

Abstract
N1-methyladenosine (m1A) is a prominent RNA modification involved in many biological processes. Accurate identification of m1A site is invaluable for better understanding the biological functions of m1A. However, limitations in experimental methods preclude the progress towards the identification of m1A site. As an excellent complement of experimental methods, a support vector machine based-method called RAMPred is proposed to identify m1A sites in H. sapiens, M. musculus and S. cerevisiae genomes for the first time. In this method, RNA sequences are encoded by using nucleotide chemical property and nucleotide compositions. RAMPred achieves promising performances in jackknife tests, cross cell line tests and cross species tests, indicating that RAMPred holds very high potential to become a useful tool for identifying m1A sites. For the convenience of experimental scientists, a web-server based on the proposed model was constructed and could be freely accessible at http://lin.uestc.edu.cn/server/RAMPred.
The N1-methyladenosine (m1A) is a prominent post-transcriptional modification found in RNA, which is catalyzed by methyltransferase1. Besides adding a methyl group to the nitrogen at the 1st position of the adenosine base, m1A also endows the modified adenosine with a positive charge2, as shown in Fig. 1. It has been found that m1A has major influences on the structure and function of tRNA and rRNA3,4,5. For example, m1A in tRNA can respond to environmental stress6,7, and m1A in rRNA can affect ribosome biogenesis8 and mediate antibiotic resistance in bacteria9. Although the functions of m1A in tRNA and rRNA were well studied, similar researches in mRNA were precluded due to the lack of effective methods for detecting m1A in mRNA2,10. Therefore, the knowledge about the positions of m1A site is important for understanding mechanisms and functions of this post-transcriptional modification.
Figure 1. An illustration to show the N1-methylation and demethylation of adenosine.

The formation of m1A is catalyzed by methyltransferases and its reversible modification is catalyzed by demethyltransferases. Besides adding a methyl group (-CH3) to the nitrogen at the 1st position of the adenosine base, N1-methylation also endows the modified adenosine with a positive charge as indicated in red in this figure.
With the development of high-throughput experimental techniques, such as MeRIP-seq2 and m1A-ID-seq10, high-resolution m1A maps are available for H. sapiens, M. musculus and S. cerevisiae transcriptomes2. These experimental results revealed that m1A sites are enriched in 5′-untranslated region and coding sequence of mRNA transcripts2,10, and also demonstrated that m1A is dynamic in response to physiological conditions and correlates positively with protein production2.
Experimental methods did play a role in promoting the research progress on identifying m1A sites. However, their resolutions are not fully satisfactory, i.e. they cannot pinpoint which adenosine residue is actually modified10. Therefore, it is necessary to develop new methods for studying the distribution of m1A site. As excellent complements to experimental techniques, computational methods will speed up genome-wide m1A detection.
The high-resolution experimental data provided unprecedented opportunities and made it feasible to develop computational methods for accurately predicting m1A sites. However, to the best of our knowledge, there is no computational tool available for the identification of m1A. Hence, in the present study, we propose a support vector machine based-method to identify the m1A sites in the H. sapiens, M. musculus and S. cerevisiae genomes. By using the nucleotide chemical property and nucleotide composition, the sequence-order effects and nucleotide chemical properties are integrated together in the proposed model. It is encouraging that the proposed methods obtained promising performances in jackknife tests, cross cell line tests and cross species tests. For the convenience of scientific community, a web-server for the proposed model is provided at http://lin.uestc.edu.cn/server/RAMPred.
Result and Discussion
m1A sites identification
In statistical prediction, three cross-validation methods, i.e., independent dataset test, sub-sampling (or n-fold cross-validation) test, and jackknife test, are often used to evaluate the anticipated success rate of a predictor. Among the three methods, the jackknife test is deemed the least arbitrary and most objective11. Therefore, the jackknife test has been increasingly adopted by researchers to examine the quality of various computational models12,13,14,15,16. Thus, the jackknife test was used to examine the performance of the proposed model. In the jackknife test, each sample in the training dataset is in turn singled out as an independent test sample and all the properties are calculated without including the one being identified.
