

Abstract

Oncogenic MLL fusion proteins aberrantly recruit Dot1L, a histone methyltransferase, to ectopic loci, leading to local hypermethylation of H3K79 and misexpression of HoxA genes driving MLL-rearranged leukemias. Inhibition of the methyltransferase activity of Dot1L in this setting is predicted to reverse aberrant H3K79 methylation, leading to repression of leukemogenic genes and tumor growth inhibition. In the context of our Dot1L drug discovery program, high-throughput screening led to the identification of 2, a weak Dot1L inhibitor with an unprecedented, induced pocket binding mode. A medicinal chemistry campaign, strongly guided by structure-based consideration and ligand-based morphing, enabled the discovery of 12 and 13, potent, selective, and structurally completely novel Dot1L inhibitors.
Keywords: Dot1L, protein lysine methyltransferase, inhibitor, mixed lineage leukemia, protein structure-based design
Protein lysine methyltransferases (PKMT) are a class of 51 enzymes that catalyze the transfer of a methyl group from cofactor S-adenosylmethionine (SAM) to the lysine ε-amino group of proteins.1−3 Typically, PKMTs mono-, di-, or trimethylate lysine residues of histone proteins, thereby controlling chromatin structure and transcriptional accessibility of genes. However, a number of other nonhistone substrate proteins have also been identified.4
Dot1L is a 165 kDa protein comprising 1537 amino acids. The catalytic domain of Dot1L, residing in the N-terminal 400 amino acids, is structurally distinct to the SET-domain containing protein lysine methyltransferases and clusters more closely with the catalytic domain of a related class of proteins, the protein arginine methyltransferases (PRMT).3,5 Dot1L is the only known enzyme to methylate lysine 79 of histone 3 (H3K79), with the H3K79me2 mark being associated with active transcription.6−8 Under physiological conditions, Dot1L is critical for normal hematopoiesis;9,10 however, misdirected catalytic activity is believed to be causative for a subset of acute leukemias.11,12 Several oncogenic fusion proteins including MLL-ENL, MLL-AF4, and MLL-AF9 aberrantly recruit Dot1L to ectopic loci, leading to local hypermethylation of H3K79 and misexpression of genes (including HoxA), which drive the leukemic phenotype. Inhibition of the methyltransferase activity of Dot1L in MLL-rearranged leukemias (mixed lineage leukemia, MLL) is predicted to reverse ectopic H3K79 methylation, leading to repression of leukemogenic genes (HoxA9, Meis1) and tumor growth inhibition.13 The recent quest for Dot1L inhibitors has been spearheaded by Epizyme and culminated in the discovery of EPZ-5676, a SAM-competitive, nucleoside-containing Dot1L inhibitor, which is currently being evaluated in MLL patients in Phase 1b clinical trials.14 The agent is administered by uninterrupted, continuous intravenous (i.v.) infusion due to its physicochemical properties.15 Other research groups, based at Baylor College and the Structural Genomics Consortium, have identified additional Dot1L inhibitors structurally related to EPZ-5676.16−19
Herein, we describe the discovery of SAM-competitive, structurally novel Dot1L inhibitors, which bind to an induced pocket adjacent to the SAM binding site. The compounds display cellular potency and molecular properties suitable for further optimization.
A variety of approaches to discover chemical starting points in the Dot1L program were taken and included virtual, biophysical, and biochemical screens. In our high-throughput screening (HTS) campaign, a luminescence-based coupled assay20 was used to measure Dot1L-catalyzed nucleosome methylation and miniaturized to allow screening of the Novartis Compound Collection in 1536-well plates (primary hit rate 0.47%, threshold 50% inhibition at 26.7 μM compound concentration). For IC50 and Ki determinations, a scintillation proximity assay (SPA) format was employed as a more direct readout, quantifying enzyme activity via the amount of the transfer of 3H-labeled methyl group from SAM to biotinylated nucleosomes. Experiments described herein were carried out at SAM concentration equal to KM, while nucleosome concentration was in excess relative to KM (Supporting Information). In the HTS, a commercial compound sample, assigned to structure 1, was identified as an inhibitor of Dot1L (IC50 = 14 μM) (Figure 1). Analysis of the screening sample revealed that the active principle is a regioisomeric impurity, compound 2 (IC50 = 4.4 μM), whereas purified 1 had no inhibitory activity up to 100 μM.
