

ABSTRACT
Prions are infectious proteins and over the past few decades, some prions have become renowned for their causative role in several neurodegenerative diseases in animals and humans. Since their discovery, the mechanisms and mode of transmission and molecular structure of prions have begun to be established. There is, however, still much to be elucidated about prion diseases, including the development of potential therapeutic strategies for treatment. The significance of prion disease is discussed here, including the categories of human and animal prion diseases, disease transmission, disease progression and the development of symptoms and potential future strategies for treatment. Furthermore, the structure and function of the normal cellular prion protein (PrPC) and its importance in not only in prion disease development, but also in diseases such as cancer and Alzheimer's disease will also be discussed.
KEYWORDS: bovine spongiform encephalopathy, infectious protein, Creutzfeldt-Jakob disease, PRNP, Alzheimer's disease, prion treatment
INTRODUCTION
Some prion proteins have become well known for their causative role in a range of neurodegenerative diseases in humans and other mammals. Human prion diseases include Creutzfeldt-Jakob Disease (CJD), Gerstmann-Sträussler-Scheinker disease (GSS), Familial Fatal Insomnia (FFI), kuru and variant CJD (vCJD).1,2 Animal prion diseases include Scrapie in sheep, goats and moufflons (subspecies of wild sheep),3 Transmissible Mink Encephalopathy, Chronic Wasting Disease in deer (cervids), Bovine Spongiform Encephalopathy (BSE) in cattle, Exotic Ungulate Encephalopathy in nyala and kudu (sub-species of antelope). Feline Spongiform Encephalopathy in cats, Non-human Primate Transmissible Encephalopathy in lemurs.4 Over the past decade, significant research has been conducted to establish not only the types of prion diseases and their infection mechanisms, but to find an effective therapeutic strategy for treatment.
PRION DISEASE
Prion diseases have a number of common histopathological characteristics and neurological symptoms. These include spongiform degeneration of the central nervous system (CNS), formation of amyloid plaques, reactive gliosis (enlarged glial cells appearing after CNS injury) and neuronal loss.5 Other atypical properties characteristic of prion diseases are long incubation periods (which can extend from several months to several years), lack of inflammation and lack of disease-specific immune response.2
While at first these neuronal degeneration diseases were thought to be caused by “slow” viruses due to their long incubation periods,6 neither viral particles nor nucleic acids could be detected to support the hypothesis that these are viral diseases.2 The viral hypothesis also failed to account for the finding that 95% of human neuronal degeneration disease cases are not linked to infection and lacked the typical histopathological features of viral encephalitis. Hence, the viral disease hypothesis failed to adequately account for the findings that emerged from studies of these diseases.2,7 It was also discovered that between 10 and 15% of these neuronal degeneration diseases are dominantly inherited, including all cases of GSS and FFI and 10% of cases of CJD. The latter are referred to as familial CJD (fCJD). In summary, the data showed that these neuronal degenerative diseases can be both infectious and inherited.7
During the attempts to uncover the molecular basis of prion diseases over the past few decades, it was uncovered that the causative agent of scrapie, is a 27-30kDa protease-resistant protein designated as PrP 27-30.8 PrP 27-30 was found to be encoded by a single mammalian gene located on human chromosome 20.9 This gene was designated PRNP. Other animals have a homolog of the human PRNP gene and this gene was named Prnp. Furthermore, it was discovered that PrP27-30 was derived from a 30-35kDa protein in scrapie-infected animals, designated prion protein scrapie (PrPSc). PrPSc was discovered to be a derivative of the normal protease-sensitive form of PrP that was designated cell-surface glycoprotein (PrPC).10 Upon further investigation, it was found that all dominantly inherited forms of prion diseases are linked to mutations of or insertions in the PRNP gene.10
Development of Prion Disease
Neurodegenerative prion diseases such as CJD, Kuru and BSE, result from the post-translational conversion of normal, glycosylphosphatidylinositol (GPI)-anchored PrPC to a misfolded aggregated and pathogenic form, PrPSc.11 This conversion followed by an accumulation of PrPSc within the central nervous system resulting in disease12 (Fig. 1).
FIGURE 1.
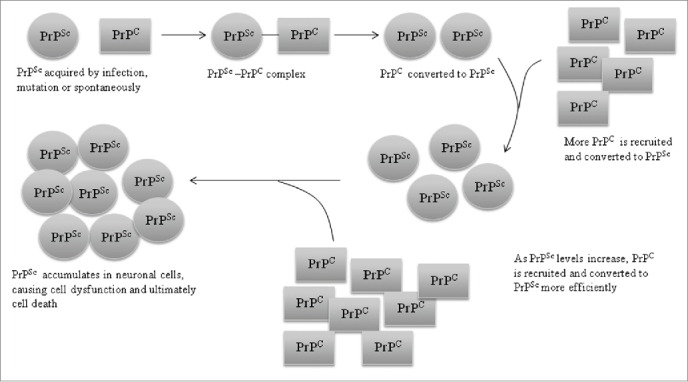
PrPse propagation in the CNS. Schematic representation of the propagation of PrPsc in neuronal cells of the CNS. PrPSc acquired by infection, mutation or spontaneous conversion of cellular PrP (PrPC) combines with PrPc thereby converting it to PrPC is recruited and converted ti PrPSc. As PrPSc levels increase PrPC recruitment and conversion become more efficient, leading to an accumulation of PrPSc in neuronal cells. PrPSc accumulation causes cell dysfunction followed by death.
As shown in Figure 1, the newly formed PrPSc acts as a template to facilitate conversion of PrPC to PrPSc, causing the accumulation of PrPSc.13 This process takes place regardless of the origin of the PrPSc, i.e. whether it is from an external source or a produced internally due to mutations in the PRNP gene (as in the case in fCJD and FFI)7 or due to a spontaneous conversion of wild-type PrPC to PrPSc, which is reffered to as ‘de novo generation/synthesis’.14
PrPSc is characterized by its resistance to protease digestion, insolubility and its high content of β-sheet structure15,16 (43% β-sheets compared to 30% α-helical). In contrast, PrPC is an soluble protein high in α-helices (42%), low in β-sheet (3%) and highly susceptible to protease digestion.13 PrPC contains 2 different domains that play different roles in the conversion of PrPC to PrPSc. The first is a stable and ordered ‘core’ domain which contains a GPI lipid anchor that tethers PrPC to the plasma membrane, 3 α-helices (helices A, B and C) (Fig. 2), 2 asparagines-linked oligosaccharides and a protein binding site capable of lowering the energy barrier for conversion of PrPC to PrPSc when PrPC binds to protein X (a species specific cofactor necessary for conversion of PrPC to PrPSc).17,18 The second domain is a ‘variable’ or disordered domain which interacts with PrPSc and changes PrPC conformation from the unstructured form to the β-sheets of PrPSc (Fig. 2).16 During conversion of PrPC to PrPSc, helix A of the core domain of PrPC (Fig. 2) also gets converted into β-sheets.7
FIGURE 2.
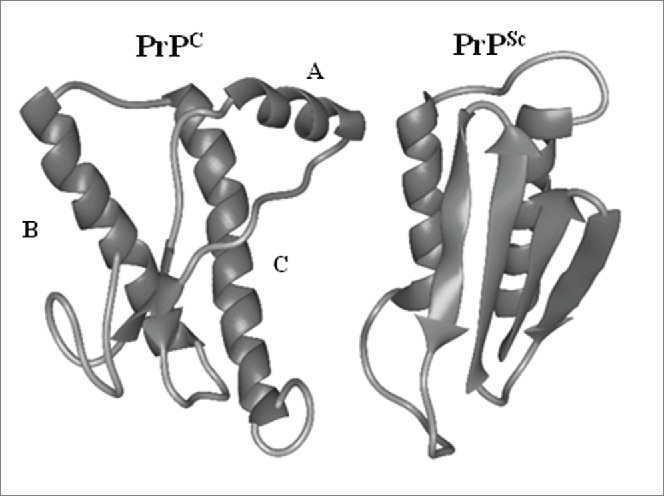
Comparison of protein structure of PrPC and PRPSc. Shown is a schematic representation of the secondary structure of PrPC in comparison to PrPSc as inferred from a recombinant fragment of Syrian hamster PrP comprising amino cid residues 90-231.A,B and C indicate α-helices.
Types of Prion Disease
Prion diseases in humans and animals have thus far been classified into 3 broad categories, based on the properties of the corresponding pathogenic PrP proteins that accumulates in the brain and on their neuroanatomical features of the prion-infected brain7 as summarized in Table 1.
TABLE 1.
