

ABSTRACT
Peas carrying the cyv1 recessive resistance gene are resistant to clover yellow vein virus (ClYVV) isolates No.30 (Cl-No.30) and 90-1 (Cl-90-1) but can be infected by a derivative of Cl-90-1 (Cl-90-1 Br2). The main determinant for the breaking of cyv1 resistance by Cl-90-1 Br2 is P3N-PIPO produced from the P3 gene via transcriptional slippage, and the higher level of P3N-PIPO produced by Cl-90-1 Br2 than by Cl-No.30 contributes to the breaking of resistance. Here we show that P3N-PIPO is also a major virulence determinant in susceptible peas that possess another resistance gene, Cyn1, which does not inhibit systemic infection with ClYVV but causes hypersensitive reaction-like lethal systemic cell death. We previously assumed that the susceptible pea cultivar PI 226564 has a weak allele of Cyn1. Cl-No.30 did not induce cell death, but Cl-90-1 Br2 killed the plants. Our results suggest that P3N-PIPO is recognized by Cyn1 and induces cell death. Unexpectedly, heterologously strongly expressed P3N-PIPO of Cl-No.30 appears to be recognized by Cyn1 in PI 226564. The level of P3N-PIPO accumulation from the P3 gene of Cl-No.30 was significantly lower than that of Cl-90-1 Br2 in a Nicotiana benthamiana transient assay. Therefore, Cyn1-mediated cell death also appears to be determined by the level of P3N-PIPO. The more efficiently a ClYVV isolate broke cyv1 resistance, the more it induced cell death systemically (resulting in a loss of the environment for virus accumulation) in susceptible peas carrying Cyn1, suggesting that antagonistic pleiotropy of P3N-PIPO controls the resistance breaking of ClYVV.
IMPORTANCE Control of plant viral disease has relied on the use of resistant cultivars; however, emerging mutant viruses have broken many types of resistance. Recently, we revealed that Cl-90-1 Br2 breaks the recessive resistance conferred by cyv1, mainly by accumulating a higher level of P3N-PIPO than that of the nonbreaking isolate Cl-No.30. Here we show that a susceptible pea line recognized the increased amount of P3N-PIPO produced by Cl-90-1 Br2 and activated the salicylic acid-mediated defense pathway, inducing lethal systemic cell death. We found a gradation of virulence among ClYVV isolates in a cyv1-carrying pea line and two susceptible pea lines. This study suggests a trade-off between breaking of recessive resistance (cyv1) and host viability; the latter is presumably regulated by the dominant Cyn1 gene, which may impose evolutionary constraints upon P3N-PIPO for overcoming resistance. We propose a working model of the host strategy to sustain the durability of resistance and control fast-evolving viruses.
INTRODUCTION
Host plants protect themselves from virus infection by activating defense systems mediated by immune receptors (e.g., nucleotide-binding-site–leucine-rich-repeat [NB-LRR] proteins) (1). Plants have many NB-LRR immune receptors, each of which recognizes specific viral proteins. The activated immune response is referred to as a hypersensitive response (HR) and is often accompanied by cell death. When an HR is induced, the virus is localized in and around the infection locus. NB-LRR immune receptors are encoded by resistance genes that are genetically dominant.
Another important defense against plant virus infection is genetically recessive resistance (2). The viral life cycle totally relies on host cells, and viruses require host factors in order to multiply within cells and move to neighboring cells. Therefore, the lack of a specific host-coopted factor required for the viral life cycle leads to host resistance against the virus, which would be recessively inherited. Many natural recessive resistance genes against viruses have been identified in diverse crops (2). Extensive studies have been carried out on viruses belonging to the Potyvirus genus, the major genus of the Potyviridae family, which is one of the two largest plant virus genera and is found in most climatic regions worldwide (3). These viruses infect a broad range of host plants, including both monocots and dicots. They cause considerable crop damage, resulting in severe economic losses. Most of the recessive resistance genes against members of the genus Potyvirus have been identified as encoding eukaryotic initiation factors such as eIF4E. Host eIF4E binds the viral VPg protein that is covalently attached to the 5′ end of the viral genomic RNA, and the complex initiates the translation of viral proteins (4). For example, cyv2 in pea confers recessive resistance to clover yellow vein virus (ClYVV), which causes severe damage to important legume crops, including French bean, broad bean, and pea. A previous study showed that the resistance to ClYVV conferred by cyv2 is mediated by eIF4E (5).