By encoding RNA sequence using nucleotide chemical property and nucleotide composition, each 41-bp long sequence in the dataset was transferred to a 164 (4 × 41)-dimensional vector (see Materials and Methods) and was used as the input of SVM. The model thus obtained is called RAMPred (RNA N1-adenosine methylation predictor). The jackknife test results of RAMPred for identifying m1A sites in H. sapiens, M. musculus and S. cerevisiae genomes were enumerated in the first four columns of Table 1. In addition, in order to objectively evaluate the performance of RAMPred in identifying m1A sites, the receiver operating characteristic curves and precision recall curves for H. sapiens, M. musculus and S. cerevisiae were also plotted and were shown in Fig. 2. The AUROC and AUPRC values examining the performance of RAMPred were calculated and provided in the last two columns of Table 1. As we can see from Table 1 and Fig. 2, the prediction accuracies of RAMPred were considerably high for identifying m1A sites in all the three species.
Table 1. Predictive results of the method for identifying m1A sites in different species.
| Species | Sn (%) | Sp (%) | Acc (%) | MCC | AUROC | AUPRC |
|---|---|---|---|---|---|---|
| Human | 98.38 | 99.89 | 99.13 | 0.98 | 0.98 | 0.98 |
| Mouse | 97.46 | 100 | 98.73 | 0.97 | 0.99 | 0.99 |
| S. cerevisiae | 95.65 | 100 | 97.83 | 0.96 | 0.98 | 0.98 |
Figure 2. A graphical illustration to show the performance of RAMPred for identifying m1A sites in H. sapiens (red line), M. musculus (blue line) and S. cerevisiae (green line) genomes.

The performances are illustrated by means of the ROC curves (A) and precision-recall curves (B).
The chemical properties or nucleotide composition may have different roles in the prediction of m1A site. In order to investigate the contribution of each feature for m1A site identification, we built a series of models and validated them on the benchmark dataset. Their predictive accuracies obtained from jackknife test for identifying m1A sites in H. sapiens, M. musculus and S. cerevisiae genomes were shown in Fig. 3. It was observed that, among the four kind of features (namely ring structure, hydrogen bond, chemical functionality and nucleotide composition), the model based on the ring structure yields the highest accuracy. However, it’s lower than that obtained by using their combinations (Fig. 3). These results indicate that ring structure has the largest contribution for m1A site identification in the current method, and the other three features (hydrogen bond, chemical functionality and nucleotide composition) play complementary roles for the prediction.
Figure 3. The predictive accuracies obtained from the jackknife test for identifying m1A sites in H. sapiens, M. musculus and S. cerevisiae genome by using different kinds of parameters.
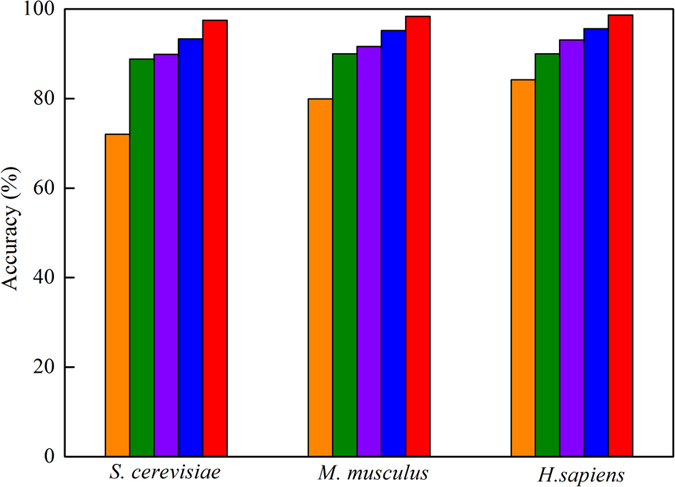
Orange, green, purple, blue and red histograms stand for the accuracies obtained by the model trained by using nucleotide composition, chemical functionality, hydrogen bond, ring structure and the combination of all the four kinds of features, respectively.