Summary of the 3 broad categories of prion diseases and the defining characteristics of each
| Category | Prion Disease | Defining characteristics | References |
|---|---|---|---|
| 1 | Scrapie, sporadic, familial and iatrogenic CJD (sCJD, fCJD, iCJD), BSE, kuru and sporadic and familial fatal insomnia (sFI and fFI) | Vacuolar (spongiform) degeneration of gray matter, accumulation of protease resistant PrPSc in gray matter, little or no PrP amyloid plaque formation. | 7 |
| 2 | Dominantly inherited syndromes (GSS) | Deposition of numerous PrP immunopositive amyloid (abnormal protein) plaques in multiple cortical and subcortical brain regions. PRNP mutation. | 7,19 |
| 3 | Variant CJD (vCJD) | PrPSc amyloid deposition, vacuolation of gray matter, accumulation of protease resistant PrPSc in neuropil (space between neuronal and glial cell bodies comprised of dendrites, axons, synapses microvasculature and glial cell processes). | 7,19-22 |
The oral route of infection is the major mode of transmission for most cases of acquired prion diseases in humans. The spread of the diseases via this route, both naturally and experimentally, has been described in many species. Kuru, for example, was shown to be spread among the Fore people who populate the eastern highlands of Papua New Guinea through ingestion of infected brain tissue during cannibalistic rituals which resulted in a particularly high incidence of the disease 23-25
The sCJD accounts for more than 90% of all cases of sporadic prion disease. However, not all CJD is sporadic. Beginning in the 1980s when a considerable number of cattle were killed by BSE concerns about the spread of BSE to humans grew dramatically in the United Kingdom.26 The concern was that BSE could spread to humans through ingestion of infected beef,27 a fear which was realized in 1995.28 A new form of CJD was first reported by the UK National CJD Research and Surveillance Unit in 1996, and is now known as variant CJD (vCJD).28 It is believed that vCJD possesses distinct clinical and neuropathological characteristics from sporadic CJD (sCJD) and other forms of prion disease in human.29 The main distinguishing neuropathological feature of vCJD is an extensive deposition of PrPSc amyloid in the brain in the form of large ‘florid’ plaques.30 Other phenotypes include a younger average age of onset, and gliosis of the thalamis. Epidemiological studies supported the possibility that the outbreak of BSE in cattle in the UK in the same period may have been responsible for the emergence of vCJD. Subsequently, experimental transmission studies of vCJD and BSE in mice proved that the vCJD agent, unlike the agent responsible for sCJD, had biological properties closely similar to those of the BSE agent.20,30 Therefore, vCJD has been confirmed as a novel prion disease and the only human prion disease acquired from another species. Subsequent studies suggested that the pathogen could be present in blood during the incubation period for vCJD,31,32 and that exposure to such blood could result in the infection of humans or other animals. Therefore, the UK National CJD Surveillance Unit reported that vCJD is caused by exposure to BSE and that the primary source of exposure is the consumption of infected meat products.29
Unlike vCJD, Iatrogenic CJD is an entirely person to person transmitted disease, identified to be transmitted through a number of mechanisms including contaminated surgical instruments,33 and dura mater grafts,34 blood transfusions35 and injection of products from cadaveric pituitary glands.36,37 The first case of iCJD was described in 1974 where a corneal graft recipient died after acquiring a dementia-like likness.38 With improved screening and sterilisation techniques, rates of iCJD continue to decrease with new cases that occur often the result of longer incubation periods following infection acquired in the 1980s.39,40
Whereas the causes of prion diseases that are genetically inherited or acquired by infection have been extensively studied and now quite well known, the cause of sporadic prion diseases is still a topic of speculation.41 Among the many mechanisms that have been proposed to provoke sporadic prion disease, the mechanism that is supported by the most compelling evidence is a mechanism in which malfunction of the so called quality control complex in cells is responsible for initiation of the disease.42 The cellular quality control complex acts like a set of enzymes which assist newly synthesized polypeptides to “grow” to their proper conformation and also play a role in disposal of those polypeptides that fail to adopt the native structure. Therefore, a lowered efficiency of the quality control complex, perhaps due to aging or if the quality control complex becomes overwhelmed by excessive protein production, may result in errors in protein folding occurring and the production of misfolded proteins.
One of the inherited prion diseases is Gerstmann–Sträussler–Scheinker disease (GSS). GSS is caused by a pathological mutation in the prion protein gene (PRNP) located on chromosome 20.It is a very rare disease with autosomal dominant inheritance. The age of onset of GSS is relatively early but the disease progresses slowly with an average illness duration of 49–57 months (until death).43,44 The typical GSS syndrome includes prominent ataxia, gait disturbances, cognitive decline and spasticity in the lower extremities.45,46 Rarely the syndrome may also include painful dysesthesias and visual disturbances, dystonia and myoclonus and dementia.45,47,48 A GSS case attributable to an A133V mutation in PRNP resulted in an uncommon phenotype similar to that of progressive supranuclear palsy.49 Several other pathological variations that cause GSS have been described. The most common mutation in PRNP is P102L, which is found in more than 80% of cases.50 The P102L mutation was found in the original GSS pedigree.51 Other mutations in PRNP known to cause GSS include P105L, P105S, A117V, G131V, Y145*, H187R, D202N, Q212P, Q217R, M232T, and base-pairs insertions at positions 96, 192, or 216.52
Recently, several GSS cases with novel PRNP mutations were reported. A 61-year-old British-born woman with no history of neurodegenerative disorder among her first-degree relatives in Australia presented with a rapidly progressive dementia. Sequencing of the PRNP gene demonstrated a V176G mutation. Subsequent Western blot analysis resulted in the detection of an 8 kDa atypical protease-resistant PrP band.53 Sequencing of the PRNP gene of another GSS patient (this one in North America) revealed a 24-nucleotide insertion that when translated would result in a protein product with an 8-amino acid insertion.54 Independent neuropathological studies of 2 GSS pedigrees with the P102L mutation obtained divergent findings.55,56 The authors note that the variable clinical presentation of GSS patients (even those with the same PRNP mutation) makes diagnosis of GSS challenging and in some families the presence of GSS may be missed. Routine clinical and laboratory investigations, sequence analysis of the PRNP gene and post-mortem examination are recommended in all cases with a family history of any type of neuropsychiatric syndrome.
Treatment of Prion Disease
There are currently no treatments that have proven effective for curing prion diseases in humans or animals. However, monoclonal antibodies that recognize PrPSc and PrPC have been shown to inhibit prion replication and delay prion disease development in animals models.57 These antibodies block PrPSc replication by accelerating the degradation of PrPC (i.e., through reduction of the half life of the PrPC protein).58
Another strategy has been the use of low molecular weight compounds. Compounds that have been undergoing clinical investigation as a possible therapy for prion disease include heterocyclic compounds, e.g. various tricyclic derivatives of acridine and phenothiazine, particularly quinacrine and chlorpromazine.59,60 Treatments such as this have been shown to be effective in inhibiting the formation of nascent PrPSc from PrPC in ScN2a (human neuroblastoma) cells.59,61 These chemicals have been in clinical use for many years, quinacrine as an antimalarial drug and chlorpromazine as an antipsychotic drug, and both are capable of crossing the blood-brain barrier. Treatments with these chemicals are therefore considered to be attractive options for use essentially “as is” in treatment of prion infections.59,62
Quinacrine has been shown the ability to inhibit the formation of PrPSc in ScN2a cells with an IC50 of 0.3–0.4 µM.59,61
A study by Barrett et al.62 re-evaluated the potential of quinacrine and chlorpromazine as treatments for prion diseases. The efficacies of these drugs were assessed in vitro using 2 cell line models (ScN2a and ScGT1) and in vivo using a mouse model. While the results obtained from the previous studies using the ScN2a cell line were replicated in this study for the ScN2a cell line, this study obtained different results from previous studies for the ScGT1 cell line. In contrast to the findings from the previous studies using ScGT1 cells, this study found that only higher doses of chlorpromazine and quinacrine (10 times the concentration previously described, 4 µM) decreased PrPSc accumulation in ScGT1 cells in a single treatment. Lower doses of quinacrine (0.4 µM) cured ScGT1 cells only over longer treatment times (i.e. daily treatment for 3 weeks) and the effect was not permanent as PrPSc infection was re-stablished after approximately 3 months. Furthermore, quinacrine and chlorpromazine failed to inhibit PrPSc accumulation in vivo (either individually or even in combination). It was also noted that quinacrine treatment did not affect the proteinase K-resistance or accumulation of PrPSc in the spleens of mice inoculated with scrapie. Thus overall the studies show that quinacrine and chlorpromazine, individually or in combination, do not have therapeutic anti-prion effects in animal models and highlights the need for new and more effective therapeutics.
Doxycycline has been used as a compassionate treatment for CJD patients and was observed to increase mean survival times by 4-7 months in comparison to historical controls. Furthermore, a patient with variably protease-sensitive prionopathy was treated from an early stage and for 4 years in total with doxycycline and not only lived one year longer than the longest surviving patient with that subgroup of prion disease, but also had less severe and widespread lesions.63 However, while indeed promising these beneficial effects were not confirmed in randomized, double-blind trials.64 In experimental rodent models of prion disease, treatment with doxycycline at early stages (i.e., pre-clinical onset) showed good efficacy, while there was little or no apparent effect once clinical signs had emerged.65 This suggests that treatment with doxycycline may be most useful as a preventive measure, for example for those patients who are carriers of the PRNP mutation that causes Familial Fatal Insomnia,66 and this trial is currently underway.67
A recent study has shown that treatment of prion disease in humans with non-human prion proteins may be a viable treatment option. This approach is based on the knowledge that conversion of PrPC to PrPSc has a strong dependence on protein sequence homology between the prion inoculum and host PrPC.68-70 Skinner et al.71 showed that animals treated with a heterologous prion protein (bacterially expressed and purified recombinant hamster prion protein), demonstrated reduced prion-disease-associated pathology, decreased accumulation of protease-resistant disease associated prion protein and delayed onset of clinical symptoms (including motor deficits), as well as significantly increased mean survival times in comparison to mock-treated control animals.
THE NORMAL CELLULAR PRION PROTEIN
PrPC, as mentioned previously, is the normal cellular form of the causative agent of PrPSc and is required for the development of all the above-mentioned prion diseases. Expression of PrPC begins in embryogenesis, and expression reaches its highest level in adulthood. In adults, PrPC is highly expressed in the neurons of the nervous system,72 and lower, or no expression is observed in other peripheral organs.73 While the 3-dimensional structure and a number of putative roles of PrPC have been reported the exact biochemical function of PrPC is yet to be elucidated.
Processing, 3D Structure and Putative Physiological Role of PrPC
3D Structure of PrPC
PrPC is generally located on the cell membrane and associates with cholesterol-rich microdomains (rafts) in cultured non-neuronal and neuronal cells.74 The immature PrPC protein is approximately 253amino acid residues long and 32-35kDa in mass and comprises of an unstructured N-terminal region and a structured C-terminal domain. The C-terminal domains consists of 3 α-helices, a β-sheet comprising 2 antiparallel β-stands75 and a signal sequence for attachment of the GPI anchor.76 The unstructured N-terminal domain contains an octarepeat and a hydrophobic region.
Processing of PrPC
In order to form a mature protein PrPC undergoes a number of posttranslational modifications and these are initiated by the removal of the N-terminal and C-terminal signal peptides which is coincident with import of the nascent chain into the endoplasmic reticulum and attachment of the GPI anchor. Two N-linked glycans are also attached and this is followed by a disulphide bond between Cys178 and Cys213.77,78 This disulphide bond is important as it connects the C-terminal α-helices, and serves to stabilize the fold of the PrPC protein.79 PrPC (which is 210 amino acid residues in length77) is then targeted to the outer leaflet of the plasma membrane by the GPI-anchor.77,78 PrPc can also undergo 2 endoproteolytic cleavage events.80 The normal constitutive cleavage, known as α-cleavage81 (Fig. 3B), occurs in the brain and in cultured cells between residues 110 and 111. This cleavage is stimulated by agonists of the protein kinase C pathway82 and results in the formation of a 9kDa soluble N-terminal fragment and a 17kDa C-terminal fragment that remains attached to the cell membrane via the GPI anchor.83-85 The second cleavage, known as β-cleavage81 (Fig. 3C), is mediated by reactive oxygen species (ROS)81,86 and leads to the formation of a 19kDa GPI-anchored C-terminal fragment and a 7kDa N-terminal fragment.84,85,87
FIGURE 3.
