription factors c-MYC and KLF4 are commonly known as proto-oncogenes19; similar signalling pathways are active in cancer development and upon generation of iPSCs18,20. Down-regulation of tumour suppressor genes, such as p53, enhances reprogramming efficiency21, while premature termination of cell reprogramming in vivo leads to cancer development22. It is even speculated that in vivo cancer progression could be initiated by reprogramming-like events23. Despite all these findings, only a few reports have been able to convincingly demonstrate successful reprogramming of cancer cells24.
According to the current view, general transcription machinery is a dynamic and cell context-specific structure25. Transcription factor TFIID as a subunit of the general transcription machinery consists of TATA-binding protein (TBP) and up to 14 TBP-associated factors (TAFs)26,27,28. Most of the TAF subunits in TFIID complex are needed for self-renewal of human embryonic stem cells (hESCs)29, while a few of TFIID subunits are required for cell differentiation30,31,32. TAF4 is one of the major structural and regulatory components of TFIID. Previous studies have found that TAF4 is subjected to extensive cell- and tissue-specific splicing33,34,35. Our recent data show that splicing events in the region encoding the co-activator hTAF4-TAFH domain control the differentiation of human neural progenitors (NHNPs)35,36 and human adipose-derived mesenchymal stem cells (hMSCs)34. Targeted proteolysis of Taf4 was demonstrated to be necessary for differentiation of mouse F9 embryonic carcinoma cells37 and myogenic differentiation of myoblasts38, whereas enforced expression of TAFH domain blocked differentiation of F9 cells towards early endodermal lineages39. Moreover, inactivation of Taf4 in mouse epidermis resulted in hyperplasia and development of aggressive melanomas in the dermis compartment40,41. In keratinocytes, the absence of Taf4 led to ectopic expression of melanocyte-specific melan-A and melanoma-associated antigen 9 (MAGEA9) and development of invasive melanomas40. Interestingly, an upstream enhancer containing binding sites for pluripotency transcription factors Oct4 and Nanog was identified in mouse Taf4 gene42. Most recent findings described TAF4 as one of the critical components in converting human fibroblasts into iPSCs43. Thus, with important functions in maintenance of TFIID stability and integrity, TAF4 represents a unique tool for manipulating the whole TFIID composition and in promoting specific cellular programs.
Here, we provide a new concept for cell reprogramming, where instead of changing the transcription regulatory networks by forced expression of lineage-specific transcription factors or use of different miRNAs, we advocate for targeted AS of the core components of RNA Pol II transcription machinery. As an example, we targeted the activity of TAF4 by TAFH-specific RNAi to examine the potential of this approach for reprogramming of human dermal fibroblasts. Data presented here allow us to conclude that targeting AS of TAF4 affects the entire TFIID complex, providing a unique model system to induce iMels and reprogram tumour cells to less or more aggressive cancer phenotypes.
Results
Differential activity patterns of hTAF4-TAFH are characteristic to dermal fibroblasts, melanocytes and melanoma cells
Previously, we have demonstrated that exons encoding the hTAF4-TAFH domain are subjected to extensive AS in hMSCs and NHNP cells34,35. To assess whether AS of TAF4 exons V, VI and VII encoding the hTAF4-TAFH (Fig. 1a) is prevailing in cells of neural crest (NC) origin, we examined the expression of TAF4 ASVs in facial dermal fibroblasts, melanocytes and melanomas.
Figure 1. Expression of TAF4 ASVs in human dermal fibroblasts, melanocytes, primary melanoma and immortalised melanoma cells.
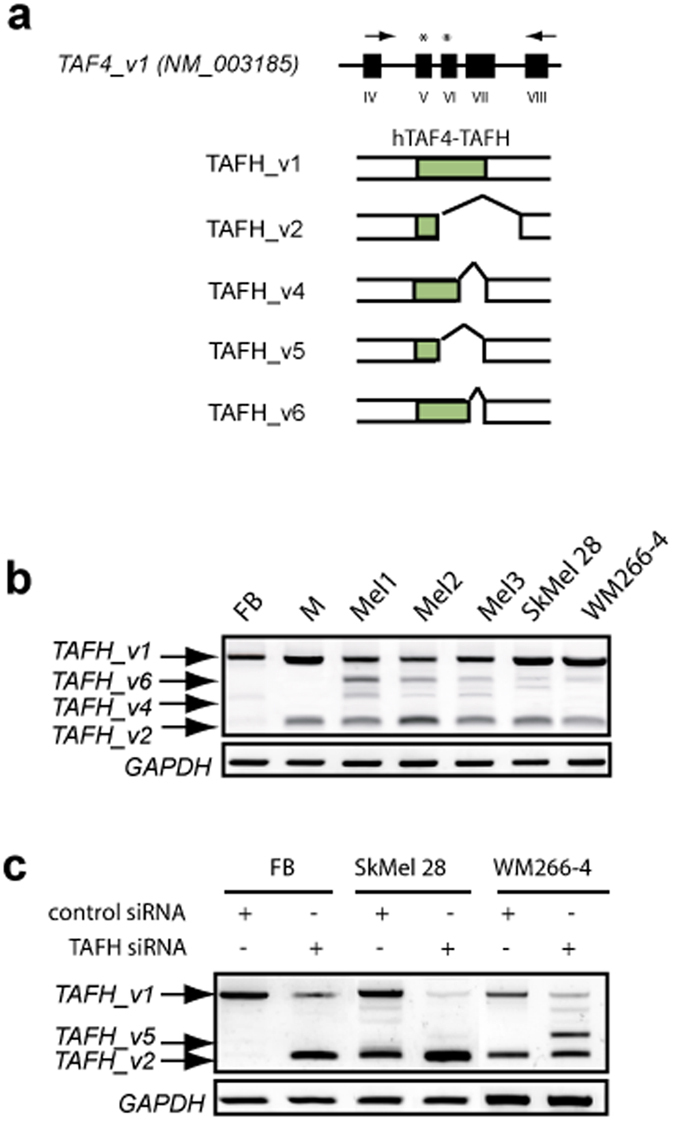
(a) Schematic representation of a fragment of TAF4 gene relevant to the current study. Black blocks with numbers indicated below show exons. Arrows indicate the location of TAFH-specific RT-PCR primers and asterisks correspond to the respective TAFH siRNAs. TAF4 isoforms (TAFH_v1, 2, 4, 5, 6) with splicing-affected hTAF4-TAFH domains (in green) are depicted below the gene structure. TAF4 isoforms are referred according to the previously published data34. (b) RT-PCR analysis of primary dermal fibroblasts (FB), melanocytes (M), primary melanomas (Mel1-3), and melanoma SkMel28 and WM266-4 cells using TAFH-specific primers. Canonical (TAFH_v1) and major alternative splice variants (TAFH_v2, _v4 and _v6) are indicated. (c) RT-PCR analysis of TAFH (TAF4ex5 and TAF4ex6) siRNA-treated fibroblasts, SkMel28 and WM266-4 melanoma cells showed effective disruption of exons encoding hTAF4-TAFH domain. GAPDH mRNA expression is used as a loading control.