Figure 1.

Protein structure-guided optimization of HTS hit 1.
The crystal complex structure with compound 2 was solved using the catalytic domain of Dot1L (Figure 2, PDB code 5drt). To our surprise, 2 binds to a newly formed pocket adjacent to the SAM binding site, inducing a fundamentally different conformation of Dot1L compared to the conformation of SAM-bound Dot1L (Figure 2B).21 A flexible loop comprising amino acids 126–140, which forms the “lid” of the SAM binding pocket in the SAM-bound state, is completely rearranged and partially disordered and forms a part of the induced binding pocket of 2. We and others subsequently observed that only SAM and SAH are able to induce a closed lid (e.g., PDB code 3uwp), whereas inhibitors rearrange and often engage the flexible loop.16,22 Other residues, such as Leu143, Met147, Phe239, and Tyr312, also undergo conformational shifts compared to the SAM-bound complex and are forming the bottom lining of the binding pocket of 2. Remarkably, 2 forms five direct hydrogen bonds with Dot1L (Figure 2A). The central urea NHs are coordinating Asp161, whose side chain rotation is cushioned by crystal waters. The interaction of the urea carbonyl with Asn241 varies between 3.2 and 3.8 Å across several cogeneric cocrystal structures. The urea of 2 interacts with Dot1L in a comparable fashion to the urea-containing Dot1L inhibitor EPZ-4777.16 The second hydrazide amide forms a hydrogen bond to Gly163 perpendicular to the urea plane and stacks onto the Ser164-Gly165 backbone amide. The tetrazole forms a hydrogen bond with Gln168, and the tetrazole-phenoxy motif engages the flexible loop through hydrophobic contacts, while the p-chlorophenyl substituent slides between the Asn241-containing loop and an α-helix, occupying a similar position as the tert-Bu group of EPZ-5676 (Figure 2C).
Figure 2.
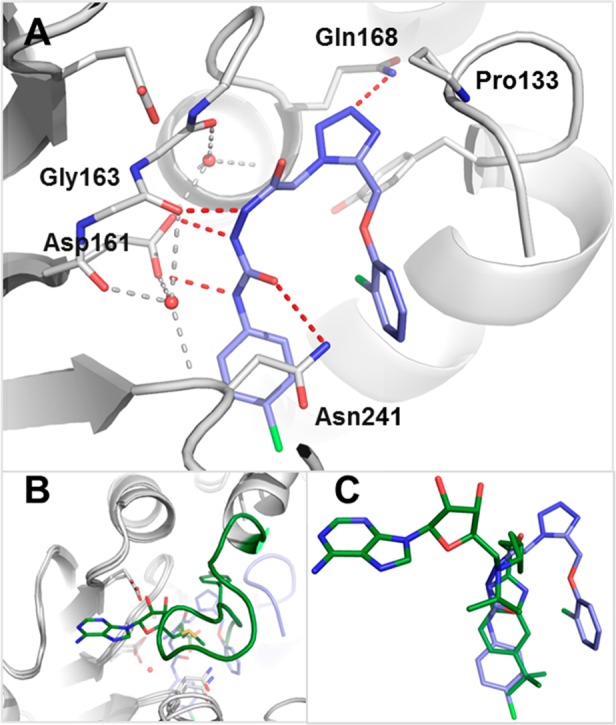
(A) X-ray cocrystal structure of Dot1L (gray) with 2 (blue) (PDB code 5drt). Amino acid side chains engaged in key interactions with the ligand are illustrated as sticks. Key polar interactions of Dot1L and 2 are shown as red dotted red lines, the tetrahedral coordination of the two waters ligating Asp161 as gray dotted lines. (B) Comparison of the flexible loop (amino acids 126–140) in Dot1L bound to 2 (blue, protein gray) (PDB code 5drt) and SAM (green) (PDB code 3qow). (C) Overlay of ligands 2 (blue) (PDB code 5drt) and EPZ-5676 (green) (PDB code 4hra) bound to Dot1L in the same view as 2A. PyMol was used for structural visualization and figure preparation.25
Structural analysis of the Dot1L–compound 2 complex revealed a large hydrophobic pocket, which is only partially filled by the p-chlorophenyl substituent. Structure-based chemistry optimization led to the discovery of the significantly more potent compound 3 (IC50 = 20 nM) in which the p-chlorophenyl substituent was replaced by an N-arylated indole system (Figure 1). The indole core is deeply buried in a hydrophobic cleft, while the N-phenyl moiety engages in a face-edge interaction with Phe131 thereby further stabilizing the flexible loop of Dot1L (Figure 3, PDB code 5dry). The o-Cl substituent does not only preorganize the bioactive conformation of 3 but also fills a hydrophobic nook of Dot1L.