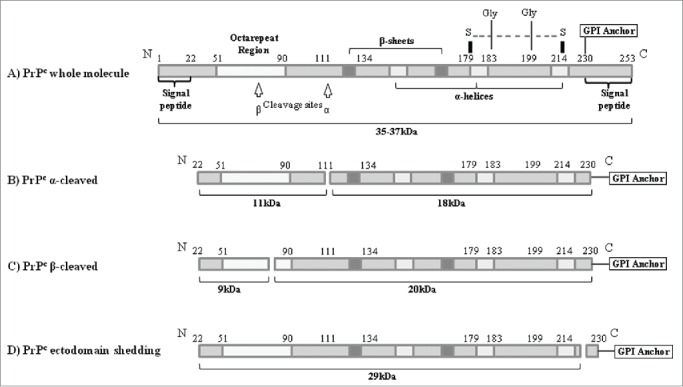
Schematic representation of the protein structure and the proteolytic processing of the PrPC in human cells. (A) Whole PrPC moleule. Post-translational modification is initiated by the removal of N-terminal and C-terminal signal p eptides. (B) Normal constitutive cleavage occurring in brain and culture cells. (C) β-cleavage mediated by reactive oxygen species (ROS). (D) Ecotodomain Shecking of the PrPC where nearly whole PrPc molecule is released from the cellular membrane.
PrPC can then undergo a third cleavage (known as ectodomain shedding) in which PrPC is cleaved at a site close to the GPI anchor thus releasing the nearly full-length PrPC protein from the plasma membrane into the extracellular medium. This proteolytic cleavage has been shown to be performed by the sheddase ADAM10.88-90 Release of PrPC from the cell surface has not only been demonstrated in cell culture, but also in neuronal and lymphoid cells in vivo.88,91-93
Copper Regulation
PrPC is a metal ion-binding protein. It binds copper and zinc with high affinity and manganese and nickel cations with a lower affinity.94-97 Copper binding involves the histidine residues located within the octarepeat region of the N-terminal domain98,99 although recent studies reveal additional copper-binding sites.100 Since the N-terminal domain is also involved in the binding of PrPC to a number of protein ligands, it has been hypothesized that copper binding may play a structural role and influence the binding of PrPC to these other proteins.101
In support of a possible physiological role for PrPC in copper homeostasis, it has been shown that PrPC-deficient (PRNP null) mice exhibit a 50% lower copper concentration in synaptosomal fractions in comparison to wild type mice. This suggests that PrPC may be involved in the regulation of copper concentrations in the synaptic region of the neuron, e.g., by playing a role in the uptake of copper into presynaptic cells.101 Furthermore, PrPC endocytosis has been shown to be stimulated when copper is added to cultured neuroblastoma cells,102 suggesting that PrPC internalisation may be involved in the transport of copper from extracellular to intracellular compartments. It may also indicate that PrPC functions as a copper buffer, binding the copper and transferring it to another membrane transporter.103
Qin and colleagues104 reported that in murine neuro-2a and human HeLa cells, endogenous PrPC rapidly reacts with Cu2+. Cu2+ elevates PrPC expression through transcriptional up-regulation mediated by the ataxia-telangiectasia mutated (ATM) transcription factor. Elevation of PrPC expression protects the cell against copper-induced oxidative stress (and therefore prevents cell death) by playing a role in the modulation of intracellular copper concentrations.
Recently, PrPC has further been shown to function as a modulator of heavy metal concentrations, protecting cells against heavy metal build-up and thus oxidative stress. It was shown in cells with full-length PrPC were more resistant to chronic overload of heavy metals (copper, sinc, nickel and manganese) than their PrP-knock out counterparts.105
Signal Transduction
PrPC has been hypothesized to modulate various signaling pathway components involved in proliferation, cell adhesion, transmembrane signaling, differentiation, and trafficking. For example, PrPC has been shown to have a functional link to phosphatidylinositol 3 kinase (PI-3), a protein kinase involved in cell survival, with in vitro (cell line) and in vivo (mouse) studies showing cells that express PrPC have higher PI-3 levels than those without PrPC.106 PrPC has further been shown to transduce neuroprotective signals through the cyclic AMP-dependent protein kinase/protein kinase A (PKA) pathway107 as well as Fyn108 and many others.
Immune System
PrPC has recently been reported to play a role in the development, activation and proliferation of T lymphocytes.109 While PrPC is widely expressed in the immune system including in human T and B lymphocytes, natural killer cells, platelets, monocytes, and dendritic cells, it has been found to be up-regulated during T-lymphocyte activation and at even more upregulated during natural killer cell differentiation.110 Follicular dendritic cells show high expression of PrPC, however, mice with follicular dendritic cell specific PrPC knock down, showed no alteration in on maturation or function of follicular dendridic cells,111 indicating the high expression or PrPC is non-essential. In contrast, PrPC expression has been shown to be important for macrophage function, modulating phagocytosis in vitro and in vivo.112
PrPC has been found to physically interact with a signal transduction protein with an important role in T lymphocyte activation and proliferation: zeta-chain-associated protein (ZAP)-70.109 In addition, the expression of interleukin-2 is increased when PrPC is expressed.113 These observations suggest that PrPC is involved in the development, activation and proliferation of T-lymphocytes. Furthermore, a recent study showed a soluble recombinant form of PrPC activates human natural killer cells via the ERK and JNK signaling pathways, facilitating IL-15 induced proliferation of natural killer cells, as well as inducing phosphoryation of ERK1/2 and JNK.114
Protection from Programmed Cell Death
When PRNP was knocked out in mice, there was no alteration of life span or observed change in the phenotype of mice,115 indicating that PrPC has a non-critical function or its function is taken over by another protein in its absence. However, further research into the function of PrPC in the CNS demonstrated that its absence in hippocampal neurons resulted in apoptotic (programmed) cell death.116
PrPC also has a structural similarity to the BH2 domain of B-cell lymphoma (Bcl)-2 family members, resulting in the suggestion that PrPC may also function as a member of this family of proteins.117 It was demonstrated that in vitro, PrPC protects human neurons against Bcl-2-associated X protein (Bax)-mediated cell death,118 a pro-apoptotic protein that accelerates cell death by initiating the release of apoptogenic factors by mitochondria.119 When Bcl-2 and Bax are co-expressed, hyperactivation of Bax-induced apoptosis is prevented. Similarly, co-expression of PrPc with Bcl-2 also prevented Bax-induced cell death, implying that PrPC may play a role in protection of neurons against Bax-induced cell death.117,120
Role of PrPC in Central Nervous System Functions
Electrophysiological in vivo studies on PrP-null mice have found that PrPC influences a number of processes within the CNS. These studies demonstrated a number of functional abnormalities in the hippocampus. First there was reduced gamma-aminobutyric acid type A/GABAa receptor-mediated synaptic transmission (GABAa is a ligand-gated ion channel and voltage-dependent calcium channel that play a role in synaptic transmission and regulation of neuronal excitability). Another defect observed in PrPC neurons was attenuation of long-term potentiation. Long-term potentiation is the long-lasting signal transmission between neurons involved in memory. Furthermore, the PrPC neurons exhibited slow after hyperpolarization (i.e. prolonged phase of an action potential in which the membrane potential of a neuron falls below resting potential). Other defects in PrPC-null neurons included: disruption of calcium currents, activation of potassium currents, reduction in the amplitude of inhibitory postsynaptic potentials121-125 and abnormal reorganization of the mossy fiber circuitry in the hippocampus.122
Impairment in hippocampus-dependent spatial learning and altered excitatory and inhibitory neurotransmission in PrP-null mice further support the proposed role of PrPC in synaptic function.126 Studies have also found changes in motility, anxiety and equilibrium in adult mice that were attributable to reduced levels of PrPC.127
In addition, PrPC has been shown to bind to the neural cell adhesion molecule (NCAM) which is a signaling receptor128 in the nervous system that takes part in a number of developmental processes including cell migration, synaptic plasticity and neurite outgrowth.129-131 In summary, these various studies suggests that PrPC may play a role in CNS development128,132 through effect on directed cell migration of neural progenitor cells and the spatial coordination of the outgrowth of neurites as well as playing a role in neuronal survival.133
Potential Role or PrPC in Cancer Development, Progression And Multi-Drug Resistance
Evidence supporting a role of prions and/or prion-like proteins in cancer is becoming increasingly significant. PrPC has been shown to be highly expressed in a number of cancers including pancreatic, gastric, breast, prostate and colorectal and multi-drug resistant forms of breast and gastric. Consistant with this, over-expression of PrPC has also been reported in a number of human gastric cancer cell lines including SCG7901 and AGS.73,134,135 PrPC was also found to be expressed in gastric carcinoma tissues using immunohistochemical staining.73,134 Du et al.73 discovered that PrPC is expressed more strongly in gastric adenocarcinoma tissues than in adjacent non-tumorous tissues and is weakly, or not expressed, in normal gastric mucosa.