In addition, to ensure that the predictive accuracy of RAMPred is not sensitive to the selection of negative data, we repeated the random sampling procedure ten times. In each time, a prediction model was built based on the positive dataset and the generated negative dataset. For saving computational time, the four metrics as defined in Eq. 4 for the other nine models in identifying m1A sites via the 10-fold cross validation test were reported in Supplementary Tables S1–S3 for H. sapiens, M. musculus and S. cerevisiae, respectively. We found that the predictive accuracy is not affected by the selection of negative data, demonstrating the reliability and robustness of the model proposed in this study.
Comparison with Other classifiers
To the best of our knowledge, there is no published computational method for identifying m1A sites. We could not provide the comparison analysis with existing results to confirm that RAMPred is superior to other methods. However, to further testify its superiority, the predictive results of RAMPred were compared with that of other commonly used classifiers, i.e., J48 Tree, Random Forest, Naïve Bayes and BayesNet as implemented in WEKA17. For saving computational time, the 10-fold cross validation test results of different classifiers for identifying m1A in the benchmark dataset were reported in Supplementary Table S4. It is shown that the four metrics as defined in Eq. 4 for RAMPred are all higher than those of J48 Tree, Random Forest, Naïve Bayes and BayesNet.
Recently, Chen et al. proposed the iRNA-Methyl tool to identify post-transcriptional RNA modifications18. In iRNA-Methyl, RNA sequence was formulated with the “pseudo dinucleotide composition” (PseDNC)19,20,21 into which three RNA physical-chemical properties (i.e. enthalpy, entropy and free energy) were incorporated18. To demonstrate the effectiveness of nucleotide chemical properties and nucleotide composition for m1A site identification, the PseDNC-based SVM model was also developed. The 10-fold cross validation test results of the PseDNC-based SVM model in identifying m1A site by using the same benchmark dataset are given in Supplementary Table S5, from which we can see that the performance of RAMPred is superior to the PseDNC-based SVM model for identifying m1A site. All these results indicate that RAMPred can be effectively used to identify m1A sites.
Cross cell line and cross species validation
m1A is a dynamic modification in response to certain stress conditions and its level varies among different tissues2. Since the training dataset of RAMPred were collected from different species and cell lines (see Materials and Methods), it is interesting to see to what extent a model trained by using the data from one tissue or specie recognizes the m1A sites from other tissues or species. To demonstrate this point, we trained cell line-specific and species-specific models based on the m1A site data from different cell lines and species, and then validated them on the independent datasets from other cell lines or species. The cross cell line and cross species independent test results are given in Fig. 4.
Figure 4. The heat map showing the cross cell line and cross species prediction accuracies.

The accuracies were evaluated through the independent tests. More specifically, once a cell line or species model was established on its own training dataset, it was tested on the data from the same cell line (or species) as well as those from the other cell lines (or species). In cross species analysis, we considered three kinds of species (i.e., H. sapiens, M. musculus and S. cerevisiae). In cross cell line analysis, we considered three kinds of human cell lines (i.e., HeLa, HEK293 and HepG2) and two kinds of mouse cell lines (i.e., Liver and MEFs).
It was found that the mammalian models trained using data from H. sapiens and M. musculus genomes can accurately identify each other’s m1A sites and the performances are pretty good. Although the performances of the mammalian models for identifying m1A sites in yeast genome are acceptable, they are lower than that obtained by the model trained using data from yeast own data. This result indicates that the construction of species-specific predictor for identifying m1A sites is necessary for yeast. It was also found that the cross cell line prediction performances are satisfactory and are equivalent to the intra-cell line performances in the three human cell lines (i.e., HeLa, HEK293 and HepG2) and two mouse cell lines (i.e., Liver and MEFs), indicating that there is no need to construct cell line-specific models to identify m1A sites in mammalian genomes.
Web-Server and User Guide
To enable applications of the proposed method and for the conveniences of community, a freely accessible online web-server called RAMPred was established. To maximize the user’s convenience, a step-by-step guide of the web-server on how to use RAMPred is given as following.
Firstly, browse the web server at http://lin.uestc.edu.cn/server/RAMPred and you will see the top page of RAMPred on your computer screen, as shown Fig. 5. Click on the Read Me button to see a brief introduction about the predictor and the caveat when using it. Click on the Data button to download the benchmark datasets used to train RAMPred. Click on the Citation button to find the relevant papers that document the detailed development and algorithm of RAMPred.