Figure 3.
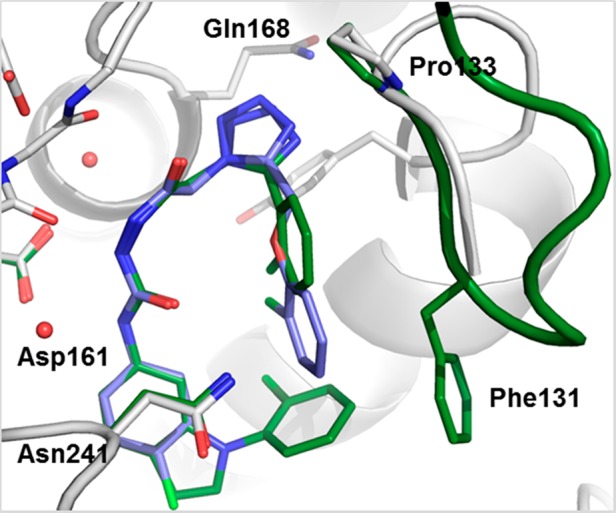
Overlay of X-ray cocrystal structures of Dot1L with 2 (ligand blue, protein gray) (PDB code 5drt) and 3 (ligand green, parts of the protein green) (PDB code 5dry). Prominent amino acid side chains are illustrated as sticks.
Inhibitor 3 shows low permeability as assessed in the parallel artificial membrane permeation (PAMPA) assay, which might not be surprising, considering its number of hydrogen bond acceptors and donors (Supporting Information). It is likely that the weak flux of 3 contributes to the lack of oral bioavailability in rats and weak cellular activity.
Considering the poor physicochemical properties, the complex functional group arrangement, and high molecular weight (MW = 521), we regarded 3 unsuitable for further stepwise optimization. Therefore, we decided to pursue a more radical fragmentation approach in which the entire aryl-tetrazole system was eliminated leading to urea 4, which showed residual biochemical potency (IC50 = 40 μM) (Figure 4). Compound 4 was viewed as a more suitable starting point for additional chemistry activities. Initial work focused on the identification of urea replacements with the goal to reduce the risk of low solubility and poor permeability often associated with ureas. A series of functional groups were discovered as urea isosteres capable of coordinating the central Asp161 side chain of Dot1L. Diamino-pyrimidines 5 and 6 (in their protonated forms), amino-pyrimidone 7, amino-pyridazine 8 (with its polarized C–H group), and amino-imidazolone 9 inhibit Dot1L with improved potency compared to urea 4 (Figure 4). It is worth noting that the methyl group in the 2-position of the indole and the insertion of a nitrogen atom into the N-phenyl substituent typically contribute to the potency by a 5-fold reduction of the IC50. Remarkably, the N-methyl group of 6 increases inhibitory activity against Dot1L by almost 20-fold over 5, possibly through a preorganization effect (discussed below).
Figure 4.

Fragmentation of 3 to urea 4. Discovery of urea replacements 5–9.