PRNP was found to be over-expressed in pancreatic ductal adenocarcinoma (PDAC) in a microarray study by Han et al.136 Another study investigated 7 human PDAC cell lines to determine whether PrPC is overexpressed. While PrPC is over-expressed in the PDAC cell lines, it exists in the form of a pro-protein (Pro-PrP) and is neither glycosylated nor GP- anchored.137
RT-PCR analysis conducted on surgically removed colorectal cancer specimens revealed that PRNP expression is up-regulated in colorectal carcinoma when compared to normal colorectal tissue. This suggests a role for PrPC in the development of colorectal cancer.138 Examination of PrPC expression in colorectal cancer was conducted using formalin-fixed paraffin-embedded colonic neoplastic tissue sample from 110 patients. This study also found that PrPC protein expression increased in cancerous colorectal tissues in comparison to normal colorectal tissues139 and, moreover, that the differential expression in these tissues was even greater than that observed for PRNP mRNA levels in the previous study.138
Study of differential gene expression profiles revealed that the PRNP gene is upregulated in the adriamycin-resistant gastric carcinoma cell line (SGC7901/ADR) when compared to its parental cell line SGC7901.140 This indicates that PrPC may have a role in the development of multi-drug resistance (MDR) phenotypes in gastric carcinomas and has led to a focus on PrPC expression levels in gastric cancer and the mechanisms of PrPC action within MDR gastric carcinoma relvant to its acquisition of MDR phenotypes.73
PrPC has been shown to promote the metastasis of colorectal cancer by mediating epithelial-mesenchymal transition (EMT). It was shown that PrPC expression is associated with the invasive front of colorectal cancer where cells display the characteristics of EMT and that PrPC expression facilitates tumor invasion. Furthermore, functional assays showed that ectopic PrPC expression promotes metastatic potential while knock-down reduces motility of cells. Additionally, knock down of PrPC in implanted colorectal cancer cells in orthotopic xenograft mouse model abolished the number of distant metastases. It was shown that PrPC accelerates tumor metastasis by upregulation of SATB1 expression via the Fyn-SP1 pathway.141 SATB1, a matrix attachment region binding protein important for tissue-specific gene expression, has been shown to alter the gene expression profile of cancer cells to induce an aggressive phenotype and promote metastasis. Depletion of SATB1 reduces cancer progression and results in a metastatic cells undergoing transition to a normal phenotype.142 The molecular signaling events downstream of PrPC were abolished when the Fyn tyrosine kinase was inhibited. This indicates a requirement for Fyn kinase signaling for PrPC-mediated SATB1 expression. Furthermore, SP1 inhibition also reduced SATB1 expression and the metastatic potential of colorectal cancer cells. Overall, this study showed for the first time a link between PrPC expression levels and colorectal cancer metastasis and furthermore showed that the signaling is through a PrPC -Fyn-SP1-SATB1 axis. This signaling pathway may represent a potential target for the future development of anti-metastasis drugs.141
Once it had been confirmed that PrPC is overexpressed in gastric cancer tissues relative to adjacent non-tumor tissues and normal gastric mucosa,73 studies were performed to assess whether PrPC expression level influences the invasive and metastatic properties of gastric cancer. While PrPC was found to be more highly expressed in metastatic cancer than non-metastatic cancer, there was no significant correlation between PrPC expression levels at the primary site and at the metastatic site of the same metastatic gastric cancer. This suggests that an increase in PrPC expression is an early determinant of metastasis and may be useful as a prognostic factor. PrPC expression was also shown to promote adhesive, invasive and metastatic properties of gastric cancer cells in vivo. The N-terminal region of PrPC exhibits an invasion-promoting effect through the activation of the mitogen-activated protein kinase (MAPK)/ extracellular-signal-regulated kinase (ERK) pathway or the MEK/ERK pathway [11]. The MEK/ERK pathway controls cellular processes such as proliferation, apoptosis, survival and differentiation.143 In part, MEK/ERK signaling controls these processes by the transactivation of expression of MMP11 (matrix metalloproteinase 11) [11]. MMP11 in turn is promotes matrix degradation, inflammation and tissue remodeling.144
Potential Role of PrPC in Alzheimer's
Alzheimer's disease accounts for 60% to 70% of all cases of dementia.145 There are 2 core pathological hallmarks of Alzheimer's disease: amyloid plaques and neurofibrillary tangles. The cause of the disease is complex, but the amyloid hypothesis proposes that extracellular amyloid plaques comprising the protein amyloid β (Aβ) is the fundamental cause of the disease. The deposition of Aβ in the brain could trigger neuronal dysfunction and cause neuronal cell death.146 Several studies have shown a physical interaction between Aβ oligomers and PrPC. These studies have provided evidence that the Aβ-PrPC interaction may play an important role in the Aβ toxicity at the synapse by perforating membranes, increasing intracellular calcium ion concentrations and release of synaptic vesicles.147 Recently, it has been shown that sodium dextran sulphate inhibits the binding of Aβ to PrPC and furthermore that simultaneous treatment with sodium dextran sulphate protects long-term potentiation in mouse hippocampal slices from the toxic effects of Aβ oligomers.148
Like Alzheimer's disease,149,150 human prion diseases are associated with a number of psychiatric and behavioral symptoms such as depression and anxiety.151 It has been speculated that the monoaminergic system plays a role in the development of some of these symptoms. It has been shown that PrPC-null mice display depressive-like behavior,152 and recently, PrPC has been shown to regulate the functions of the monoaminergic systems. The authors suggest that the loss of PrPC function in neurodegenerative diseases, attributable to the accumulation of PrPSc in prion diseases and binding of Aβ oligomer to PrPC in Alzheimer's disease interferes with the monoaminergic signaling thus resulting in the neuropsychotic manifestations of these diseases.153 In support of this hypothesis, the behavioral phenotypes of PrPC-null mice have similarities to the behavioral effects observed following injection of wild type Aβ peptide oligomers.154 Overall, recent studies indicate that PrPC may be a potential target for the future development of drugs to treat depression and depression-related disorders.
CONCLUSION
Investigations into prion disease and suitable treatments continue to advance. However, a cure for these diseases still appears to be a distant possibility. Additionally, although decades of research have uncovered a vast range of information about PrPC, there is still much that remains to uncover, particularly in relation to the exact molecular function of PrPC. While PrPC does not appear to be essential, with some studies showing PRNP knock out mice displaying no apparent deficiencies, and others showing minor behavioral changes, PrPC does appear to have a range of important functions. Even more interestingly, the role of PrPC in the development and progression of cancer as well as other diseases such as Alzheimer's disease, is receiving increasing research attention. While the exact role of PrPC in these other diseases remains undetermined, existing research has already shown PrPC to be a potential target for the future development of drug treatments for these other diseases.
ABBREVIATIONS
- BSE
Bovine Spongiform Encephalopathy
- CJD
Creutzfeldt-Jakob Disease
- Fcjd
familial CJD
- FFI
Familial Fatal Insomnia
- GSS
Gerstmann-Sträussler-Scheinker disease
- iCJD
iatrogenic CJD
- PrP
Prion Protein
- PrPC
Cellular Prion Protein
- PrPSc
Prion protein scrapie
- sFI
sporadic fatal insomnia
- vCJD
variant CJD
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
Authors confirm there is no conflict of interest.
Funding
This work was supported by grants from the Australian Research Council (DP110100389) to ALM. Molecular Basis of Disease Program of the Menzies Health Institute Strategic Project Grant to Trina Stewart and ALM.
REFERENCES
- [1].Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 2001; 24:519-50; PMID:11283320; http://dx.doi.org/ 10.1146/annurev.neuro.24.1.519 [DOI] [PubMed] [Google Scholar]
- [2].Prusiner SB. Prions. Proc Natl Acad Sci 1998; 95:13363-13383; PMID:9811807; http://dx.doi.org/ 10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jeffrey M, Gonzalez L. Classical sheep transmissible spongiform encephalopathies: pathogenesis, pathological phenotypes and clinical disease. Neuropathol Appl Neurobiol 2007; 33(4):373-94; PMID:17617870; http://dx.doi.org/ 10.1111/j.1365-2990.2007.00868.x [DOI] [PubMed] [Google Scholar]
- [4].Imran M, Mahmood S. An overview of animal prion diseases. Virol J 2011; 8:493; PMID:22044871; http://dx.doi.org/ 10.1186/1743-422X-8-493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fields BN, Knipe DM, Howley PM. Virology: Third edition. 3rd ed New York: Lippincott-Laven Publishers; 1996. [Google Scholar]
- [6].Sigurdsson B, Rida A. Chronic encephalitis of sheep with general remarks on infections which develop slowly and some of their special characteristics. Br Vet J 1954; 110:341-54. [Google Scholar]
- [7].DeArmon S, Bouzamondo E. Fundamentals of prion biology and diseases. Toxicology 2002; 181-182:9-16; PMID:12505278; http://dx.doi.org/ 10.1016/S0300-483X(02)00249-4 [DOI] [PubMed] [Google Scholar]