Using transcript-specific RT-PCR approach, we exclusively examined the splicing of TAF4 exons encoding hTAF4-TAFH domain. TAFH_v1 mRNAs encoding intact hTAF4-TAFH were detected in all analysed cells, while melanocytes, primary melanoma tissues and cultured melanoma cells next to TAFH_v1 mRNAs expressed TAFH_v2 mRNAs encoding proteins with no hTAF4-TAFH domain at comparable to TAFH_v1 levels (Fig. 1b). In primary melanoma tumours and cultured cells, TAFH_v2 was expressed along with TAFH_v4 and TAFH_v6, which all result in isoforms with no hTAF4-TAFH activity (Fig. 1a,b).
For the detailed analysis of hTAF4-TAFH function, we elaborated RNAi approach with TAFH-specific siRNAs targeting exons V or VI of TAF4 to affect the expression of different ASVs (Fig. 1a). Upon TAFH RNAi, silencing of TAFH_v1 mRNAs both in fibroblasts and melanoma SkMel28 and WM 266-4 cells promoted the expression of transcripts encoding isoforms without hTAF4-TAFH (Fig. 1c) that was also verified by changed patterns of TAF4 isoform expression (Fig. 2e). Expression analysis data along with TAFH RNAi intrigued us to study the effects of hTAF4-TAFH activity in NC-derived cells in more detail.
Figure 2. hTAF4-TAFH activity controls conversion of facial dermal fibroblasts to melanocyte-like cells.

(a) Gradual rise in TAFH siRNA concentration from 20 to 100 nM changed the colour of pelleted fibroblasts to dark brown, indicating to the melanin production in these cells upon treatment. (b) Melanin content assay. Fibroblasts were transfected with different amounts of TAFH siRNAs. The relative production of melanin was calculated by measuring cell absorption spectra values at 405 nm in TAFH and control siRNA-treated fibroblasts and normalised to the amount of total protein. For reference control, melanin synthesis was induced by 0.1 mM IBMX (3-isobutyl-1-methylxanthine) in SkMel28 melanoma cells and compared to the value in control siRNA-treated fibroblasts (dashed line). (c) RT-qPCR analysis showed increased expression of melanocyte-specific genes in TAFH siRNA-transfected fibroblasts relatively to the control siRNA-treated cells at 24 h after treatment. Data shown are relatively to the levels of gene expression in terminally differentiated melanocytes, where 0% is the gene expression in primary fibroblasts, and 100% shows the levels of gene expression in primary melanocytes. Data were received from three independent experiments, with p at least < 0.01. (d) Immunofluorescence staining analysis of TAFH or control siRNA-treated fibroblasts at 48 h post-treatment reveals induced expression of MITF in the nuclear compartment of the cells. For positive control, nuclear localisation of MITF expression was verified in SkMel28 and WM266-4 melanoma cells. Scale bar, 40 μm. (e) TAFH siRNA-treatment induced up-regulation of p53 in fibroblasts: phosphorylated p53Ser15 (left) and TP53 mRNAs (right). mRNA data are normalised to GAPDH levels and shown as mean ± SD of three independent experiments with p value < 0.001 (***).
Silencing of hTAF4-TAFH converts facial dermal fibroblasts to iMels
We have established that abrogation of hTAF4-TAFH activity promotes differentiation of hMSCs and NHNPs34,35. In the current work, hTAF4-TAFH silencing in fibroblasts by RNAi resulted in highly obvious visual colour change of cell pellets from pale to dark brown in a siRNA dose-dependent manner (Fig. 2a). This finding was in a good agreement with RT-PCR analysis data establishing that expression of TAF4 ASVs in TAFH RNAi-treated fibroblasts resembled that of melanocytes. Namely, TAFH_v1 was expressed at low and TAFH_v2 at high levels (Fig. 1c). Upon TAFH RNAi, the induction of melanogenesis in dermal fibroblasts was further confirmed by the results of melanin content assay (Fig. 2b) and expression analysis of MITF and genes of melanin biosynthesis pathway (Fig. 2c,d). Specifically, treatment of fibroblasts with TAFH-specific siRNAs induced the concentration-dependent increase in the content of melanin relative to the control siRNA-treated cells (Fig. 2b). Conclusively, RT-qPCR analysis of TAFH RNAi-treated fibroblasts compared to the control siRNA-treated cells showed the following (Fig. 2c): a) induced expression of SNAI2 and PAX3, early transcription factors of melanocyte development; b) stimulation of expression of total MITF mRNA and melanocyte-specific alternative form MITF-M mRNA; c) increased expression of MITF target genes TYR, TYRP1 and DCT. When RT-qPCR data of TAFH RNAi-treated fibroblasts were normalised to the levels of gene expression in primary fibroblasts (arbitrarily referenced as 0%) or melanocytes (taken for 100%), it became apparent that the expression of melanocyte-specific genes in induced melanocytes (iMels) was highly similar to that in melanocytes (Fig. 2c). While immunofluorescence (IF) analysis further confirmed the nuclear expression of MITF in TAFH RNAi-treated fibroblasts similar to melanoma cells (Fig. 2d). Altogether, these data suggested that hTAF4-TAFH activity perturbs the spontaneous conversion of dermal fibroblasts to iMels.
Silencing of hTAF4-TAFH activity in fibroblasts induced the expression of TP53 mRNAs and accumulation of phospho-p53 (Fig. 2e) similar to the hMSCs34, indicating that the function of p53 in the process of hTAF4-TAFH-associated reprogramming is conserved across different cell types.