Figure 5. A semi-screenshot for the top-page of the RAMPred web-server at http://lin.uestc.edu.cn/server/RAMPred.
Secondly, select the organism or species by checking on the corresponding open circle. To get the anticipated prediction accuracy, the species button must be consistent with the source of query sequences: if the query sequences are from H. sapiens, check on the ‘H. sapiens’ button; if from M. musculus, check on the ‘M. musculus’ button; if from S. cerevisiae, check on the ‘S. cerevisiae’ button. Either type or copy/paste the query RNA sequences into the input box at the center of Fig. 5. The input sequence should be in FASTA format. For the examples of RNA sequences in FASTA format, click the Example button right above the input box. The predicted results will be shown on the computer screen by clicking on the Submit button.
Conclusions
By using nucleotide chemical property and nucleotide composition, for the first time, we developed a support vector machine-based model to identify m1A sites in H. sapiens, M. musculus and S. cerevisiae genomes. The jackknife test results on the rigorous benchmark datasets demonstrate that the proposed method RAMPred is very promising for identifying m1A sites in the three eukaryotic genomes.
To identify the key features for m1A site identification, we compared the predictive results obtained by using different kind of parameters and found that the ring structure has the largest contribution for m1A site identification. This result holds for all the three genomes and is consistent with the following fact. N1-methylation on RNA adenosine occurs at the Watson-Crick interface and is catalyzed by methyl-transferases that need to recognize and bind with specific genomic regions22. Therefore, nucleotide ring structure could facilitate the π-cation/π-π/van der Waals contact between methyl-transferases and RNA sequence.
In order to rigorously evaluate its performance, we also tested the proposed method by performing cross cell line and cross species validations. It is encouraging to see that the cross cell line performances are quite good, indicating that our method is stable for identifying m1A site in mammalian genomes. We also noticed that the performances of mammalian based models for identifying yeast m1A sites are lower than that of the yeast specific one and vice versa.
As an epigenetic modification, RNA methylation is a complicate progress. Besides sequence context and nucleotide chemical properties, other factors may be also helpful for m1A site identification. For example, it has been demonstrated that m1A correlates with elevated translation, and enriched in 5′-untranslated region and coding sequence, and also overrepresented in the start codon upstream of the first splice site2,10. In addition, high-resolution experimental data with quantitative information about m1A modification are also highly desirable, which would aid the representation of the sequence context surrounding the m1A sites. For better understanding of the biological function of N1-methylation on RNA adenosine, we will combine all these factors and develop new models to improve the predictor’s performance for m1A sites identification in the future work.
Materials and Methods
Datasets
Based on MeRIP-seq technique, Dominissini and his colleagues obtained the m1A peaks in H. sapiens, M. musculus and S. cerevisiae genomes2. By mapping these peaks to H. sapiens (hg.19), M. musculus (mm10) and S. cerevisiae genome, respectively, we obtained m1A site containing sequences for these three genomes. It was observed via preliminary trials that when the length of the sequences in the benchmark dataset is 41 bp with the m1A in the center, the corresponding predictive results were most promising. Accordingly, we focus on RNA sequence with 41 nucleotides in the current study.
To overcome redundancy and reduce the homology bias, sequences with more than 80% sequence similarity were removed by using the CD-HIT program23. After such a screening procedure, we obtained 6,366, 1,064 and 483 m1A site containing sequences and deemed them as the positive samples of H. sapiens, M. musculus and S. cerevisiae, respectively. If the sequence identity is set to a lower percentage, such as 40%, the result will be more objective and reliable. However, in this study we did not use such a stringent criterion because the currently available data do not allow this. Otherwise, the number of samples will be too few to have statistical significance.
The negative samples in each species were obtained by choosing the 41-nt long sequences satisfying the rule that the adenosine in the center was not detected by the MeRIP-seq technique. By doing so, we could obtain a great number of negative samples in each species. Therefore, the number of negative samples will be dramatically larger than those of positive samples. In machine-learning problems, imbalanced datasets can significantly affect the performance evaluation of learning methods. To balance out the numbers between positive and negative samples in model training, we randomly picked out 6,366, 1,064 and 483 sequences to form the negative samples for H. sapiens, M. musculus and S. cerevisiae, respectively. To demonstrate the robustness of the proposed model, we repeated the random sampling procedure ten times and obtained ten random samples of negative datasets for downstream training and prediction for each species.