- [8].Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136-144; PMID:6801762; http://dx.doi.org/ 10.1126/science.6801762 [DOI] [PubMed] [Google Scholar]
- [9].Sparkes RS, Simon M, Cohn VH, Fournier R, Lem J, Klisak I, Heinzmann C, Blatt C, Lucero M, Mohandas T, et al.. Assignment of the human and mouse prion protein genes to homologous chromosomes. Proc Natl Acad Sci 1986; 83:7358-7362; PMID:3094007; http://dx.doi.org/ 10.1073/pnas.83.19.7358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Meyer R, McKinley M, Bowman K, Braunfeld M, Barry R, Prusiner S. Separation and properties of cellular and scrapie prion proteins. Proc Natl Acad Sci 1986; 83:2310-2314; PMID:3085093; http://dx.doi.org/ 10.1073/pnas.83.8.2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cali Kim J, Surewicz I, Kong K, Raymond Q, Atarashi G, Race R, Qing B, Gambetti L, Caughey P, et al.. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 2010; 285(19):14083-7; PMID:20304915; http://dx.doi.org/ 10.1074/jbc.C110.113464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sassa Y, Kataoka N, Inoshima Y, Ishiguro N. Anti-PrP antibodies detected at terminal stage of prion-affected mouse. Cell Immunol 2010; 263(2):212-218; PMID:20417929; http://dx.doi.org/ 10.1016/j.cellimm.2010.03.018 [DOI] [PubMed] [Google Scholar]
- [13].Pan K, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci 1993; 90:10962-10966; PMID:7902575; http://dx.doi.org/ 10.1073/pnas.90.23.10962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Benetti F, Legname G. De novo mammalian prion synthesis. Prion 2009; 3(4):213-9; PMID:19887900; http://dx.doi.org/ 10.4161/pri.3.4.10181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Prusiner S, McKinley M, Bowman K, Bolton D, Bendheim P, Groth D, Glenner G. Scrapie prions aggregate to form amyloid-like birefringentrods. Cell Immunol 1983; 35:349-358. [DOI] [PubMed] [Google Scholar]
- [16].Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell 1998; 93:337-348; PMID:9590169; http://dx.doi.org/ 10.1016/S0092-8674(00)81163-0 [DOI] [PubMed] [Google Scholar]
- [17].Kaneko K, Zulianello L, Scott M, Copper C, Wallace A, James T, Cohen F, Prusiner S. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci 1997; 94:10069-10074; PMID:9294164; http://dx.doi.org/ 10.1073/pnas.94.19.10069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Telling G, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen F, DeArmond S, Prusiner S. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995; 83(1):79-90; PMID:7553876; http://dx.doi.org/ 10.1016/0092-8674(95)90236-8 [DOI] [PubMed] [Google Scholar]
- [19].Ghetti B, Piccardo P, Frangione B, Bugiani O, Giaccone G, Young K, Prelli F, Farlow M, Dlouhy S, Tagliavini F. Prion protein amyloidosis. Brain Pathol 1996; 6(2):127-145; PMID:8737929; http://dx.doi.org/ 10.1111/j.1750-3639.1996.tb00796.x [DOI] [PubMed] [Google Scholar]
- [20].Scott MR, Will R, Ironside J, Nguyen H-O, Tremblay P, DeArmond S, Prusiner S. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci 1996; 96:15137-15142; http://dx.doi.org/ 10.1073/pnas.96.26.15137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Will RG, Alpers MP, Dormont D, Schonberger LB, Tateishi J. Infectious and sporadic prion diseases in Prion Biology and Diseases. Prusiner S.B., Editor. Cold Spring Harbor Laboratory Press: Cold Spring Harbor; 1999. pp. 465-507. [Google Scholar]
- [22].Spocter MA, Hopkins WD, Barks SK, Bianchi S, Hehmeyer AE, Anderson SM, Stimpson CD, Fobbs AJ, Hof PR, Sherwood CC. Neuropil distribution in the cerebral cortex differs between humans and chimpanzees. J Comp Neurol 2012; 520(13):2917-29; PMID:22350926; http://dx.doi.org/ 10.1002/cne.23074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mead S, Stumpf MP, Whitfield J, Beck JA, Poulter M, Campbell T, Uphill JB, Goldstein D, Alpers M, Fisher EM, et al.. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science 2003; 300(5619):640-3; PMID:12690204; http://dx.doi.org/ 10.1126/science.1083320 [DOI] [PubMed] [Google Scholar]
- [24].Heath CA, Barker RA, Esmonde TF, Harvey P, Roberts R, Trend P, Head MW, Smith C, Bell JE, Ironside JW, et al.. Dura mater-associated Creutzfeldt-Jakob disease: experience from surveillance in the UK. J Neurol Neurosurg Psychiatry 2006; 77(7):880-2; PMID:16627534; http://dx.doi.org/ 10.1136/jnnp.2005.073395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Alpers MP. Kuru in the 21st century–an acquired human prion disease with very long incubation periods. Lancet 2006; 367(9528):2068-74; PMID:16798390; http://dx.doi.org/ 10.1016/S0140-6736(06)68930-7 [DOI] [PubMed] [Google Scholar]
- [26].Houston F, McCutcheon S, Goldmann W, Chong A, Foster J, Sisó S, González L, Jeffrey M, Hunter N. Prion diseases are efficiently transmitted by blood transfusion in sheep. Blood 2008; 112(12):4739-4745; PMID:18647958; http://dx.doi.org/ 10.1182/blood-2008-04-152520 [DOI] [PubMed] [Google Scholar]
- [27].Huang FP, MacPherson GG. Dendritic cells and oral transmission of prion diseases. Adv Drug Deliv Rev 2004; 56(6):901-13; PMID:15063597; http://dx.doi.org/ 10.1016/j.addr.2003.09.006 [DOI] [PubMed] [Google Scholar]
- [28].Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996; 347(9006):921-5; PMID:8598754; http://dx.doi.org/ 10.1016/S0140-6736(96)91412-9 [DOI] [PubMed] [Google Scholar]
- [29].Ironside JW, McCardle L, Horsburgh A, Lim Z, Head MW. Pathological diagnosis of variant Creutzfeldt-Jakob disease. Apmis 2002; 110(1):79-87; PMID:12064259; http://dx.doi.org/ 10.1034/j.1600-0463.2002.100110.x [DOI] [PubMed] [Google Scholar]
- [30].Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, et al.. Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature 1997; 389(6650):498-501; PMID:9333239; http://dx.doi.org/ 10.1038/39057 [DOI] [PubMed] [Google Scholar]
- [31].Ward HJT, Everington D, Cousens SN, Smith-Bathgate B, Leitch M, Cooper S, Heath C, Knight RS, Smith PG, Will RG. Risk factors for variant Creutzfeldt-Jakob disease: A case-control study. Ann Neurol 2006; 59(1):111-120; PMID:16287153; http://dx.doi.org/ 10.1002/ana.20708 [DOI] [PubMed] [Google Scholar]
- [32].Turner ML, Ironside JW. New-variant Creutzfeldt-Jakob disease: the risk of transmission by blood transfusion. Blood Rev 1998; 12(4):255-268; PMID:9950096; http://dx.doi.org/ 10.1016/S0268-960X(98)90007-8 [DOI] [PubMed] [Google Scholar]
- [33].Gibbs CJ Jr., Asher DM, Kobrine A, Amyx HL, Sulima MP, Gajdusek DC. Transmission of Creutzfeldt-Jakob disease to a chimpanzee by electrodes contaminated during neurosurgery. J Neurol Neurosurg Psychiatry 1994; 57(6):757-8; PMID:8006664; http://dx.doi.org/ 10.1136/jnnp.57.6.757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hamaguchi T, Sakai K, Noguchi-Shinohara M, Nozaki I, Takumi I, Sanjo N, Sadakane A, Nakamura Y, Kitamoto T, Saito N, et al.. Insight into the frequent occurrence of dura mater graft-associated Creutzfeldt-Jakob disease in Japan. J Neurol Neurosurg Psychiatry 2013; 84(10):1171-5; PMID:23595947; http://dx.doi.org/ 10.1136/jnnp-2012-304850 [DOI] [PubMed] [Google Scholar]
- [35].Davidson LR, Llewelyn CA, Mackenzie JM, Hewitt PE, Will RG. Variant CJD and blood transfusion: are there additional cases? Vox Sang 2014; 107(3):220-5; PMID:24916465; http://dx.doi.org/ 10.1111/vox.12161 [DOI] [PubMed] [Google Scholar]
- [36].Billette de Villemeur T, Gelot A, Deslys JP, Dormont D, Duyckaerts C, Jardin L, Denni J, Robain O. Iatrogenic Creutzfeldt-Jakob disease in three growth hormone recipients: a neuropathological study. Neuropathol Appl Neurobiol 1994; 20(2):111-7; PMID:8072642; http://dx.doi.org/ 10.1111/j.1365-2990.1994.tb01169.x [DOI] [PubMed] [Google Scholar]
- [37].Delisle MB, Fabre N, Rochiccioli P, Doerr-Schott J, Rumeau JL, Bes A. [Creutzfeldt-Jakob disease after treatment with human extracted growth hormone. A clinicopathological study]. Rev Neurol (Paris) 1993; 149(10):524-7; PMID:8023064 [PubMed] [Google Scholar]
- [38].Duffy P, Wolf J, Collins G, DeVoe AG, Streeten B, Cowen D. Letter: Possible person-to-person transmission of Creutzfeldt-Jakob disease. N Engl J Med 1974; 290(12):692-3; PMID:4591849 [PubMed] [Google Scholar]
- [39].Brown P, Brandel JP, Preece M, Sato T. Iatrogenic Creutzfeldt-Jakob disease: the waning of an era. Neurology 2006; 67(3):389-93; PMID:16855204; http://dx.doi.org/ 10.1212/01.wnl.0000231528.65069.3f [DOI] [PubMed] [Google Scholar]
- [40].Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, Will RG, Ladogana A, Pocchiari M, Leschek EW, Schonberger LB. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis 2012; 18(6):901-7; PMID:22607808; http://dx.doi.org/ 10.3201/eid1806.120116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Palmer CM. A week that shook the meat industry: The effects on the UK beef industry of the BSE crisis. Br Food J 1996; 98(11):17-25; http://dx.doi.org/ 10.1108/00070709610153650 [DOI] [Google Scholar]
- [42].Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: Molecular insights and diagnosis. Lancet Neurol 2012; 11(7):618-628; PMID:22710755; http://dx.doi.org/ 10.1016/S1474-4422(12)70063-7 [DOI] [PubMed] [Google Scholar]
- [43].Ben-Gedalya T, Cohen E. Quality control compartments coming of age. Traffic 2012; 13(5):635-642; PMID:22280095; http://dx.doi.org/ 10.1111/j.1600-0854.2012.01330.x [DOI] [PubMed] [Google Scholar]
- [44].Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A, Giulivi A, Coulthart M, et al.. Genetic prion disease: the EUROCJD experience. Hum Genet 2005; 118(2):166-174; PMID:16187142; http://dx.doi.org/ 10.1007/s00439-005-0020-1 [DOI] [PubMed] [Google Scholar]