Altogether, these data argue that TAFH RNAi is an efficient strategy to generate iMel cells from facial dermal fibroblasts.
hTAF4-TAFH activity restricts differentiation potential of melanoma cells
Based on the data from iMel cells, we expected hTAF4-TAFH activity to control the differentiation of melanoma cells. To examine this, we compared TAFH RNAi effects in two metastatic SkMel28 and WM 266-4 melanoma cells. In both cells, TAFH siRNA treatment suppressed cell proliferation by day 5 (Fig. 3a). Moreover, RT-qPCR analysis revealed that hTAF4-TAFH activity was essential for NC- and stem cell-specific gene expression in melanoma cells (Fig. 3b). Upon TAFH RNAi, expression of pluripotency-associated NANOG and NC-specific MSX2, PAX7, SOX10, and SNAI1 mRNAs was significantly reduced, whereas the levels of stem cell factors KLF4 and OCT4 remained steady (Fig. 3b). These results suggested that depletion of hTAF4-TAFH activity was needed to initiate differentiation of cancer cells. We tested this hypothesis by using culture conditions supporting lineage-specific differentiation. As expected, silencing of hTAF4-TAFH activity in melanoma cells promoted the initiation of mixed routes of differentiation towards chondrogenic, adipogenic and neurogenic lineages. In differentiation-supported conditions, TAFH RNAi-treated melanoma cells started to express chondrogenic-specific SOX9, NKX3.2 and RUNX2; adipogenic PPARG; and neurogenic NTRK2, NF-M and SYP mRNAs at 48 h post-stimulation of differentiation (Fig. 3b).
Figure 3. hTAF4-TAFH activity regulates differentiation of melanoma cells.

(a) Data of WST-1 assay of TAFH and control siRNA-treated WM266-4 and SkMel28 cells subjected to 0 h, 24 h and 5 days of proliferation. In WST-1 proliferation assay, the amount of formazan that occurs during the enzymatic cleavage of the tetrazolium salt WST-1 by cellular mitochondrial dehydrogenases present in viable cells, were measured and normalised to the values in day one of proliferation. The data represented as mean ± SD of three independent experiments. (b) RT-qPCR analysis showed that upon TAFH RNAi, the expression of pluripotency-associated (KLF4, OCT4, NANOG) and neural crest (MSX2, PAX7, SOX10, SNAI1) mRNAs were down-regulated or maintained, while the expression of chondrogenic (SOX9, NKX3.2, RUNX2), adipogenic (PPARG) and neuronal (NTRK2, MF-M, SYP) mRNAs were up-regulated in WM266-4 and SkMel28 cells. For differentiation studies, TAFH siRNA-treated for 24 h cells were stimulated to differentiate and analysed at 48 h post-treatment. All data are normalised to GAPDH expression and relatively to the expression in control siRNA-treated cells and represented in LN scale. Mean ± SD values from three independent experiments with statistical significance of ***p < 0.001 and **p < 0.01 are shown. Similar statistical findings are indicated with *** in the middle of neighbouring bars. (c) Immunofluorescence staining of chondrocyte-specific SOX9 (red) in TAFH or control siRNA-treated WM266-4 and SkMel28 melanoma cells stimulated to chondrogenesis is shown at 7 day of differentiation. Nuclei are stained blue with DAPI (4′, 6-diamidino-2-phenylindole). Results are shown at 20× magnification. (d) Alcian blue staining of TAFH or control siRNA-treated WM266-4 and SkMel28 melanoma cells that were stimulated to chondrogenic differentiation for 14 days. Images were taken at 10× magnification.
Our recent findings established that silencing of hTAF4-TAFH specifically supported the differentiation of hMSCs along chondrogenic lineages34. Considering this and given that in general the cartilaginous differentiation of melanomas is regarded to be a rare event44,45, we decided to analyse TAFH siRNA-treatment effects on melanoma differentiation in chondrogenesis-supporting media conditions in more detail. Immunoanalysis data revealed that at 7 days of differentiation, TAFH RNAi-treated melanomas showed high levels of expression of chondrocyte-specific SOX9 proteins (Fig. 3c) and prominent staining with Alcian blue that evidenced the synthesis of cartilage-specific ECM-proteins (Fig. 3d). All these data further argued that hTAF4-TAFH rendered TAF4 to control chondrogenic differentiation in melanoma cells.
In sum, TAFH RNAi strategy was beneficial for reprogramming of melanoma cells to recapitulate different stages of cancer progression and unmask the differentiation potential of melanomas along lineages supported by the environmental surrounding.
hTAF4-TAFH controls migration of iMels and invasiveness of melanoma cells
Given that differentiation is accompanied by enhanced migratory activity of lineage-committed cells, we analysed the effects of TAFH RNAi on cell motility. For that, migration and invasion potential of iMels and melanoma cells was evaluated by measuring their migration activity towards 10% FBS across polycarbonate membrane. For invasion experiments, membranes were coated with ECM proteins derived from primary fibroblasts that were initially cultivated on the same transwell membranes. TAFH RNAi induced a robust (7-fold) rise in migration of iMels and also supported the invasion of melanoma cells (Fig. 4a). Consistent with these findings, both, iMels and cancer cells, showed high expression of CDH2 and matrix metallopeptidase 3 (MMP3) mRNAs and low expression of CDH1 and keratin 14 (KRT14) mRNAs at 72 h after TAFH RNAi (Fig. 4b). Western blot analysis further confirmed the induction of expression of N-cadherin (CDH2) and inhibition of E-cadherin (CDH1) upon effective hTAF4-TAFH silencing in both cell types (Fig. 4c).
Figure 4. hTAF4-TAFH activity controls expression of E- and N-cadherins and cell motility of dermal fibroblasts and melanoma cells.

(a) Cell migration and invasion assays showed that TAFH RNAi accelerated the motility of fibroblasts and SkMel28 and WM266-4 melanoma cells. Control and TAFH siRNA-treated cells were grown for 72 h following serum-starvation for 16 h before induction of migration. Migrated towards 10% FBS as a chemoattractant cells were stained with 0.1% crystal violet and photographed (10× magnification). The number of migrated cells was determined by counting from five independent microscopic fields and represented as mean ± SD with ***p < 0.001. (b) Relative expression of indicated genes (TAF4, CDH1, CDH2, KRT14 and MMP3) in dermal fibroblasts and melanoma cells after treatments with TAFH siRNAs as analysed by RT-qPCR and compared to control siRNA treatments. Dashed line shows basal levels of gene expression in control siRNA-treated cells. At least three independent experiments were performed and represented as mean ± SD with **p < 0.01 and ***p < 0.001. (c) Western blot analysis of total protein extracts showed changes in the expression of CDH1 (E-cadherin) and CDH2 (N-cadherin) upon TAFH RNAi as compared with control siRNA-treated cells. Equal loading was confirmed by GAPDH expression.
The results from the migration and invasion assays established that suppression of hTAF4-TAFH activity was critical for high motility of iMels and invasiveness of melanoma cells.