According to Dominissini and his colleagues’ work2, the m1A site containing sequences in H. sapiens were from three cell lines, namely, HeLa (cervical adenocarcinoma), HepG2 (hepatocellular carcinoma) and HEK293 (embryonic kidney) cell lines, and those sequences in M. musculus were from two cell lines, namely, primary mouse embryonic fibroblasts (MEFs) and liver cell lines. To further validate the performance of the proposed method, we also built cell line specific datasets for H. sapiens and M. musculus, respectively. The numbers of positive and negative samples of the cell line specific datasets were shown in Fig. 6. All the data are available at http://lin.uestc.edu.cn/server/RAM/data.
Figure 6. A graph to show the number of positive and negative samples in H. sapiens (top panel) and M. musculus (down panel) different cell lines.

The left panel is the number of positive samples in each cell line, which is enumerated in brackets of the pie chart. The right panel is the number of negative samples in each cell line, which is enumerated in brackets of the pie chart.
Representation of RNA sequences
Stimulated by its success in identifying post-transcriptional RNA modifications24,25, nucleotide chemical property and nucleotide composition were used to represent RNA sequences for identifying m1A sites in the present work. Below is the brief elaboration on how to encode RNA sequences using nucleotide chemical property and nucleotide composition.
RNA is transcribed with four nucleotides, namely, adenine (A), guanine (G), cytosine (C) and uracil (U). These four bases have different chemical properties. In terms of ring structures, adenine and guanine are purines that have two rings, while cytosine and uracil are pyrimidines that have one ring. When forming secondary structures, guanine and cytosine form strong hydrogen bonds, whereas adenine and uracil form weak hydrogen bonds. In terms of chemical functionality, adenine and cytosine can be classified into the amino group, while guanine and uracil into the keto group.
In order to include these chemical properties in RNA encoding, three coordinates (x, y, z) were used to represent the chemical properties of the four nucleotides and were assigned 1 or 0 values24,26. The x coordinate stands for the ring structure, y for the hydrogen bond, and z for the chemical functionality. Hence, each nucleotide in RNA sequence can be encoded by (xi, y i, zi), where24,25
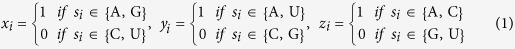 |
Thus, nucleotides A, C, G and U can be represented by the coordinates (1, 1, 1), (0, 0, 1), (1, 0, 0) and (0, 1, 0), respectively.
For the purpose of including nucleotide composition surrounding the m1A sites as well2, the density di of any nucleotide nj at position i in a RNA sequence was defined by the following formula.
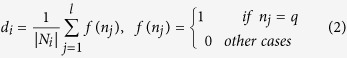 |
where l is the sequence length, |Ni| is the length of the i-th prefix string {n1, n2, …, ni} in the sequence, q ∈ {A, C, G, U}.
Therefore, by integrating nucleotide chemical properties and nucleotide composition, the sequence with a length of l will be encoded by a (4 × l)-dimensional vector. An example of encoding RNA sequence using nucleotide chemical properties and nucleotide composition is shown in Fig. 7.
Figure 7. Scheme of encoding RNA sequence using nucleotide chemical properties and nucleotide composition.

Support Vector Machine
Support vector machine (SVM) is a powerful and popular method for pattern recognition and is widely used in the realm of bioinformatics18,27,28,29. The basic idea of SVM is to transform the input data into a high dimensional feature space and then determine the optimal separating hyperplane. In the current study, the LibSVM package 3.18 ( http://www.csie.ntu.edu.tw/~cjlin/libsvm/) was used to implement SVM. Due to its effectiveness and speed in training process, the radial basis kernel function (RBF) was used to obtain the classification hyperplane in the current study. In the SVM operation engine, the grid search method was applied to optimize the regularization parameter C and kernel parameter γ using a grid search approach as defined by
 |
Performance evaluation
The performance of the proposed method was evaluated by using the following four metrics, namely sensitivity (Sn), specificity (Sp), Accuracy (Acc) and the Mathew’s correlation coefficient (MCC), which are expressed as
 |
where TP, TN, FP, and FN represent true positive, true negative, false positive, and false negative, respectively.