- [45].Webb TEF, Poulter M, Beck J, Uphill J, Adamson G, Campbell T, Linehan J, Powell C, Brandner S, Pal S, et al.. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain 2008; 131:2632-2646; PMID:18757886; http://dx.doi.org/ 10.1093/brain/awn202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Arata H, Takashima H, Hirano R, Tomimitsu H, Machigashira K, Izumi K, Kikuno M, Ng AR, Umehara F, Arisato T, et al.. Early clinical signs and imaging findings in Gerstmann-Straussler-Scheinker syndrome (Pro102Leu). Neurology 2006; 66(11):1672-1678; PMID:16769939; http://dx.doi.org/ 10.1212/01.wnl.0000218211.85675.18 [DOI] [PubMed] [Google Scholar]
- [47].Yamada M, Tomimitsu H, Yokota T, Tomi H, Sunohara N, Mukoyama M, Itoh Y, Suematsu N, Otomo E, Okeda R, et al.. Involvement of the spinal posterior horn in Gerstmann-Straussler-Scheinker disease (PrP P102L). Neurology 1999; 52(2):260-265; PMID:9932941; http://dx.doi.org/ 10.1212/WNL.52.2.260 [DOI] [PubMed] [Google Scholar]
- [48].Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H. Mutations of the prion protein gene - Phenotypic spectrum. J Neurol 2002; 249(11):1567-1582; PMID:12420099; http://dx.doi.org/ 10.1007/s00415-002-0896-9 [DOI] [PubMed] [Google Scholar]
- [49].Kretzschmar HA, Kufer P, Riethmüller G, DeArmond S, Prusiner SB, Schiffer D. Prion protein mutation at Codon-102 in an Italian family with gerstmann-straussler-scheinker syndrome. Neurology 1992; 42(4):809-810; PMID:1348851; http://dx.doi.org/ 10.1212/WNL.42.4.809 [DOI] [PubMed] [Google Scholar]
- [50].Rowe DB, Lewis V, Needham M, Rodriguez M, Boyd A, McLean C, Roberts H, Masters CL, Collins SJ. Novel prion protein gene mutation presenting with subacute PSP-like syndrome. Neurology 2007; 68(11):868-870; PMID:17353478; http://dx.doi.org/ 10.1212/01.wnl.0000256819.61531.98 [DOI] [PubMed] [Google Scholar]
- [51].Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, Westaway D, Ott J, Prusiner SB. Linkage of a Prion Protein Missense Variant to Gerstmann-Straussler Syndrome. Nature 1989; 338(6213):342-345; PMID:2564168; http://dx.doi.org/ 10.1038/338342a0 [DOI] [PubMed] [Google Scholar]
- [52].Kretzschmar HA, Honold G, Seitelberger F, Feucht M, Wessely P, Mehraein P, Budka H. Prion Protein Mutation in Family 1st Reported by Gerstmann, Straussler, and Scheinker. Lancet 1991; 337(8750):1160-1160; PMID:1674033; http://dx.doi.org/ 10.1016/0140-6736(91)92826-N [DOI] [PubMed] [Google Scholar]
- [53].Aguzzi A, Calella AM. Prions: Protein aggregation and infectious diseases. Physiol Rev 2009; 89(4):1105-1152; PMID:19789378; http://dx.doi.org/ 10.1152/physrev.00006.2009 [DOI] [PubMed] [Google Scholar]
- [54].Montagnese F, Barca E, Musumeci O, Mondello S, Migliorato A, Ciranni A, Rodolico C, De Filippi P, Danesino C, Toscano A. Clinical and molecular aspects of 30 patients with late-onset Pompe disease (LOPD): Unusual features and response to treatment. J Neurol 2015; 262(4):968-978; PMID:25673129; http://dx.doi.org/ 10.1007/s00415-015-7664-0 [DOI] [PubMed] [Google Scholar]
- [55].Hinnell C, Coulthart MB, Jansen GH, Cashman NR, Lauzon J, Clark A, Costello F, White C, Midha R, Wiebe S, et al.. Gerstmann-Straussler-Scheinker disease due to a novel prion protein gene mutation. Neurology 2011; 76(5):485-487; PMID:21282596; http://dx.doi.org/ 10.1212/WNL.0b013e31820a0ab2 [DOI] [PubMed] [Google Scholar]
- [56].Riudavets MA, Sraka MA, Schultz M, Rojas E, Martinetto H, Begué C, de Halac IN, Poleggi A, Equestre M, Pocchiari M, et al.. Gerstmann-Straussler-Scheinker syndrome with variable phenotype in a new kindred with PRNP-P102L Mutation. Brain Pathol 2014; 24(2):142-147; PMID:23944754; http://dx.doi.org/ 10.1111/bpa.12083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].White A, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature; 2003. 422(6927):80-3; PMID:12621436; http://dx.doi.org/ 10.1038/nature01457 [DOI] [PubMed] [Google Scholar]
- [58].Perrier V, Solassol J, Crozet C, Frobert Y, Mouton-Gilles C, Grassi J, Lehmann S. Anti-PrP antibodies block PrPsc replication in prion-infected cell cultures by accelerating PrPc degradation. J Neurochem 2004; 89:454-463; PMID:15056288; http://dx.doi.org/ 10.1111/j.1471-4159.2004.02356.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Korth C, May B, Cohen F, Prusiner S. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci 2001; 98(17):9836-41; PMID:11504948; http://dx.doi.org/ 10.1073/pnas.161274798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Korth C, Peters P, Emerging pharmacotherapies for Creutzfeldt– Jakob disease. Arch Neurol 2006; 63(4):497-501; PMID:16606761; http://dx.doi.org/ 10.1001/archneur.63.4.497 [DOI] [PubMed] [Google Scholar]
- [61].Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol 2000; 74(10):4894-4897; PMID:10775631; http://dx.doi.org/ 10.1128/JVI.74.10.4894-4897.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Barrett A, Tagliavini F, Forloni G, Salmona M, Colombo L, Luigi D, Limido L, Suardi S, Rossi G, Auvre F, et al.. Evaluation of quinacrine treatment for prion diseases. J Virol 2003; 77(15):8462-8469; PMID:12857915; http://dx.doi.org/ 10.1128/JVI.77.15.8462-8469.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Assar H, Topakian R, Weis S, Rahimi J, Trenkler J, Höftberger R, Aboulenein-Djamshidian F, Ströbel T, Budka H, Yull H, et al.. A case of variably protease-sensitive prionopathy treated with doxycyclin. J Neurol Neurosurg Psychiatry 2015; 86(7):816-8; PMID:25575846; http://dx.doi.org/ 10.1136/jnnp-2014-309871 [DOI] [PubMed] [Google Scholar]
- [64].Haik S, Marcon G, Mallet A, Tettamanti M, Welaratne A, Giaccone G, Azimi S, Pietrini V, Fabreguettes JR, Imperiale D, et al.. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014; 13(2):150-8; PMID:24411709; http://dx.doi.org/ 10.1016/S1474-4422(13)70307-7 [DOI] [PubMed] [Google Scholar]
- [65].Vetrugno V, Puopolo M, Cardone F, Capozzoli F, Ladogana A, Pocchiari M. The future for treating Creutzfeldt–Jakob disease. Exp Opin Orphan Drugs 2015; 3(1):57-74; http://dx.doi.org/ 10.1517/21678707.2015.994605 [DOI] [Google Scholar]
- [66].Forloni G, Artuso V, Roiter I, Morbin M, Tagliavini F. Therapy in prion diseases. Curr Top Med Chem 2013; 13(19):2465-76; PMID:24059336; http://dx.doi.org/ 10.2174/15680266113136660173 [DOI] [PubMed] [Google Scholar]
- [67].Forloni G, Tettamanti M, Lucca U, Albanese Y, Quaglio E, Chiesa R, Erbetta A, Villani F, Redaelli V, Tagliavini F, et al.. Preventive study in subjects at risk of fatal familial insomnia: Innovative approach to rare diseases. Prion 2015; 9(2):75-9; PMID:25996399; http://dx.doi.org/ 10.1080/19336896.2015.1027857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rigter A, Bossers A. Sheep scrapie susceptibility-linked polymorphisms do not modulate the initial binding of cellular to disease-associated prion protein prior to conversion. J Gen Virol 2005; 86(Pt 9):2627-34; PMID:16099922; http://dx.doi.org/ 10.1099/vir.0.80901-0 [DOI] [PubMed] [Google Scholar]
- [69].Horiuchi M, Priola SA, Chabry J, Caughey B. Interactions between heterologous forms of prion protein: binding, inhibition of conversion, and species barriers. Proc Natl Acad Sci U S A 2000; 97(11):5836-41; PMID:10811921; http://dx.doi.org/ 10.1073/pnas.110523897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Priola SA, Vorberg I. Molecular aspects of disease pathogenesis in the transmissible spongiform encephalopathies. Methods Mol Biol 2004; 268:517-40; PMID:15156065 [DOI] [PubMed] [Google Scholar]
- [71].Skinner PJ, Kim HO, Bryant D, Kinzel NJ, Reilly C, Priola SA, Ward AE, Goodman PA, Olson K, Seelig DM. Treatment of prion disease with heterologous prion proteins. PLoS One 2015; 10(7):e0131993; PMID:26134409; http://dx.doi.org/ 10.1371/journal.pone.0131993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Westergard L, Christensen H, Harris D. The cellular prion protein (PrPC): Its physiological function and role in disease. Vet Res 2007; 39(4):629-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Du J, Pan Y, Shi Y, Guo C, Jin X, Sun L, Liu N, Qiao T, Fan D. Overexpression and significance of prion protein in gastric cancer and multidrug-resistant gastric carcinoma cell line SGC7901/ADR. Int J Cancer 2005; 113(2):213-20; PMID:15386405; http://dx.doi.org/ 10.1002/ijc.20570 [DOI] [PubMed] [Google Scholar]
- [74].Vey M, Pilkuhn S, Wille S, Nixon R, DeArmond S, Smart E, Taraboulos A, Prusiner S. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. PNAS 1996; 93(25):14945-14949; PMID:8962161; http://dx.doi.org/ 10.1073/pnas.93.25.14945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Alfonso DS, Adriana Z, Philippe D. Structural and hydration properties of the partially Unfolded States of the prion protein. Biophys J 2007; 93(4):1284-92; PMID:17483173; http://dx.doi.org/ 10.1529/biophysj.107.108613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mehrpour M, Codogno P, From physiology to cancer biology. Cancer Lett 2010; 290(1):23; http://dx.doi.org/ 10.1016/j.canlet.2009.07.009 [DOI] [PubMed] [Google Scholar]
- [77].Linden R, Martins V, Prado M, Cammarota M, Izquiero I, Brentani R. Physiology of the prion protein. Physiol Rev 2008; 88:673-728; PMID:18391177; http://dx.doi.org/ 10.1152/physrev.00007.2007 [DOI] [PubMed] [Google Scholar]
- [78].Stewart R, Harris D. Mutational analysis of topological determinants in prion protein (PrP) and measurement of transmembrane and cytosolic PrP during prion infection. J Biol Chem 2003; 278:45960-45968; PMID:12933795; http://dx.doi.org/ 10.1074/jbc.M307833200 [DOI] [PubMed] [Google Scholar]
- [79].Welker E, Raymond LD, Scheraga HA, Caughey B. Intramolecular versus intermolecular disulfide bonds in prion proteins. J Biol Chem 2002. 277(36):33477-81; PMID:12082114; http://dx.doi.org/ 10.1074/jbc.M204273200 [DOI] [PubMed] [Google Scholar]