Enforced expression of TAF4 supports cancer stem cell-like features in melanoma cells
To further assess the role of hTAF4-TAFH activity in cell reprogramming, we investigated whether melanoma cells were amenable to reprogramming towards induced cancer stem cell (iCSC)-like cells by using forced expression of TAF4. Overexpression of TAF4 resulted as expected in the heightened levels of TAF4 protein (Fig. 5a) and significantly increased TAF4_v1 mRNA expression (Fig. 5b), and brought along phenotypic changes of SkMel28 melanoma towards cells with drastically reduced invasive potential (Fig. 5c). These cells had lost their ability to migrate because of the high hTAF4-TAFH activity that switched cells to predominant expression of E-cadherin (Fig. 5a). These changes were also observed at mRNA levels (Fig. 5b). Consistently, CDH2 and MMP3 levels were reduced, and CDH1 and KRT14 mRNA levels increased in these cells (Fig. 5b). On the other hand, enforced expression of TAF4 promoted expression of pluripotency- associated KLF4, OCT4 and NANOG, and NC-related MSX2, PAX7, SOX10 and SNAI1 (Fig. 5b), reaffirming the critical role of hTAF4-TAFH activity in the maintaining of multipotency in melanoma cells.
Figure 5. Effects of enforced expression of TAF4 on invasion and multipotency of melanoma cells.

(a) Western blot analysis of whole cell extracts confirmed the high levels of expression of TAF4 and E-cadherin (CDH1), and low expression of N-cadherin (CDH2) in SkMel28 cells transfected with TAF4 recombinant expression vector at 72 h post-transfection. Equal loading was confirmed by GAPDH expression analysis. (b) RT-qPCR analysis of pluripotency, NC (neural crest) and migration-associated genes upon enforced expression of TAF4 in SkMel28 cells showed that high levels of TAF4 expression support multipotency and inhibit invasion. Data are normalised to GAPDH expression relatively to control plasmid-treated cells and represented in LN scale. At least three independent experiments were performed for each analysis and represented as mean±SD with ***p < 0.001, and **p < 0.01. (c) Cell invasion assays showed that high levels of TAF4 suppress the invasion potential of SkMel28 cells. The number of invaded cells was determined by counting from five independent microscopic fields, normalised to cell numbers that were transfected with control vector (Contr) and represented as mean±SD with ***p<0.001 determined by Student t-test.
All in all, data of melanoma cell reprogramming by enforced expression of TAF4 evidenced the fast induction of multipotent features in tumours just by altering the hTAF4-TAFH activity levels in cells.
TAFH RNAi results in changed splicing of transcripts encoding TFIID complex subunits
Finally, we examined whether TAF4-associated cell reprogramming might affect the expression of other members of TAF complex. Firstly, we analyzed the expression of TAF4b, TAF4 paralog, in response to TAF4-TAFH siRNA treatments (Supplementary Figure S1). Western blot (Fig. S1a) and RT-PCR (Fig. S1b) data established that levels of TAF4b remained unaffected upon TAFH silencing. Next, RT-PCR analysis revealed that human fibroblasts and melanoma cells, in general, expressed a wide variation of ASVs encoding TFIID complex subunit proteins (Fig. 6A and data not shown). Transcript-specific RT-PCR analysis showed that many of these had retained ORFs and thus the potential to code different isoforms (Fig. 6A and Supplementary Figure S2). In fibroblasts, silencing of hTAF4-TAFH activity led to the reduced expression of distinct ORF-retaining TAF2, TAF6 and TAF12 ASVs. In melanomas, TAFH RNAi induced the differential expression of TBP, TAF1, TAF2, TAF6, TAF10, and TAF12 ASVs. More specifically, TAFH RNAi in melanoma cells led to the high levels of TAF6_v3 encoding the protein isoform with altered histone fold domains (HFD) similar to the apoptosis-dependent form of TAF6, namely TAF6δ46 (Fig. 6a and Supplementary Figure S2). Furthermore, in SkMel28 melanoma cells, TAFH RNAi promoted the expression of TBP_v2, which encodes protein isoforms with no poly-glutamine Armadillo-like helical region that is functionally important for hetero-oligomeric interactions47 (Fig. 6a and Supplementary Figure S2). Splicing of TAF1, the largest and multifunctional TFIID subunit, was also affected by perturbations in TAFH structure, introducing alterations into its protein kinase and histone acetyltransferase domains (Fig. 6a and Supplementary Figure S2). Altogether, these data argued for the tight control of TFIID composition and function at the level of AS of TAF4. However, yet more data is needed to draw more definite conclusions.
Figure 6. hTAF4-TAFH governs AS events of different TFIID subunits and global differentiation and migratory potential of NC-derived cells.

(A) RT-PCR analysis of TBP, TAF1, TAF2, TAF6, TAF10 and TAF12 in response to TAFH RNAi in primary fibroblasts and cultured melanoma WM 266-4 and SkMel28 cells was performed by using transcript-specific primers. Respective ASVs that were sequence verified are denoted on the left and characterised in more detail in Figure S2. (B) High levels of proteins with intact hTAF4-TAFH (TAFH) are inherent to the stem and stem-like cells with low capacity to migrate, while abolished hTAF4-TAFH activity (∆TAFH protein isoforms) is characteristic to highly motile cells committed to differentiate. High levels of expression are shown in bold. Yellow colour indicates to the expression of TAF4 proteins in normal NC-derived cells (facial fibroblasts and melanocytes), whereas purple area relates to melanoma cells. Data of TAFH overexpression in fibroblasts are taken from43. Scheme is adapted from71. iMel–induced melanocytes; iCSC-like cells—induced cancer stem cell-like cells.
In whole, our data demonstrated for the first time that subunits of TFIID complex were targets of extensive cell-specific AS. Changes in hTAF4-TAFH activity affected the expression of TFIID complex components as a part of the molecular control of cellular reprogramming and cancer development.
Discussion
This study concludes that splicing patterns retaining high levels of hTAF4-TAFH activity lead to cell phenotypes with high multipotency but low capacity to migrate, while splicing events resulting in low hTAF4-TAFH activity enforce cells to migrate and differentiate (Fig. 6B). Simultaneous presence of TAF4 isoforms with high and low hTAF4-TAFH activities in normal cells and in cancer suggests that dominance of any of these forms is likely to have dramatic effects upon development and in tumour progression.