The optimization opportunities for urea replacements 6 and 7 were evaluated in detail. In line with our structural understanding, the diamino-pyrimidine of 6 and the amino-pyrimidone of 7 undergo polar interactions with Asp161 (Figure 5). The N-aryl 2-methylindole core of both analogues is shape-complementary to the hydrophobic pocket formed by the Asn241-containing loop and the α-helix. Importantly, the carbonyl group of amino-pyrimidone 7 and the N-methyl group of diamino-pyrimidine 6 interact with Dot1L in distinct ways: The N-methyl group of 6 does not undergo any specific contacts with Dot1L, but rather plays a role in preorganizing the ligand by pointing into the N-aryl moiety (van der Waals contact). In contrast, the carbonyl group of 7 engages in polar interactions with Asn241 and the backbone NH of Tyr136, thereby tightly stabilizing the flexible loop of Dot1L (Figure 5). Consequently, potential growth vectors in ortho- and meta-position of the N-pyridyl moiety of pyrimidone 7 are blocked by the loop, whereas they are open for pyrimidine 6 (Figure 5).
Figure 5.
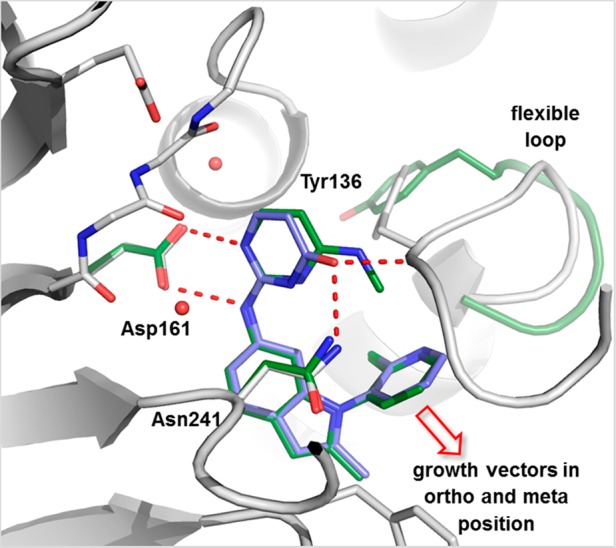
Overlay of X-ray cocrystal structures of Dot1L with 6 (ligand green, parts of the protein green) and 7 (ligand blue, protein gray).23 Amino acid side chains engaging in key interactions with the ligands are illustrated as sticks; polar contacts are highlighted as dotted red lines. Growth vectors (solid red arrow) open in ortho- and meta-positions for 6, but blocked for 7 due to tighter engagement of the flexible loop.
Both growth vectors, extruding from either the ortho- or the meta-position, were explored for 6. Guided by above-mentioned protein-structural considerations, the direct attachment of a pyridone in meta-position, leading to compound 10 (Figure 6), turned out to have a beneficial impact on potency (IC50 = 10 nM). Growing 6 from the ortho-position was best achieved with an aryloxy group while eliminating the Cl substituent in the “other” ortho-position, as, e.g., in compound 11 (IC50 = 150 nM) (Figure 6).
Figure 6.

Optimization of 6 by growing from the meta-position, leading to 10, or from the ortho-position, leading to 11.
Analysis of the X-ray cocrystal structures of 10 and 11 bound to Dot1L revealed some noteworthy aspects (Figure 7, PDB codes 5dsx and 5dt2): both compounds stack tightly with Phe243 and engage the flexible loop through their diaryl and diaryl ether groups, respectively. Furthermore, the pyridone carbonyl of 10 forms a hydrogen bond to the side-chain NH2 of Asn241. The N-phenyl group of 11 moves deep into the hydrophobic pocket, taking the place of the o-Cl substituent of 10.
Figure 7.