There is another recessive gene, cyv1, that confers resistance to ClYVV in pea (6). ClYVV isolate No.30 (Cl-No.30) cannot infect pea cultivar PI 429853 carrying cyv1, but ClYVV isolate 90-1 Br (Cl-90-1 Br2) can infect PI 429853 systemically (7) (Table 1). Our previous analysis revealed that P3N-PIPO, which consists of the N-terminal amino acids of P3 followed by a small peptide called PIPO, encoded in the +2 reading frame (8–10), is a major determinant for breaking of cyv1 resistance (7). We suggested that the higher level of accumulation of P3N-PIPO in Cl-90-1 Br2 than in Cl-No.30 contributes to the breaking of resistance (7). P3N-PIPO has an essential role in virus cell-to-cell movement (10). Three independent groups, including our own, recently showed that P3N-PIPO is produced mainly by transcriptional slippage within the P3 gene (11–13).
TABLE 1.
Symptoms induced by ClYVV, BYMV, and WClMV expressing P3N-PIPO in peaa
| Pea cultivar (genotype) | Symptom induced by: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| ClYVV |
BYMV CS | WClMVb |
|||||||
| I89-1 | 90-1 Br2 | 90-1 | No.30 | ClYVV P3N-PIPO of isolate: |
BYMV CS P3N-PIPO | GFP | |||
| 90-1 Br2 | No.30 | ||||||||
| PI 429853 (Cyn1c cyv1) | SCDd | No/CSd | No infectiond | No infectiond | No infection | Cell death | Cell death | Delayed cell death | No |
| PI 226564 (Cyne) | LSCD | LSCD | SCD | M/CSf,g | M/CSf | Cell death | Cell death | No | No |
| PI 118501 (Cyn1) | LSCD | LSCD | LSCD | LSCDf,g | M/CSg | Cell death | Cell death | Delayed cell death | No |
LSCD, lethal systemic cell death; SCD, systemic cell death; M, mosaic; CS, chlorotic spot; No, no symptoms.
Cell death induction in inoculated leaves was observed until 8 days after inoculation.
Assumed to carry Cyn1.
See reference 7.
Putative weak Cyn1 allele (20).
See reference 20.
See reference 21.
Plant viruses continually evolve and gain virulence, which enables them to inhibit or escape plant defense/resistance systems. Although higher virulence is favorable for virus adaptation, extremely high virulence (i.e., induction of lethal systemic cell death) seems to be a disadvantage for virus survival because the virus loses an environment in which to propagate if the host cells are dead. Several examples of excessively high virulence have been reported. Soybean mosaic virus (SMV) strain G7 (SMV-G7) induces cell death systemically and kills the host plant (14). Turnip mosaic virus (TuMV) isolate TuR1 induces lethal systemic cell death in Arabidopsis thaliana accession Landsberg erecta (Ler) (15). It was shown previously that systemic cell death caused by SMV or TuMV is a form of an HR that is regulated by Rsv1 or TuNI in soybean or A. thaliana Ler, respectively (16, 17). It was suggested that both Rsv1 and TuNI encode NB-LRR resistance proteins (18, 19). These reports and others led us to propose that extremely high virulence, an unfavorable state for the virus, is controlled genetically by host plants.
Extremely high virulence of ClYVV has been observed for many pea cultivars: ClYVV systemically induces cell death, resulting in plant death within 2 weeks when used to infect young plants (20–22) (Table 1). We previously reported that cell death induced by Cl-No.30 in pea cultivar PI 118501 is a form of an HR-like response accompanied by the activation of the salicylic acid (SA) pathway, which is one of the hallmarks of an HR (20). However, Cl-No.30 infection is not localized and induces cell death systemically. A genetic study in pea suggested that this cell death is controlled by a single dominant locus called Cyn1 (21). Cyn1 has been suggested to be an NB-LRR gene whose product recognizes Cl-No.30 and induces an HR associated with cell death (20, 21).
We previously revealed that Cl-90-1 Br2, a mutant isolate that originated from Cl-90-1, breaks cyv1 recessive resistance in pea (7) (Table 1). In the present study, we revealed that Cl-90-1 Br2 induced lethal systemic cell death and found that P3N-PIPO, but not P3, was a major determinant of the induction of cell death in pea cultivar PI 226564. This was discovered by infection of PI 226564 plants with chimeric ClYVVs and by transient assays using white clover mosaic virus (WClMV) vectors. We showed that P3N-PIPO is quantitatively and/or qualitatively involved in cell death induction in pea by using chimeric and mutant ClYVVs and by transient assays in Nicotiana benthamiana. Chimeric P3N-PIPO expression analysis by using WClMV indicated that the PIPO peptide was not the sole determinant of cell death induction. Finally, we showed a consistent gradation of virulence among ClYVV isolates in cyv1-carrying peas (recessive resistance) and two susceptible pea lines. This study, combined with data from our previous studies (7), shows that P3N-PIPO is involved in both breaking of recessive resistance and disease expression. We suggest that the pleiotropic effects of P3N-PIPO in determining ClYVV virulence in pea result in a trade-off between the breaking of recessive resistance (cyv1) and maintenance of host viability, which is presumably controlled by a Cyn1-mediated immune response.