Current cell separation and cultivation methods have been limiting the use of melanocytes in clinics48. Generation of functional melanocytes from a highly accessible cell source such as dermal fibroblasts by cell reprogramming method as described herein would establish an easily scalable supply of autologous melanocytes, which could be beneficial for developing cell-based treatments of congenital pigment disorders, or in the studies of the etiopathogenesis of melanomas. Previous data have substantiated that reprogramming of dermal fibroblasts into functional melanocytes implies the use of certain combinations of exogenously added factors including transcription factors MITF, SOX10 and PAX316. Instead of changing the transcriptional networks by enforced expression of specific transcription factors or microRNAs, this study provides a new concept for manipulating cell fate by targeted AS of general transcription machinery components. Here we show that targeted splicing of exons encoding hTAF4-TAFH domain alters TAF4 activity and converts fibroblasts directly into iMel cells. This is the first study to demonstrate that TAFH RNAi approach is an effective tool for generating the iMel cells with characteristics of normal melanocytes. Moreover, our findings establish a link between hTAF4-TAFH activity and activation of p53 to accompany with effective reprogramming of fibroblasts into iMels. Involvement of p53 in tanning response49 and hyperpigmentation of epidermis has been described50. Simultaneous activity of MITF and p53 in pigmented skin has suggested a model of crosstalk between these two cellular pathways as necessary for protection of skin against UV harms49. Thus, TAF4-p53 interactions may be crucial for effective reprogramming of cells. However, this hypothesis remains yet to be tested.
Recent studies along with ours have demonstrated that differentiated cells do not express canonical TAF4 protein at high levels30,38,51. Herein we show that malignantly transformed cells, melanoma in particular, represent a unique model where both, canonical and alternative TAF4 isoforms, are expressed at comparable levels providing an attractive context for manipulating cancer cells in vitro. Similar to hMSCs34, neural progenitor cells35, and facial dermal fibroblasts, silencing of hTAF4-TAFH activity in melanoma promotes these cells to differentiate. Melanoma is a highly divergent neoplasia, with prominent genetic and clonal heterogeneity52,53. The phenomenon of reversal differentiation is well described for NC-derived lineages54,55. In the current study, we have demonstrated the critical role of hTAF4-TAFH in driving the fibroblast-like (mesenchymal) features in melanoma cells, as silencing of hTAF4-TAFH activity promoted melanoma cells to differentiate along mesenchymal lineages. In addition, enforced expression of hTAF4-TAFH revealed that melanoma cells avidly acquire phenotypes of highly plastic NC stem cell-like cells56 and characteristics of iCSCs57,58, supporting the expression of pluripotency KLF4, NANOG and OCT4 and genes associated with multipotent NC phenotype. It is highly conceivable that similar changes in AS of TAF4 with consequences to reprogramming of cancer cells can spontaneously occur in vivo. In sum, our study is the first to pinpoint to the role of hTAF4-TAFH activity of TAF4 as one of the mechanisms in driving heterogeneity in melanoma, where cells with low hTAF4-TAFH activity differentiate along different lineages and cells with high activity of hTAF4-TAFH retain multipotency.
Our current data suggest that integrity of hTAF4-TAFH domain is critical for cell migration. We established that changes in the activity of hTAF4-TAFH can spontaneously convert dermal fibroblasts into highly motile iMels and support invasion of melanoma cells. Low levels of hTAF4-TAFH activity induce E- to N-cadherin switches in these cells. Changes in the expression of these ECM proteins are a characteristic of epithelial-to-mesenchymal transition (EMT), which is a crucial process in development and in cancer progression59. Current findings provide a new understanding of how one of the core processes of EMT is under the control of hTAF4-TAFH activity.
By analysing the expression of ASVs of different subunits of TFIID in response to TAFH RNAi, we found that cell-specific AS is highly common for components of TFIID complex. The majority of TAF subunits are expressed in tissue- and development-specific manner. The functions of TAF4, TAF8 and TAF10 have been found to be critical for maintaining pluripotency43, whereas TAF2, TAF7 and TAF12 have come across as ESC determinants60. Silencing of TAF3 and TAF5, on the other hand, results in loss of OCT4 function61,62. Differential expression of TAF subunits drives lineage-specific differentiation29. For example, TAF3 is predominantly expressed in myocytes38; expression of TAF1, TAF3, TAF4 and TAF9 is absent in liver63; TAF8 is essential for adipogenic but not for myogenic differentiation64. Our data extend these findings by showing that TAF isoforms generated by cell-specific AS control the composition and activity of cellular TFIID complexes through dynamic regulation of their tissue- and development-specific functions. Flexible arrangement of TFIID complexes in different cellular contexts offers an efficient means to control cell fate by targeted AS of a single component of TFIID, such as established by us TAF4 subunit. Previous studies have shown that TAF4b can compensate for the loss of TAF4 at early stages of embryogenesis65, consistent with the earlier findings that both proteins retain specific and non-redundant functions66. Thereby, although TAF4 and TAF4b have at some extent overlapping roles, part of their functions is specific. As TAF4-TAFH-dependent mechanism of cell reprogramming is clearly not associated with the compensatory functions of TAF4b, this suggests that alternative splicing of TAF4 leads to TAFH-less protein isoforms that lack the ability to substitute for canonical TAF4 activity in RNA PolII-dependent transcription, but behave as dynamic fine-tuners of specific cell differentiation programs dictated by the surrounding.
In conclusion, a more detailed understanding is required to clarify the role of TAF4 in the sequence of events driving the generation of iMels and cancer progression. However, it is quite clear from our studies that targeted AS of TAF4 could be used as a new way for cell reprogramming with advantages in cell-based therapies.
Materials and Methods
Ethics Statement
Use of human biological materials for the study was pre-approved by the local ethical committee at the National Institute for Health Development, Tallinn, Estonia (Approval No 2234 from Dec 09, 2010). All experiments were performed in accordance with relevant guidelines and regulations. Written informed consent was obtained from all participants prior to the study.
Cell culture
Human primary dermal fibroblasts were freshly isolated from the facial eye-lid skin of women aged between 30 to 50 years67, expressed typical for fibroblasts specific cell-surface markers CD73, CD90, CD105, and were negative of CD45 by analysis of flow cytometry as previously described68. All fibroblasts expressed NC markers including SLUG/SNAIL, SOX9, SOX2, PAX3, PAX7 and MSX1 (Supplementary Table S3). The cells were propagated in the medium of low glucose DMEM with glutamine (DMDM-LG) (PAA Laboratories, Austria) supplemented with 1% penicillin/streptomycin (PAA Laboratories, Austria) and 10% heat-inactivated foetal bovine serum (FBS) (PAA Laboratories, Austria) in a humidified atmosphere at 37 °C and 5% CO2. Throughout the study, fibroblasts from low (<7) number passages were used in the functional assays. WM266-4 and SkMel28 human melanoma cell lines were obtained from ATCC and cultured in DMEM high glucose medium with glutamine (DMEM-HG)(PAA Laboratories, Austria) supplemented with 10% FBS and 1% penicillin/streptomycin.