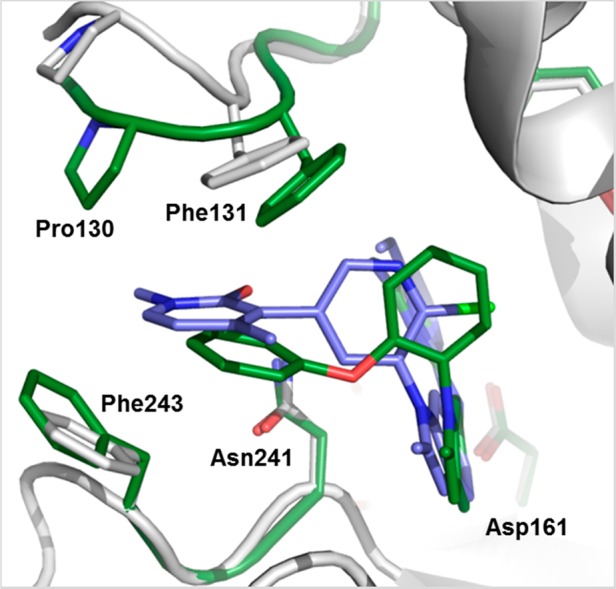
Overlay of ligands 10 (ligand blue, protein gray) (PDB code 5dsx) and 11 (ligand green, parts of the protein green) (PDB code 5dt2) bound to Dot1L based on X-ray cocrystal structures. Both aromatic extensions engage Phe243 and the Pro130-Phe131 motif of the flexible loop.
Chemistry optimization continued on both structural subtypes exemplified by compounds 10 (“meta”) and 11 (“ortho”). A number of opportunities were discovered to further improve potency. For example, the 2-methylindole core of 10 can be replaced by a 2-chloro benzothiophene core, leading to the highly potent Dot1L inhibitor 12 (IC50 = 1.4 nM) (Table 1). Along different lines, the aromatic stacking capacity of 11 can be dramatically improved by substituting the outer phenyl group of 11 by an aza-benzimidazole group, resulting in 400-fold more potent compound 13 (IC50 = 0.4 nM). Both compounds 12 and 13 are characterized by long on-target residence times as assessed by surface plasmon resonance experiments (τ = 43 min for 12 and >240 min for 13 (the detection limit of our internal SPR assay). Most importantly, 12 and 13 potently suppress H3K79 dimethylation (IC50 = 23 and 16 nM, respectively), the direct product of the Dot1L-catalyzed reaction, as well as the activity of the HoxA9 promoter (IC50 = 384 and 340 nM, respectively) in cellular systems (Table 1). We speculate that the significantly higher SAM concentration in cells (∼200 μM)24 compared to our biochemical assay (200 nM) is contributing to the considerable cell-enzyme IC50 shift. Finally, both compounds effectively inhibit proliferation of the human MLL-rearranged leukemia cell line MV4-11 carrying the oncogenic MLL-AF4 fusion (IC50 = 85 and 128 nM, respectively) (Table 1). Importantly, 12 and 13 display a favorable selectivity profile against a panel of 22 PKMTs and PRMTs with no inhibitory activity up to 50 μM.
Table 1. Biochemical, Biophysical, and Cellular Characterization of Lead Compounds 12 and 13a.

| 12 | 13 | |
|---|---|---|
| IC50 (Dot1L SPA) [nM] | 1.4 | 0.4 |
| Ki (Dot1L SPA) [nM] | 0.36 | 0.08 |
| τ (Dot1L SPR) [min] | 43 | >240 |
| IC50 (HeLa, H3K79me2 ELISA) [nM] | 23 | 16 |
| IC50 (Molm-13, HoxA9 RGA) [nM] | 384 | 340 |
| IC50 (MV4-11, prolif.) [nM] | 85 | 128 |
All data are the results of at least two assay runs with the mean value reported. The coefficient of variation was less than 60% in all cases. Biochemical IC50 values were determined at KM for SAM. Ki values were determined by applying the Morrison tight binding model (Supporting Information).