- [80].Hooper N. Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein. Biochem Soc F Trans 2005; 33:335-338; http://dx.doi.org/ 10.1042/BST0330335 [DOI] [PubMed] [Google Scholar]
- [81].Mange A, Beranger F, Peoc’h K, Onodera T, Frobert Y, Lehmann S. Alpha- and beta-cleavages of the amino-terminus of the cellular prion protein. Biol Cell 2004; 96:125-132; PMID:15050367; http://dx.doi.org/ 10.1016/j.biolcel.2003.11.007 [DOI] [PubMed] [Google Scholar]
- [82].Vincent B, Paitel E, Frobert Y, Lehmann S, Grassi J, Checler F. Phorbol ester-regulated cleavage of normal prion protein in HEK293 human cells and murine neurons. J Biol Chem 2000; 275:35612-35616; PMID:10952979; http://dx.doi.org/ 10.1074/jbc.M004628200 [DOI] [PubMed] [Google Scholar]
- [83].Pan K, Stahl N, Prusiner S. Purification and properties of the cellular prion protein from Syrian hamster brain. Protein Sci 1992; 1:1343-1352; PMID:1363897; http://dx.doi.org/ 10.1002/pro.5560011014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jimenez-Huete A, Lievens P, Vidal R, Piccardo P, Ghetti B, Tagliavini F, Frangione B, Prelli F. Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues. Am J Pathol 1998; 153:1561-1572; PMID:9811348; http://dx.doi.org/ 10.1016/S0002-9440(10)65744-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Nieznanski K, Nieznanska H, Skowronek K, Osiecka K, Stepkowski D. Direct interaction between prion protein and tubulin. Biochem Biophys Res Commun 2005; 334:403-411; PMID:16004966; http://dx.doi.org/ 10.1016/j.bbrc.2005.06.092 [DOI] [PubMed] [Google Scholar]
- [86].Watt N, Taylor D, Gillott A, Thomas D, Perera W, Hooper N. Reactive oxygen species-mediated beta-cleavage of the prion protein in the cellular response to oxidative stress. J Biol Chem 2005; 280:35914-35921; PMID:16120605; http://dx.doi.org/ 10.1074/jbc.M507327200 [DOI] [PubMed] [Google Scholar]
- [87].Taraboulos A, Raeber A, Borchelt D, Serban D, Prusiner S. Synthesis and trafficking of prion proteins in cultured cells. Mol Biol Cell 1992; 3:851-863; PMID:1356522; http://dx.doi.org/ 10.1091/mbc.3.8.851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, Hooper NM. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem 2009; 284(34):22590-600; PMID:19564338; http://dx.doi.org/ 10.1074/jbc.M109.032599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Altmeppen HC, Prox J, Puig B, Kluth MA, Bernreuther C, Thurm D, Jorissen E, Petrowitz B, Bartsch U, De Strooper B, et al.. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener 2011; 6:36; PMID:21619641; http://dx.doi.org/ 10.1186/1750-1326-6-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Stahl N, Baldwin MA, Burlingame AL, Prusiner SB. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 1990; 29(38):8879-84; PMID:1980209; http://dx.doi.org/ 10.1021/bi00490a001 [DOI] [PubMed] [Google Scholar]
- [91].Parizek P, Roeckl C, Weber J, Flechsig E, Aguzzi A, Raeber AJ. Similar turnover and shedding of the cellular prion protein in primary lymphoid and neuronal cells. J Biol Chem 2001; 276(48):44627-32; PMID:11571302; http://dx.doi.org/ 10.1074/jbc.M107458200 [DOI] [PubMed] [Google Scholar]
- [92].Tagliavini F, Prelli F, Porro M, Salmona M, Bugiani O, Frangione B. A soluble form of prion protein in human cerebrospinal fluid: implications for prion-related encephalopathies. Biochem Biophys Res Commun 1992; 184(3):1398-404; PMID:1375461; http://dx.doi.org/ 10.1016/S0006-291X(05)80038-5 [DOI] [PubMed] [Google Scholar]
- [93].Parkin ET, Watt NT, Turner AJ, Hooper NM. Dual mechanisms for shedding of the cellular prion protein. J Biol Chem 2004; 279(12):11170-8; PMID:14711812; http://dx.doi.org/ 10.1074/jbc.M312105200 [DOI] [PubMed] [Google Scholar]
- [94].Kubosaki A, Yusa S, Nasu Y, Nishimura T, Nakamura Y, Saeki K, Matsumoto Y, Itohara S, Onodera T. Distribution of cellular isoform of prion protein in T lymphocytes and bone marrow, analyzed by wild-type and prion protein gene-deficient mice. Biochem Biophys Res Commun 2001; 282:103-107; PMID:11263978; http://dx.doi.org/ 10.1006/bbrc.2001.4538 [DOI] [PubMed] [Google Scholar]
- [95].Herms J, Tings T, Gall S, Madlung A, Giese A, Siebert H, Schumann P, Windl O, Brose N, Kretzscmar H. Evidence of presynaptic location and function of the prion protein. J Neuroscience 1999; 19(20):8866-75; PMID:10516306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Collinge J, Whittinton M, Sidle K, Smith C, Clarke Palmer M, Jeffers J. Prion Protein is necessary for normal synaptic function. Nature 1994; 370:295-7; PMID:8035877; http://dx.doi.org/ 10.1038/370295a0 [DOI] [PubMed] [Google Scholar]
- [97].Lledo P, Tremblay P, DeArmond S, Prusiner S, Nicoll R. Mice deficient for prion protein exhibit normal neuronal excitability and synaptic transmission in the hippocampus. Proc Natl Acad Sci USA 1996; 93:2403-7; PMID:8637886; http://dx.doi.org/ 10.1073/pnas.93.6.2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Jackson G, Hosszu Nurray I, Gibbs N, Waltho J, Clarke A, Collinge J. Location and properties of metal binding sites on the human prion protein. Prc Natl Acad Sci USA 2001; 98(15):8531-5; http://dx.doi.org/ 10.1073/pnas.151038498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Walter ED, Stevens DJ, Visconte MP, Millhauser GL. The prion protein is a combined zinc and copper binding protein: Zn2+ alters the distribution of Cu2+ coordination modes. J Am Chem Soc 2007; 129(50):15440-1; PMID:18034490; http://dx.doi.org/ 10.1021/ja077146j [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Walter ED, Stevens DJ, Spevacek AR, Visconte MP, Dei Rossi A, Millhauser GL. Copper binding extrinsic to the octarepeat region in the prion protein. Curr Protein Pept Sci 2009; 10(5):529-35; PMID:19538144; http://dx.doi.org/ 10.2174/138920309789352056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kretzschmar H, Tings T, Madlung A, Giese A, Herms J. Function of PrPc as a copper binding protein at the synapse. Arch Virol Suppl 2000; 1:239-49. [DOI] [PubMed] [Google Scholar]
- [102].Pauly P, Harris D. Copper stimulates endocytosis of the prion protein. J Biol Chem 1998; 273:33107-10; PMID:9837873; http://dx.doi.org/ 10.1074/jbc.273.50.33107 [DOI] [PubMed] [Google Scholar]
- [103].Lasmezas C. Putative Functions of PrPc. Br Med Bulletin 2003; 66:61-70; http://dx.doi.org/ 10.1093/bmb/66.1.61 [DOI] [PubMed] [Google Scholar]
- [104].Qin K, Zhao L, Ash R, McDonough W, Zhao R. ATM mediated transcriptional elevation of prion in response to copperinduced oxidative stress. J Biol Chem 2009; 284:4582-4593; PMID:19064990; http://dx.doi.org/ 10.1074/jbc.M808410200 [DOI] [PubMed] [Google Scholar]
- [105].Prcina M, Kontsekova E, Novak M. Prion protein prevents heavy metals overloading of cells and thus protects them against their toxicity. Acta Virol 2015; 59(2):179-84; PMID:26104335; http://dx.doi.org/ 10.4149/av_2015_02_179 [DOI] [PubMed] [Google Scholar]
- [106].Vassallo N, Herms J, Behrens C, Krebs B, Saeki K, Onodera T, Windl O, Kretzschmar HA. Activation of phosphatidylinositol 3-kinase by cellular prion protein and its role in cell survival. Biochem Biophys Res Commun 2005; 332(1):75-82; PMID:15896301; http://dx.doi.org/ 10.1016/j.bbrc.2005.04.099 [DOI] [PubMed] [Google Scholar]
- [107].Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. Embo J 2002; 21(13):3317-26; PMID:12093733; http://dx.doi.org/ 10.1093/emboj/cdf324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289(5486):1925-8; PMID:10988071; http://dx.doi.org/ 10.1126/science.289.5486.1925 [DOI] [PubMed] [Google Scholar]
- [109].Mattei V, Garofalo T, Misasi R, Circella A, Manganelli V, Lucania G, Pavan A, Sorice M. Prion protein is a component of the multimolecular signalling complex involved in T cell activation. FEBS Lett 2004; 560(1-3):14-8; PMID:14987990; http://dx.doi.org/ 10.1016/S0014-5793(04)00029-8 [DOI] [PubMed] [Google Scholar]
- [110].Martinez D, Lopez-Bravo M, Metharom P, Ardavin C, Aucouturier P. Prion protein expression by mouse dendritic cells is restricted to the nonplasmacytoid subsets and correlates with the maturation state. J Immunol 2006; 177(9):6137-42; PMID:17056541; http://dx.doi.org/ 10.4049/jimmunol.177.9.6137 [DOI] [PubMed] [Google Scholar]
- [111].McCulloch L, Brown KL, Mabbott NA. Ablation of the cellular prion protein, PrPC, specifically on follicular dendritic cells has no effect on their maturation or function. Immunology 2013; 138(3):246-57; PMID:23121447; http://dx.doi.org/ 10.1111/imm.12031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].de Almeida CJ, Chiarini LB, da Silva JP, E Silva PM, Martins MA, Linden R. The cellular prion protein modulates phagocytosis and inflammatory response. J Leukoc Biol 2005; 77(2):238-46; PMID:15539455; http://dx.doi.org/ 10.1189/jlb.1103531 [DOI] [PubMed] [Google Scholar]
- [113].Bainbridge J, Walker K. The normal cellular form of prion protein modulates T cell responses. Immunol Lett 2005; 96(1):147-50; PMID:15585317; http://dx.doi.org/ 10.1016/j.imlet.2004.08.006 [DOI] [PubMed] [Google Scholar]
- [114].Seong YJ, Sung PS, Jang YS, Choi YJ, Park BC, Park SH, Park YW, Shin EC. Activation of human natural killer cells by the soluble form of cellular prion protein. Biochem Biophys Res Commun 2015; 464(2):512-8; PMID:26159919; http://dx.doi.org/ 10.1016/j.bbrc.2015.06.172 [DOI] [PubMed] [Google Scholar]
- [115].Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp H, DeArmond S, Prusiner S, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell surface PrP protein. Nature 1992; 356:577-582; PMID:1373228; http://dx.doi.org/ 10.1038/356577a0 [DOI] [PubMed] [Google Scholar]
- [116].Kuwahara C, Takeuchi A, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Matsumoto Y, Yokoyama T, Itohara S, et al.. Prions prevent neuronal cell-line death. Nature 1999; 400(6741):225-6; PMID:10421360; http://dx.doi.org/ 10.1038/22241 [DOI] [PubMed] [Google Scholar]