Transfection
Fibroblasts and melanoma cells were treated with TAFH-specific siRNAs (5′-GGUUAUACCGAGAACUUAA-dTdT-3′ and 5′-CAGCUAAUGUGAAAGAGCU-dTdT-3′ referred here as TAF4ex5 siRNA and TAF4ex6 siRNA correspondently) as described34. All experiments were performed with both siRNAs, but in case of similar results obtained, siRNAs are designated in Figures as TAFH siRNAs. All synthetic Silencer® Pre-designed siRNAs together with Negative Control #2 siRNAs were purchased from Ambion, Invitrogen (UK). Cultured cells were transfected with 25 or 50 nM of siRNAs using Lipofectamin RNAiMAX reagent (Invitrogen, UK) according to the manufacturer’s protocol. For over-expression studies, 5 × 105 melanoma cells were transfected with 0.5 μg of full-length TAF4 cDNA in pcDREAM2.1 (GenScript, Piscataway, NJ, USA) using Lipofectamine 3000 (Life Technologies). pmaxGFP® vector was used for transfection control. Cells were harvested at indicated time points and used in functional assays.
RNA isolation, RT-PCR and Real-Time PCR
Human primary melanoma and normal tissue RNA panels were purchased from BioChain Institute (USA). For cultured cells, total RNA was purified using TRIzol® reagent (Invitrogen, Life Technologies, UK) following manufacturer’s recommendations. cDNA was synthesised from DNase-treated (Ambion, UK) RNA using Superscript III RT (Invitrogen, UK) and a mix of oligodT and random hexamers, according to manufacturer’s recommendations. RT-PCR was carried out using HOT FIREpol® Master Mix (Solis Biodyne, Estonia). RT-qPCR was performed in triplicates using LightCycler® SYBR Green I Master Mix (Roche Applied Science) and the LightCycler® 480 Real-Time PCR System (Roche Applied Science). The fold of change was calculated relatively to control siRNA treatments after normalization to GAPDH expression, using 2−ΔΔCt method, where ΔCt is (gene of interest Ct) − (GAPDH Ct), and ΔΔCt is (ΔCt treated)—(ΔCt control). Primer sequences are listed in Supplementary Table S4.
Immunofluorescence and immunoblot analyses
TAFH siRNA-treated and untreated human dermal primary fibroblasts and melanoma cells were grown on 22-mm2 glass slides to about 70% confluence. Anti-MITF (ab59232, Abcam) as primary, and Alexa Fluor 546 (Molecular Probes, Invitrogen, Eugene, OR, USA) as secondary antibodies were used for immunofluorescence analysis. Images were visualised using Nikon Eclipse 80i fluorescence microscope (Nikon Instruments Inc., USA).
For Western blot analysis, treated and untreated cells were collected by trypsin-EDTA (PAA Laboratories, Austria). Cell fractionation was carried out using NE-PER Nuclear and Cytoplasm Extraction Reagents (Thermo Scientific, Pierce, Rockford, IL, USA). Total protein concentration of nuclear and whole cell extracts was measured using BCA Protein Assay kit (Thermo Scientific, Pierce, Rockford, IL, USA). The following primary antibodies were used: anti-TAF4 (BD Biosciences, 612054), anti-CDH1 (Abcam, ab53033), anti-CDH2 (Abcam, ab12221), anti-phospho-TP53Ser15 (Cell Signaling, 9284), and anti-GAPDH (Sigma, G8795). Secondary HRP-conjugated antibodies were purchased from Abcam. Proteins were visualised using SuperSignal West Dura Chemiluminescent Substrate (Thermo Scientific, Pierce, Rockford, IL, USA).
Melanin content assay
Melanin content was determined according to the modified method of Hosoi et al.69. Human dermal fibroblasts were cultured and subjected to mock or siRNA treatments. Melanoma SkMel28 cells were treated with 1 mM dibutyryl-cAMP for 6 days as a control for melanin synthesis. After 24 h, cells were washed by PBS, harvested by trypsinisation and counted. The cell pellet was solubilised in 200 μl of 0.2 M NaOH containing 10% DMSO per 1 × 106 cells at 80 °C for 1 h. The absorbance of each well was measured at 405 nm using SPECTRAmax 340 PC spectrophotometer (Molecular Devises, USA) and the values were normalised to the total amount of protein.
Proliferation assay
WM266-4 and SkMel28 melanoma cells were grown in 96-well flat bottom tissue culture plates up to 80% confluence and treated with control or TAFH siRNAs. WST-1 analysis (Roche Applied Science) was performed according to manufacturer’s instructions. Absorption at 450 nm was measured using SPECTRAmax 340 PC Microplate Reader (Molecular Devises LLC, USA). The data were analysed using Softmax Pro 3.12 software and normalised to the values at the day of plating. Cell viability was evaluated every 24 h, up to 5 days post-treatment.
Differentiation
For melanoma differentiation studies, 24 h after siRNA transfection, cells were cultured in conditions supporting adipogenic, chondrogenic or neuronal differentiation as previously described34,35. The extent of chondrogenic, adipogenic and neuronal differentiation was analysed by RT-qPCR of lineage-specific marker genes. To confirm effective chondrogenic differentiation, immunofluorescence detection by chondrocyte-specific SOX9 (AB5535, Millipore) was performed 7 days after culturing of siRNA-treated melanoma cells in chondrogenic differentiation-supporting conditions. Alcian blue staining (Sigma) was performed 2 weeks after differentiation stimulation. Cells were fixed in 4% PFA, washed with distilled water, stained with Alcian blue and analysed under 10× magnification for glycosaminoglycan expression.
Cell migration and invasion assays
Cell migration and invasion were analysed by in vitro migration assays, using 8 μm-pore polycarbonate transwell filters (QCMTM Colorimetric Cell Migration Assay, Millipore, USA). For melanoma invasion assay, transwell filters were coated with extracellular matrix components (ECM) extracted from human fibroblasts70. For that, fibroblasts were plated onto transwell filters, cultivated until the confluence, and ECM were extracted by using cell lysis buffer (10 mM TRIS-HCl, 1 mM EDTA, at pH7.4) for 16 h. For migration assay, transwell filters were left uncoated. For migration/invasion studies, fibroblasts or melanoma cells were treated with TAFH siRNAs for 72 h and thereafter starved in serum-free medium for 16 h. Cells were seeded at density of 1 × 105 cells per insert in DMEM-LG medium without the addition of FBS, whereas the bottom chamber was filled with DMEM-LG supplemented with 10% FBS. The total number of cells that migrated to the lower chamber was quantified after 24 h of incubation of cells at 37 °C with 5% CO2. Migrated cells on the lower surface of the filter membrane were fixed and stained by cell stain solution with 0.1% crystal violet before counting. The number of migrated and invaded cells per well was counted in five randomly selected microscope fields in three independent experiments using Nikon Eclipse 80i fluorescence microscope (Nikon Instruments Inc., USA).