By virtue of its in vitro potency and selectivity as well as good molecular properties (e.g., improved permeability vs 3, Supporting Information), 12 was evaluated in PK experiments in rats (Table 2). After intravenous bolus administration (1 mg/kg), the compound showed high total blood clearance, high volume of distribution, and a moderate half-life. After oral administration in suspension (15 mg/kg), blood levels were detected up to 24 h. The maximum total concentration (225 nM) was reached after 3.5 h; total exposure was 2370 nM·h. Oral bioavailability at 15 mg/kg was 40%.
Table 2. Pharmacokinetic Parameters of 12 in Male Sprague Dawley Rats Following IV and PO Dosing.
| 1 mg/kg iva | 15 mg/kg pob | |
|---|---|---|
| Cl [mL/min/kg] | 84 | NA |
| Vd [L/kg] | 10 | NA |
| t1/2 [h] | 1.7 | NA |
| AUC0-inf [nM·h] | 396 | 2370 |
| Cmax [nM] | NA | 225 |
| BAV [%] | NA | 40 |
NMP/PEG200 (30:70).
MEPC5/Water (10:90).
Compound 12 can be conveniently prepared from commercially available 5-nitrobenzo[b]thiophene-2-carboxylic acid 14 (Scheme 1). Thermal decarboxylation, followed by bromination and a lithiation-chlorination sequence, yielded intermediate 15. Nitro reduction to 16 and nucleophilic aromatic substitution with chloro-pyrimidine 24 delivered key intermediate 17, suitable for late state derivatization in the 3-position of the benzothiophene core. Suzuki coupling of 17 with the customized boronic ester 20 produced lead compound 12 in a convergent fashion (Scheme 1).
Scheme 1. Synthetic Route to Compound 12.

Reagents and conditions: (a) Cu, quinoline, 2 h, 150 °C, 81%; (b) Br2, CHCl3, 30 h, 60 °C, 93%; (c) NCS, TMPMgCl·LiCl, THF, toluene, −75 °C, 80%; (d) RaNi, H2, EtOH, rt, 96%; (e) 24, p-TsOH·H2O, DMF, 15 h, 80 °C, 66%; (f) 20, Na2CO3 2 M aq, Pd(amphos)Cl2, MeCN, 30 min, 80 °C, 25%; (g) 22, Na2CO3 2 M aq, PdCl2(dppf)·DCM, DME, 80 °C, 68%; (h) NaNO2, KI, HCl 4 M, H2O, MeCN, 5 °C to rt, 36%; (i) Bis(pinacolato)diboron, KOAc, PdCl2(dppf)·DCM, dioxane, 10 h, 110 °C, 49%; (j) Bis(pinacolato)diboron, KOAc, PdCl2(dppf)·DCM, dioxane, 3 h, 100 °C, 80%; (k) Boc2O, DMAP, DCM, rt, 98%.
In conclusion, we have discovered potent, selective, and orally bioavailable Dot1L inhibitors that have a novel binding mode and are structurally unique. Future work on further improving potency and in vivo exposure based on lead compounds 12 and 13 is warranted.
Acknowledgments
We are grateful to our medicinal chemistry colleagues Ross Strang, Christian Ragot, Rainer Tschan, Michael Hediger, and Stephan Kläusler for their scientific contributions. We thank Kim Twesten and Anton Kessler for developing and running the high-throughput screening assay, and acknowledge supply of Dot1L enzyme and nucleosome substrate by the laboratories of Lukas Leder, Dirk Erdmann, and Kehao Zhao, respectively. Jidong Zhu is thanked for valuable and inspiring discussions on target biology, Steffen Renner for in silico hit list analysis, and Justin Gu for verification of screening hits in an orthogonal LC–MS assay. We thank Paul Westwood, Kerstin Pollehn, Shin Numao, Claudio Thoma, and Andreas Theuer for developing and supporting project-related biophysical, biochemical, and cellular assays. The authors would like to acknowledge Elke Koch, Céline Be, Aurelie Winterhalter, Aude Izaac, Julia Klopp, and Patrick Graff for their contributions to the protein structural work. The crystallographic experiments were performed on the X10SA beamline at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland, supported by the team of Expose GmbH. We thank Sandrine Desrayaud and her team for performing the rat PK studies as well as Bernard Faller and his team for contributing to the in vitro ADME studies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00167.