The ROC (receiver operating characteristic) curve30 was also used to evaluate the performance of the current method, which plots the true positive rate (sensitivity) against the false positive rate (1-specificity). A best possible prediction method would yield a point with the coordinate (0, 1) representing 100% sensitivity and 0 false positive rate or 100% specificity. Therefore, the (0, 1) point is also called a perfect classification. A completely random guess would give a point along a diagonal from the point (0, 0) to (1, 1). The area under the ROC curve, also called AUROC, is often used to indicate the performance quality of a binary classifier: the value 0.5 of AUROC is equivalent to random prediction while 1 of AUROC represents a perfect one. To examine the performance of the proposed predictor when restricting low false positive rates, the precision-recall curve was also plotted, which plots precision (the fraction of TP in all predicted positives) against recall (sensitivity). The area under the precision-recall curve (AUPRC) was also used to examine the performance of the proposed method when restricting low false positive rates.
Additional Information
How to cite this article: Chen, W. et al. RAMPred: identifying the N1-methyladenosine sites in eukaryotic transcriptomes. Sci. Rep. 6, 31080; doi: 10.1038/srep31080 (2016).
Supplementary Material
Acknowledgments
This work was supported by Program for the Top Young Innovative Talents of Higher Learning Institutions of Hebei Province (No. BJ2014028), the Outstanding Youth Foundation of North China University of Science and Technology (No. JP201502), China Postdoctoral Science Foundation (No.2015M582533), the Scientific Research Foundation of the Education Department of Sichuan Province (11ZB122) and the Fundamental Research Funds for the Central Universities, China (Nos ZYGX2015J144, ZYGX2015Z006).
Footnotes
Author Contributions W.C. and H.L. conceived and designed the experiments; P.F. analyzed the m1A-seq data; W.C., P.F., H.T. and H.D. implemented SVM and created the back end server; W.C. and H.L. performed the analysis and wrote the paper. All authors read and approved the final manuscript.
References
- Dunn D. B. The occurrence of 1-methyladenine in ribonucleic acid. Biochimica et biophysica acta 46, 198–200 (1961). [DOI] [PubMed] [Google Scholar]
- Dominissini D. et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 530, 441–446, doi: 10.1038/nature16998 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka M. A. et al. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic acids research 41, D262–D267, doi: 10.1093/nar/gks1007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schevitz R. W. et al. Crystal structure of a eukaryotic initiator tRNA. Nature 278, 188–190 (1979). [DOI] [PubMed] [Google Scholar]
- Saikia M., Fu Y., Pavon-Eternod M., He C. & Pan T. Genome-wide analysis of N1-methyl-adenosine modification in human tRNAs. Rna 16, 1317–1327, doi: 10.1261/rna.2057810 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. T. et al. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS genetics 6, e1001247, doi: 10.1371/journal.pgen.1001247 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M. & Alfonzo J. D. Posttranscriptional RNA Modifications: playing metabolic games in a cell’s chemical Legoland. Chemistry & biology 21, 174–185, doi: 10.1016/j.chembiol.2013.10.015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer C. et al. Yeast Rrp8p, a novel methyltransferase responsible for m1A 645 base modification of 25S rRNA. Nucleic acids research 41, 1151–1163, doi: 10.1093/nar/gks1102 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesta J. P. & Cundliffe E. Site-specific methylation of 16S rRNA caused by pct, a pactamycin resistance determinant from the producing organism, Streptomyces pactum. Journal of bacteriology 173, 7213–7218 (1991).1657884 [Google Scholar]
- Li X. et al. Transcriptome-wide mapping reveals reversible and dynamic N-methyladenosine methylome. Nature chemical biology, doi: 10.1038/nchembio.2040 (2016). [DOI] [PubMed] [Google Scholar]