- [117].Roucou X, Gains M, LeBlanc A. Neuroprotective functions of prion protein. J Neurosci Res 2004; 75(2):153-61; PMID:14705136; http://dx.doi.org/ 10.1002/jnr.10864 [DOI] [PubMed] [Google Scholar]
- [118].Bounhar Y, Zhang Y, Goodyer C, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem 2001; 276(42):39145-9; PMID:11522774; http://dx.doi.org/ 10.1074/jbc.C100443200 [DOI] [PubMed] [Google Scholar]
- [119].Martinou J, Green D. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2001; 2(1):63-7; PMID:11413467; http://dx.doi.org/ 10.1038/35048069 [DOI] [PubMed] [Google Scholar]
- [120].Roucou X, LeBlanc A. Cellular prion protein neuroprotective function: implications in prion diseases. J Mol Med 2005; 83:3-11; PMID:15645198; http://dx.doi.org/ 10.1007/s00109-004-0605-5 [DOI] [PubMed] [Google Scholar]
- [121].Collinge J, Whittington M, Sidle K, Smith C, Palmer M, Clarke A, Jefferys J. Prion protein is necessary for normal synaptic function. Nature 1994; 370:295-297; PMID:8035877; http://dx.doi.org/ 10.1038/370295a0 [DOI] [PubMed] [Google Scholar]
- [122].Colling S, Collinge J, Jefferys J. Hippocampal slices from prion protein null mice: disrupted Ca(2+)-activated K+ currents. Neurosci Lett 1996; 209:49-52; PMID:8734907; http://dx.doi.org/ 10.1016/0304-3940(96)12596-9 [DOI] [PubMed] [Google Scholar]
- [123].Curtis J, Errington M, Bliss T, Voss K, MacLeod N. Age dependent loss of PTP and LTP in the hippocampus of PrP-null mice. Neurobiol Dis 2003; 13:55-62; PMID:12758067; http://dx.doi.org/ 10.1016/S0969-9961(03)00017-2 [DOI] [PubMed] [Google Scholar]
- [124].Mallucci GR, Ratte´ S, Asante EA, Linehan J, Gowland I, Jefferys JGR, Collinge J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J 2002; 21:202-210; PMID:11823413; http://dx.doi.org/ 10.1093/emboj/21.3.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Manson JC, Hope J, Clarke AR, Johnston A, Black C, MacLeod N. PrP gene dosage and long term potentiation. Neurodegeneration 1995; 4:113-114; PMID:7600180; http://dx.doi.org/ 10.1006/neur.1995.0014 [DOI] [PubMed] [Google Scholar]
- [126].Criado J, Sanchez-Alavez M, Conti B, Giacchino J, Wills D, Henriksen S, Race R, Manson J, Chesebro B, Oldstone M. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol Dis 2005; 19:255-265; PMID:15837581; http://dx.doi.org/ 10.1016/j.nbd.2005.01.001 [DOI] [PubMed] [Google Scholar]
- [127].Lobao-Soares B, Walz R, Carlotti C Jr., Sakamoto A, Calvo F, Terzian A, da Silva J, Wichert-Ana L, Coimbra N, Bianchin M. Cellular prion protein regulates the motor behaviour performance and anxiety-induced responses in genetically modified mice. Behav. Brain Res 2007; 183:87-94; PMID:17618696; http://dx.doi.org/ 10.1016/j.bbr.2007.05.027 [DOI] [PubMed] [Google Scholar]
- [128].Mange A, Milhavet O, Umlauf D, Harris D, Lehmann S. PrP dependent cell adhesion in N2a neuroblastoma cells. FEBS Lett 2002; 514(2-3):159-62; PMID:11943143; http://dx.doi.org/ 10.1016/S0014-5793(02)02338-4 [DOI] [PubMed] [Google Scholar]
- [129].Crossin K, Krushel L. Cellular signaling by neural cell adhesion molecules of the immunoglobulin superfamily. Dev Dyn 2000; 218:260-279; PMID:10842356; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- [130].Ronn L, Berezin V, Bock E. The neural cell adhesion molecule in synaptic plasticity and ageing. Int J Dev Neurosci 2000; 18:193-199; PMID:10715574; http://dx.doi.org/ 10.1016/S0736-5748(99)00088-X [DOI] [PubMed] [Google Scholar]
- [131].Schachner M. Neural recognition molecules and synaptic plasticity. Curr Opin Cell Biol 1997; 9:627-634; PMID:9330865; http://dx.doi.org/ 10.1016/S0955-0674(97)80115-9 [DOI] [PubMed] [Google Scholar]
- [132].Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol 2005; 169(2):341-54; PMID:15851519; http://dx.doi.org/ 10.1083/jcb.200409127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Chen S, Mange A, Dong L, Lehmann S, Schachner M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol Cell Neurosci 2003; 22(2):227-33; PMID:12676532; http://dx.doi.org/ 10.1016/S1044-7431(02)00014-3 [DOI] [PubMed] [Google Scholar]
- [134].Liang J, Pan YL, Ning XX, Sun LJ, Lan M, Hong L, Du JP, Liu N, Liu CJ, Qiao TD, et al.. Overexpression of PrPC and its antiapoptosis function in gastric cancer. Tumour Biol 2006; 27(2):84-91; PMID:16582585; http://dx.doi.org/ 10.1159/000092488 [DOI] [PubMed] [Google Scholar]
- [135].Liang J, Pan Y, Zhang D, Guo C, Shi Y, Wang J, Chen Y, Wang X, Liu J, Guo X, et al.. Cellular prion protein promotes proliferation and G1/S transition of human gastric cancer cells SGC7901 and AGS. FASEB J 2007; 21(9):2247-56; PMID:17409275; http://dx.doi.org/ 10.1096/fj.06-7799com [DOI] [PubMed] [Google Scholar]
- [136].Han H, Bearss D, Browne W, Calaluce R, Nagle R, Hoff D. Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. Cancer Res 2002; 62:2890-2896; PMID:12019169 [PubMed] [Google Scholar]
- [137].Li C, Yu S, Nakamura F, Yin S, Xu J, Petrolla AA, Singh N, Tartakoff A, Abbott DW, Xin W, et al.. Binding of pro-prion to filamin A disrupts cytoskeleton and correlates with poor prognosis in pancreatic cancer. J Clin Invest 2009; 119(9):2725-36; PMID:19690385; http://dx.doi.org/ 10.1172/JCI39542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Antonacopoulou AG, Grivas PD, Skarlas L, Kalofonos M, Scopa CD, Kalofonos HP. POLR2F, ATP6V0A1 and PRNP expression in colorectal cancer: new molecules with prognostic significance? Anticancer Res 2008; 28(2B):1221-7; PMID:18505059 [PubMed] [Google Scholar]
- [139].Antonacopoulou AG, Palli M, Marousi S, Dimitrakopoulos FI, Kyriakopoulou U, Tsamandas AC, Scopa CD, Papavassiliou AG, Kalofonos HP. Prion protein expression and the M129V polymorphism of the PRNP gene in patients with colorectal cancer. Mol Carcinog 2010; 49(7):693-9; PMID:20564346 [DOI] [PubMed] [Google Scholar]
- [140].Zhao Y, You H, Liu F, An H, Shi Y, Yu Q, Fan D. Differentially expressed gene profiles between multidrug resistant gastric adenocarcinoma cells and their parental cells. Cancer Lett 2002; 185(2):211-8; PMID:12169395; http://dx.doi.org/ 10.1016/S0304-3835(02)00264-1 [DOI] [PubMed] [Google Scholar]
- [141].Wang Q, Qian J, Wang F, Ma Z. Cellular prion protein accelerates colorectal cancer metastasis via the Fyn-SP1-SATB1 axis. Oncol Rep 2012; 28(6):2029-34; PMID:22972305 [DOI] [PubMed] [Google Scholar]
- [142].Han HJ, Russo J, Kohwi Y, Kohwi-Shigematsu T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008; 452(7184):187-93; PMID:18337816; http://dx.doi.org/ 10.1038/nature06781 [DOI] [PubMed] [Google Scholar]
- [143].Kolch W. Meaningful relationships: The Regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J 2000; 351:289-305; PMID:11023813; http://dx.doi.org/ 10.1042/bj3510289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Duffy M, Maguire T, Hill A, McDermott E, O’Higgins N, Metalloproteinases: role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res 2000; 2:252-257; PMID:11250717; http://dx.doi.org/ 10.1186/bcr65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Popova SN, Tarvainen I, Capellari S, Parchi P, Hannikainen P, Pirinen E, Haapasalo H, Alafuzoff I. Divergent clinical and neuropathological phenotype in a Gerstmann-Straussler-Scheinker P102L family. Acta Neurol Scand 2012; 126(5):315-323; PMID:22211828; http://dx.doi.org/ 10.1111/j.1600-0404.2011.01628.x [DOI] [PubMed] [Google Scholar]
- [146].Burns A, Iliffe S. Alzheimer's disease. 2009; 338. [DOI] [PubMed] [Google Scholar]
- [147].Peters C, Espinoza MP, Gallegos S, Opazo C, Aguayo LG. Alzheimer's Abeta interacts with cellular prion protein inducing neuronal membrane damage and synaptotoxicity. Neurobiol Aging 2015; 36(3):1369-77; PMID:25599875; http://dx.doi.org/ 10.1016/j.neurobiolaging.2014.11.019 [DOI] [PubMed] [Google Scholar]
- [148].Aimi T, Suzuki K, Hoshino T, Mizushima T. Dextran sulfate sodium inhibits amyloid-beta oligomer binding to cellular prion protein. J Neurochem 2015; 134(4):611-7; PMID:25963375; http://dx.doi.org/ 10.1111/jnc.13166 [DOI] [PubMed] [Google Scholar]
- [149].Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D. Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry 2006; 63(5):530-8; PMID:16651510; http://dx.doi.org/ 10.1001/archpsyc.63.5.530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Green RC, Cupples LA, Kurz A, Auerbach S, Go R, Sadovnick D, Duara R, Kukull WA, Chui H, Edeki T, et al.. Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch Neurol 2003; 60(5):753-9; PMID:12756140; http://dx.doi.org/ 10.1001/archneur.60.5.753 [DOI] [PubMed] [Google Scholar]
- [151].Thompson A, MacKay A, Rudge P, Lukic A, Porter MC, Lowe J, Collinge J, Mead S. Behavioral and psychiatric symptoms in prion disease. Am J Psychiatry 2014; 171(3):265-74; PMID:24585329; http://dx.doi.org/ 10.1176/appi.ajp.2013.12111460 [DOI] [PubMed] [Google Scholar]
- [152].Gadotti VM, Bonfield SP, Zamponi GW. Depressive-like behaviour of mice lacking cellular prion protein. Behav Brain Res 2012; 227(2):319-23; PMID:21439331; http://dx.doi.org/ 10.1016/j.bbr.2011.03.012 [DOI] [PubMed] [Google Scholar]
- [153].Beckman D, Santos LE, Americo TA, Ledo JH, de Mello FG, Linden R. Prion protein modulates monoaminergic systems and depressive-like behavior in mice. J Biol Chem 2015; 290(33):20488-98; PMID:26152722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Ledo JH, Azevedo EP, Clarke JR, Ribeiro FC, Figueiredo CP, Foguel D, De Felice FG, Ferreira ST. Amyloid-beta oligomers link depressive-like behavior and cognitive deficits in mice. Mol Psychiatry 2013; 18(10):1053-4; PMID:23183490; http://dx.doi.org/ 10.1038/mp.2012.168 [DOI] [PMC free article] [PubMed] [Google Scholar]