Synthetic procedures, compound characterization, and assay protocols (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): Authors are shareholders of Novartis and/or employees of Novartis.
Supplementary Material
References
- Copeland R. A.; Solomon M. E.; Richon V. M. Protein Methyltransferases as a Target Class for Drug Discovery. Nat. Rev. Drug Discovery 2009, 8, 724–732. 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- Copeland R. A. Molecular Pathways: Protein Methyltransferases in Cancer. Clin. Cancer Res. 2013, 19, 6344–6350. 10.1158/1078-0432.CCR-13-0223. [DOI] [PubMed] [Google Scholar]
- Richon V. M.; Johnston D.; Sneeringer C. J.; Jin L.; Majer C. R.; Elliston K.; Jerva L. F.; Scott M. P.; Copeland R. A. Chemogenetic Analysis of Human Protein Methyltransferases. Chem. Biol. Drug Des. 2011, 78, 199–210. 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- Hamamoto R.; Saloura V.; Nakamura Y. Critical Roles of Non-Histone Protein Lysine Methylation in Human Tumorigenesis. Nat. Rev. Cancer 2015, 15, 110–124. 10.1038/nrc3884. [DOI] [PubMed] [Google Scholar]
- Min J.; Feng Q.; Li Z.; Zhang Y.; Xu R.-M. Structure of the Catalytic Domain of Human Dot1L, a Non-SET Domain Nucleosomal Histone Methyltransferase. Cell 2003, 112, 711–723. 10.1016/S0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- Nguyen A. T.; Zhang Y. The Diverse Functions of Dot1 and H3K79 Methylation. Genes Dev. 2011, 25, 1345–1358. 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger D. J.; Lefterova M. I.; Ying L.; Stonestrom A. J.; Schupp M.; Zhuo D.; Vakoc A. L.; Kim J.-E.; Chen J.; Lazar M. A.; Blobel G. A.; Vakoc C. R. Dot1L/KMT4 Recruitment and H3K79 Methylation Are Ubiquitously Coupled with Gene Transcription in Mammalian Cells. Mol. Cell. Biol. 2008, 28, 2825–2839. 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A.; Cuddapah S.; Cui K.; Roh T.-Y.; Schones D. E.; Wang Z.; Wei G.; Chepelev I.; Zhao K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Jo S. Y.; Granowicz E. M.; Maillard I.; Thomas D.; Hess J. L. Requirement for Dot1L in Murine Postnatal Hematopoiesis and Leukemogenesis by MLL Translocation. Blood 2011, 117, 4759–4768. 10.1182/blood-2010-12-327668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Yang Y.; Ortega M. M.; Copeland J. N.; Zhang M.; Jacob J. B.; Fields T. A.; Vivian J. L.; Fields P. E. Early Mammalian Erythropoiesis Requires the Dot1L Methyltransferase. Blood 2010, 116, 4483–4491. 10.1182/blood-2010-03-276501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt K. M.; Zhu N.; Sinha A. U.; Vempati S.; Faber J.; Krivtsov A. V.; Feng Z.; Punt N.; Daigle A.; Bullinger L.; Pollock R. M.; Richon V. M.; Kung A. L.; Armstrong S. A. MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by Dot1L. Cancer Cell 2011, 20, 66–78. 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slany R. K. The Molecular Biology of Mixed Lineage Leukemia. Haematologica 2009, 94, 984–993. 10.3324/haematol.2008.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Majer C. R.; Sneeringer C. J.; Song J.; Johnston L. D.; Scott M. P.; Smith J. J.; Xiao Y.; Jin L.; Kuntz K. W.; Chesworth R.; Moyer M. P.; Bernt K. M.; Tseng J. C.; Kung A. L.; Armstrong S. A.; Copeland R. A.; Richon V. M.; Pollock R. M. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule Dot1L Inhibitor. Cancer Cell 2011, 20, 53–65. 