Statistical analysis
At least three independent replicates were performed for each experiment. Statistical analysis was performed using an unpaired Student’s t-test with a 2-tailed p value. Differences were considered significant when the p < 0.05.
Additional Information
How to cite this article: Kazantseva, J. et al. Targeted alternative splicing of TAF4: a new strategy for cell reprogramming. Sci. Rep. 6, 30852; doi: 10.1038/srep30852 (2016).
Supplementary Material
Acknowledgments
We thank Kadri Orro and Reet Rumvolt for help with dermal fibroblast cultures, and Maila Rähn for excellent technical support. This study was supported by Protobios’s grants from Enterprise Estonia and research funding from the Estonian Ministry of Education and Research. KP was partially supported by the institutional research grant IUT19-18 from the Estonian Research Council. JK was partially supported by the grant from Enterprise Estonia (EU42735).
Footnotes
Author Contributions J.K. performed all experiments; J.K., H.S., T.N. and K.P. analysed and interpreted the data; J.K. and K.P. wrote the paper. All authors reviewed the manuscript. The authors declare no conflict of interest.
References
- Aaronson Y. & Meshorer E. Stem cells: Regulation by alternative splicing. Nature 498, 176–177 (2013). [DOI] [PubMed] [Google Scholar]
- Liu H., He L. & Tang L. Alternative splicing regulation and cell lineage differentiation. Curr Stem Cell Res Ther 7, 400–406 (2012). [DOI] [PubMed] [Google Scholar]
- Chen Y., Zhang L. & Jones K. A. SKIP counteracts p53-mediated apoptosis via selective regulation of p21Cip1 mRNA splicing. Genes Dev 25, 701–716 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm E., Pellay F.-X., Benecke A. & Bell B. TAF6delta controls apoptosis and gene expression in the absence of p53. PLoS One 3, e2721 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- David C. J. & Manley J. L. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 24, 2343–2364 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germann S., Gratadou L., Dutertre M. & Auboeuf D. Splicing programs and cancer. J Nucleic Acids 2012, 269570 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama K. et al. Identification of a novel protein isoform derived from cancer-related splicing variants using combined analysis of transcriptome and proteome. Proteomics 11, 2275–2282 (2011). [DOI] [PubMed] [Google Scholar]
- Cauffman G., Liebaers I., Van Steirteghem A. & Van de Velde H. POU5F1 isoforms show different expression patterns in human embryonic stem cells and preimplantation embryos. Stem Cells 24, 2685–2691 (2006). [DOI] [PubMed] [Google Scholar]
- Gabut M. et al. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 147, 132–146 (2011). [DOI] [PubMed] [Google Scholar]
- Han H. et al. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature 498, 241–245 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik N. & Rao M. S. A review of the methods for human iPSC derivation. Methods Mol Biol 997, 23–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R. L., Weintraub H. & Lassar A. B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000 (1987). [DOI] [PubMed] [Google Scholar]
- Vierbuchen T. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P. et al. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 475, 386–389 (2011). [DOI] [PubMed] [Google Scholar]
- Ieda M. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R. et al. Direct conversion of mouse and human fibroblasts to functional melanocytes by defined factors. Nat Commun 5, 5807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf T. Historical origins of transdifferentiation and reprogramming. Cell Stem Cell 9, 504–516 (2011). [DOI] [PubMed] [Google Scholar]
- Goding C. R., Pei D. & Lu X. Cancer: pathological nuclear reprogramming? Nat Rev Cancer 14, 568–573 (2014). [DOI] [PubMed] [Google Scholar]
- Takahashi K. & Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006). [DOI] [PubMed] [Google Scholar]
- Yu Y. et al. Stimulation of somatic cell reprogramming by ERas-Akt-FoxO1 signaling axis. Stem Cells 32, 349–363 (2014). [DOI] [PubMed] [Google Scholar]
- Liao J. et al. Inhibition of PTEN tumor suppressor promotes the generation of induced pluripotent stem cells. Mol Ther 21, 1242–1250 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi K. et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 156, 663–677 (2014). [DOI] [PubMed] [Google Scholar]
- Friedmann-Morvinski D. & Verma I. M. Dedifferentiation and reprogramming: origins of cancer stem cells . EMBO Rep 15, 244–253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Mejia V., Fraga M. F. & Menendez P. iPSCs from cancer cells: challenges and opportunities. Trends Mol Med 18, 245–247 (2012). [DOI] [PubMed] [Google Scholar]
- D’Alessio J. A., Wright K. J. & Tjian R. Shifting players and paradigms in cell-specific transcription. Mol Cell 36, 924–931 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley S. K. & Roeder R. G. Biochemistry and structural biology of transcription factor IID (TFIID). Annu Rev Biochem 65, 769–799 (1996). [DOI] [PubMed] [Google Scholar]
- Tora L. A unified nomenclature for TATA box binding protein (TBP)-associated factors (TAFs) involved in RNA polymerase II transcription. Genes Dev 16, 673–675 (2002). [DOI] [PubMed] [Google Scholar]
- Bieniossek C. et al. The architecture of human general transcription factor TFIID core complex. Nature 493, 699–702 (2013). [DOI] [PubMed] [Google Scholar]
- Maston G. A. et al. Non-canonical TAF complexes regulate active promoters in human embryonic stem cells. Elife 1, e00068 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich J. A. & Tjian R. Unexpected roles for core promoter recognition factors in cell-type-specific transcription and gene regulation. Nat Rev Genet 11, 549–558 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller F., Zaucker A. & Tora L. Developmental regulation of transcription initiation: more than just changing the actors. Curr Opin Genet Dev 20, 533–540 (2010). [DOI] [PubMed] [Google Scholar]
- Kazantseva J. & Palm K. Diversity in TAF proteomics: consequences for cellular differentiation and migration. Int J Mol Sci 15, 16680–16697 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunkhorst A. et al. Novel isoforms of the TFIID subunit TAF4 modulate nuclear receptor-mediated transcriptional activity. Biochem Biophys Res Commun 325, 574–579 (2004). [DOI] [PubMed] [Google Scholar]