- Chou K. C. Some remarks on protein attribute prediction and pseudo amino acid composition. Journal of theoretical biology 273, 236–247, doi: 10.1016/j.jtbi.2010.12.024 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H. & Li D. Identification of mitochondrial proteins of malaria parasite using analysis of variance. Amino acids 47, 329–333, doi: 10.1007/s00726-014-1862-4 (2015). [DOI] [PubMed] [Google Scholar]
- Kumar R., Srivastava A., Kumari B. & Kumar M. Prediction of beta-lactamase and its class by Chou’s pseudo-amino acid composition and support vector machine. Journal of theoretical biology 365, 96–103, doi: 10.1016/j.jtbi.2014.10.008 (2015). [DOI] [PubMed] [Google Scholar]
- Chen W., Feng P. M., Deng E. Z., Lin H. & Chou K. C. iTIS-PseTNC: a sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Analytical biochemistry 462, 76–83, doi: 10.1016/j.ab.2014.06.022 (2014). [DOI] [PubMed] [Google Scholar]
- Liu B. et al. Identification of microRNA precursor with the degenerate K-tuple or Kmer strategy. Journal of theoretical biology 385, 153–159, doi: 10.1016/j.jtbi.2015.08.025 (2015). [DOI] [PubMed] [Google Scholar]
- Liu B. et al. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 30, 472–479, doi: 10.1093/bioinformatics/btt709 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E., Hall M., Trigg L., Holmes G. & Witten I. H. Data mining in bioinformatics using Weka. Bioinformatics 20, 2479–2481, doi: 10.1093/bioinformatics/bth261 (2004). [DOI] [PubMed] [Google Scholar]
- Chen W., Feng P., Ding H., Lin H. & Chou K. C. iRNA-Methyl: Identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Analytical biochemistry 490, 26–33, doi: 10.1016/j.ab.2015.08.021 (2015). [DOI] [PubMed] [Google Scholar]
- Chen W., Feng P. M., Lin H. & Chou K. C. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic acids research 41, e68, doi: 10.1093/nar/gks1450 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. et al. PseKNC-General: a cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics 31, 119–120, doi: 10.1093/bioinformatics/btu602 (2015). [DOI] [PubMed] [Google Scholar]
- Chen W., Lei T. Y., Jin D. C., Lin H. & Chou K. C. PseKNC: a flexible web server for generating pseudo K-tuple nucleotide composition. Analytical biochemistry 456, 53–60, doi: 10.1016/j.ab.2014.04.001 (2014). [DOI] [PubMed] [Google Scholar]
- Leiros I. et al. Structural basis for enzymatic excision of N1-methyladenine and N3-methylcytosine from DNA. The EMBO journal 26, 2206–2217, doi: 10.1038/sj.emboj.7601662 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L., Niu B., Zhu Z., Wu S. & Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152, doi: 10.1093/bioinformatics/bts565 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Tran H., Liang Z., Lin H. & Zhang L. Identification and analysis of the N(6)-methyladenosine in the Saccharomyces cerevisiae transcriptome. Scientific reports 5, 13859, doi: 10.1038/srep13859 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Tang H. & Lin H. MethyRNA: a web server for identification of N6-methyladenosine sites. Journal of biomolecular structure & dynamics, 1–5, doi: 10.1080/07391102.2016.1157761 (2016). [DOI] [PubMed] [Google Scholar]
- Golam Bari A. T. M., Rokeya Reaz M. & Jeong B. S. DNA Encoding for Splice Site Prediction in Large DNA Sequence. MATCH Communications in Mathematical and in Computer Chemistry 71, 241–258 (2014). [Google Scholar]
- Feng P., Chen W. & Lin H. Prediction of CpG island methylation status by integrating DNA physicochemical properties. Genomics 104, 229–233, doi: 10.1016/j.ygeno.2014.08.011 (2014). [DOI] [PubMed] [Google Scholar]
- Feng P. M., L. H., Chen W. & Zuo Y. C. Predicting the types of J-proteins using clustered amino acids. BioMed research international 2014, 935719 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Chen W. & Ding H. AcalPred: a sequence-based tool for discriminating between acidic and alkaline enzymes. PloS one 8, e75726, doi: 10.1371/journal.pone.0075726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley J. A. & B.J., M. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143, 29–36 (1982). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.