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Basavapathruni A.; Jin L.; Boriack-Sjodin P. A.; Allain C. J.; Klaus C. R.; Raimondi A.; Scott M. P.; Waters N. J.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M. Potent Inhibition of Dot1L as Treatment of MLL-Fusion Leukemia. Blood 2013, 122, 1017–1025. 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavapathruni A.; Olhava E. J.; Daigle S. R.; Therkelsen C. A.; Jin L.; Boriack-Sjodin P. A.; Allain C. J.; Klaus C. R.; Raimondi A.; Scott M. P.; Dovletoglou A.; Richon V. M.; Pollock R. M.; Copeland R. A.; Moyer M. P.; Chesworth R.; Pearson P. G.; Waters N. J. Nonclinical Pharmacokinetics and Metabolism of EPZ-5676, a Novel Dot1L Histone Methyltransferase Inhibitor. Biopharm. Drug Dispos. 2014, 35, 237–252. 10.1002/bdd.1889. [DOI] [PubMed] [Google Scholar]
- Yu W.; Chory E. J.; Wernimont A. K.; Tempel W.; Scopton A.; Federation A.; Marineau J. J.; Qi J.; Barsyte-Lovejoy D.; Yi J.; Marcellus R.; Iacob R. E.; Engen J. R.; Griffin C.; Aman A.; Wienholds E.; Li F.; Pineda J.; Estiu G.; Shatseva T.; Hajian T.; Al-Awar R.; Dick J. E.; Vedadi M.; Brown P. J.; Arrowsmith C. H.; Bradner J. E.; Schapira M. Catalytic Site Remodelling of the Dot1L Methyltransferase by Selective Inhibitors. Nat. Commun. 2012, 3, 1288–1298. 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- Anglin J. L.; Deng L.; Yao Y.; Cai G.; Liu Z.; Jiang H.; Cheng G.; Chen P.; Dong S.; Song Y. Synthesis and Structure-Activity Relationship Investigation of Adenosine-Containing Inhibitors of Histone Methyltransferase Dot1L. J. Med. Chem. 2012, 55, 8066–8074. 10.1021/jm300917h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y.; Chen P.; Diao J.; Cheng G.; Deng L.; Anglin J. L.; Prasad B. V. V; Song Y. Selective Inhibitors of Histone Methyltransferase Dot1L: Design, Synthesis, and Crystallographic Studies. J. Am. Chem. Soc. 2011, 133, 16746–16749. 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Zhang L.; Yao Y.; Wang C.; Redell M. S.; Dong S.; Song Y. Synthesis, Activity and Metabolic Stability of Non-Ribose Containing Inhibitors of Histone Methyltransferase Dot1L. MedChemComm 2013, 4, 822–826. 10.1039/c3md00021d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáňez G.; McBean J. L.; Astudillo Y. M.; Luo M. An Enzyme-Coupled Ultrasensitive Luminescence Assay for Protein Methyltransferases. Anal. Biochem. 2010, 401, 203–210. 10.1016/j.ab.2010.03.010. [DOI] [PubMed] [Google Scholar]
- A related protein-structural rearrangement of Dot1L is described in a Companion Paper:Scheufler C.; Be C.; Fernandez C.; Moebitz H.; Ragot C.; Beyer K.; Gaul C.; Stauffer F.. Optimization of a Fragment-Based Screening Hit toward Potent DOT1L Inhibitors Interacting in an Induced Binding Pocket. ACS Med. Chem. Lett. 2016, 10.1021/acsmedchemlett.6b00167. Companion Paper. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglin J. L.; Song Y. A. Medicinal Chemistry Perspective for Targeting Histone H3 Lysine-79 Methyltransferase Dot1L. J. Med. Chem. 2013, 56, 8972–8983. 10.1021/jm4007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C. Personal communication.
- Bennett B. D.; Kimball E. H.; Gao M.; Osterhout R.; an Dien S. J.; Rabinowitz J. D. Absolute Metabolite Concentrations and Implied Enzyme Active Site Occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.5.0.4; Schrödinger, LLC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.