- Kazantseva J. et al. Alternative Splicing Targeting the hTAF4-TAFH Domain of TAF4 Represses Proliferation and Accelerates Chondrogenic Differentiation of Human Mesenchymal Stem Cells. PLoS One 8, e74799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantseva J., Tints K., Neuman T. & Palm K. TAF4 Controls Differentiation of Human Neural Progenitor Cells Through hTAF4-TAFH Activity. J Mol Neurosci 55, 160–166 (2015). [DOI] [PubMed] [Google Scholar]
- Brunkhorst A. et al. A specific role for the TFIID subunit TAF4 and RanBPM in neural progenitor differentiation. Mol Cell Neurosci 29, 250–258 (2005). [DOI] [PubMed] [Google Scholar]
- Harris T. M. & Childs G. Global gene expression patterns during differentiation of F9 embryonal carcinoma cells into parietal endoderm. Funct Integr Genomics 2, 105–119 (2002). [DOI] [PubMed] [Google Scholar]
- Deato M. D. E. & Tjian R. Switching of the core transcription machinery during myogenesis. Genes Dev 21, 2137–2149 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadloun A. et al. Retinoic acid induces TGFbeta-dependent autocrine fibroblast growth. Oncogene 27, 477–489 (2008). [DOI] [PubMed] [Google Scholar]
- Fadloun A. et al. The TFIID subunit TAF4 regulates keratinocyte proliferation and has cell-autonomous and non-cell-autonomous tumour suppressor activity in mouse epidermis. Development 134, 2947–2958 (2007). [DOI] [PubMed] [Google Scholar]
- Mengus G. et al. TAF4 inactivation in embryonic fibroblasts activates TGF beta signalling and autocrine growth. EMBO J 24, 2753–2767 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 (2008). [DOI] [PubMed] [Google Scholar]
- Pijnappel W. W. M. P. et al. A central role for TFIID in the pluripotent transcription circuitry. Nature 495, 516–519 (2013). [DOI] [PubMed] [Google Scholar]
- Ackley C. D. et al. Primary chondroid melanoma. J Cutan Pathol 28, 482–485 (2001). [DOI] [PubMed] [Google Scholar]
- McKay K. M., Deavers M. T. & Prieto V. G. Chondroid melanoma metastatic to lung and skin showing SOX-9 expression: a potential diagnostic pitfall. Int J Surg Pathol 20, 169–172 (2012). [DOI] [PubMed] [Google Scholar]
- Bell B., Scheer E. & Tora L. Identification of hTAF(II).80 delta links apoptotic signaling pathways to transcription factor TFIID function. Mol Cell 8, 591–600 (2001). [DOI] [PubMed] [Google Scholar]
- Friedman M. J. et al. Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci 10, 1519–1528 (2007). [DOI] [PubMed] [Google Scholar]
- Scott G. Selective proliferation of normal human melanocytes in vitro in the presence of phorbol ester and cholera toxin by Eisinger and Marko. Exp Dermatol 23, 18–19 (2014). [DOI] [PubMed] [Google Scholar]
- Cui R. et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell 128, 853–864 (2007). [DOI] [PubMed] [Google Scholar]
- Murase D. et al. The essential role of p53 in hyperpigmentation of the skin via regulation of paracrine melanogenic cytokine receptor signaling. J Biol Chem 284, 4343–4353 (2009). [DOI] [PubMed] [Google Scholar]
- Perletti L., Kopf E., Carré L. & Davidson I. Coordinate regulation of RARgamma2, TBP, and TAFII135 by targeted proteolysis during retinoic acid-induced differentiation of F9 embryonal carcinoma cells. BMC Mol Biol 2, 4 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E. et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 18, 510–523 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M., Morita R. & Takehara K. Clonal heterogeneity in sporadic melanomas as revealed by loss-of-heterozygosity analysis. Int J Cancer 85, 492–497 (2000). [PubMed] [Google Scholar]
- Dupin E., Real C., Glavieux-Pardanaud C., Vaigot P. & Le Douarin N. M. Reversal of developmental restrictions in neural crest lineages: transition from Schwann cells to glial-melanocytic precursors in vitro. Proc Natl Acad Sci USA 100, 5229–5233 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin N. M. & Dupin E. Multipotentiality of the neural crest. Curr Opin Genet Dev 13, 529–536 (2003). [DOI] [PubMed] [Google Scholar]
- Bailey C. M., Morrison J. A. & Kulesa P. M. Melanoma revives an embryonic migration program to promote plasticity and invasion. Pigment Cell Melanoma Res 25, 573–583 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatton T. & Frank M. H. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res 21, 39–55 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakhova O. & Sommer L. Testing the cancer stem cell hypothesis in melanoma: the clinics will tell. Cancer Lett 338, 74–81 (2013). [DOI] [PubMed] [Google Scholar]
- Thiery J. P. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2, 442–454 (2002). [DOI] [PubMed] [Google Scholar]
- Chia N.-Y. et al. A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 468, 316–320 (2010). [DOI] [PubMed] [Google Scholar]
- Loh Y.-H. et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38, 431–440 (2006). [DOI] [PubMed] [Google Scholar]
- Liu Z., Scannell D. R., Eisen M. B. & Tjian R. Control of embryonic stem cell lineage commitment by core promoter factor, TAF3. Cell 146, 720–731 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio J. A., Ng R., Willenbring H. & Tjian R. Core promoter recognition complex changes accompany liver development. Proc Natl Acad Sci USA 108, 3906–3911 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guermah M., Ge K., Chiang C. M. & Roeder R. G. The TBN protein, which is essential for early embryonic mouse development, is an inducible TAFII implicated in adipogenesis. Mol Cell 12, 991–1001 (2003). [DOI] [PubMed] [Google Scholar]
- Langer D. et al. Essential role of the TFIID subunit TAF4 in murine embryogenesis and embryonic stem cell differentiation. Nat Commun 7, 11063 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahat A. et al. TAF4b and TAF4 differentially regulate mouse embryonic stem cells maintenance and proliferation. Genes Cells 18, 225–237 (2013). [DOI] [PubMed] [Google Scholar]
- Takashima A. Establishment of fibroblast cultures. Curr Protoc Cell Biol Chapter 2, Unit 2.1 (2001). [DOI] [PubMed] [Google Scholar]
- Jääger K. & Neuman T. Human dermal fibroblasts exhibit delayed adipogenic differentiation compared with mesenchymal stem cells. Stem Cells Dev 20, 1327–1336 (2011). [DOI] [PubMed] [Google Scholar]
- Hosoi J., Abe E., Suda T. & Kuroki T. Regulation of melanin synthesis of B16 mouse melanoma cells by 1 alpha, 25-dihydroxyvitamin D3 and retinoic acid. Cancer Res 45, 1474–1478 (1985). [PubMed] [Google Scholar]
- Erkell L. J. & Schirrmacher V. Quantitative in vitro assay for tumor cell invasion through extracellular matrix or into protein gels. Cancer Res 48, 6933–6937 (1988). [PubMed] [Google Scholar]
- Ribeiro J. R., Lovasco L. A., Vanderhyden B. C. & Freiman R. N. Targeting TBP-Associated Factors in Ovarian Cancer. Front Oncol 4, 45 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.