MATERIALS AND METHODS
Preparation of plasmid constructs and infectious clones.
Sequences of the primers used for vector construction are available upon request. The construction of ClYVV chimeric clones Cl-P1HC, Cl-BB, Cl-NS, Cl-SB, Cl-RB, and Cl-RBM28R with GFP (RB/P3 and P3N-PIPOM28R in reference 7) was described previously (7). To make Cl-RB without GFP, the SalI-BamHI fragment of Cl-BB with GFP was used to replace that of Cl-RB with GFP. To make Cl-RB+P1HC without GFP (Fig. 1B), the SalI-BamHI fragment of Cl-BB with GFP was used to replace that of Cl-RB+P1HC with GFP; Cl-RB+P1HC with GFP was made by replacing the BglII (HC-Pro)-BglII (P3) fragment of Cl-BB carrying GFP with that of Cl-P1HC with GFP. To make Cl-RB+NS without GFP (Fig. 1B), the EcoRV-BglII (HC-Pro) fragment of pClYVV-Pst/CP (23) was used to replace that of Cl-RB+NS with GFP; Cl-RB+NS with GFP was made by replacing the BglII (HC-Pro)-NheI (P3) fragment of Cl-RB containing HC-Pro of Cl-No.30 and P3 of Cl-90-1 Br2 with that of Cl-NS with GFP. To replace the BglII-NheI fragment, two NheI sites within the BglII-NheI fragment of Cl-90-1 Br2 were disrupted by site-directed mutagenesis to generate BgNhΔNh. BgNhΔNh was amplified with primers 167 and 196 by using two overlapping fragments amplified with primer sets 167/201 and 202/196 as the templates. To make Cl-RB+SB without GFP (Fig. 1B), the SalI-BamHI fragment of Cl-SB with GFP was used to replace that of Cl-RB without GFP. To make Cl-90-1 Br2-P3BNo.30 without GFP (Fig. 1B), the SalI-BamHI fragment of Cl-SB with GFP was used to replace that of Cl-P1HC with GFP, thus producing Cl-P1HC+SB without GFP, and the NheI-SalI fragment of Cl-NS with GFP was then used to replace that of Cl-P1HC+SB without GFP to produce the final vector. Cl-90-1 Br2-P3BNo.30 without GFP contains the region from the NheI site to the BglII site at the 3′ end of the P3B region, but the sequence of the region of Cl-90-1 Br2-P3BNo.30 without GFP is identical to that of Cl-No.30 except for a synonymous mutation (G [Cl-No.30] to A [Cl-90-1 Br2-P3BNo.30]) at nucleotide position 801 of the P3 gene (7).
FIG 1.

Mapping of the virulence determinant of Cl-90-1 Br2 in susceptible pea cultivar PI 226564. (Aa to k) Cl-90-1 Br2 or Cl-No.30 was inoculated, and symptoms were observed in inoculated leaves (a to c), upper uninoculated leaves (d to i), and the whole plant (j and k). Photographs were taken at 14 dpi (a to i), 17 dpi (j), and 21 dpi (k). (B) Schematic representations of chimeric viruses constructed from Cl-90-1 Br2 (yellow) and Cl-No.30 (blue). Symptom severity in PI 226564 is indicated by the number of + symbols. We also show the infection profile for PI 429853 carrying the recessive gene cyv1, which was reported previously (7). Black triangles indicate positions of the GFP insertion. (Ca to c) A series of chimeric viruses tagged with GFP were inoculated, and symptoms were monitored. Photographs were taken at 12 dpi (a), 17 dpi (b), and 15 dpi (c) (except for Cl-90-1 Br2, at 12 dpi). (Da and b) Effect of a GFP insertion into ClYVV on symptom development (a) and virus accumulation (b). (a) Cl-No.30 without GFP, with GFP between P1 and HC-Pro, or with GFP between NIb and CP was inoculated onto PI 226564 plants, and their symptoms were compared. (b) Viral CP accumulation levels were compared by Western blotting using antiserum against Cl-No.30 CP. The ribulose 1,5-bisphosphate carboxylase/oxygenase large subunit (rbc-L) band from the SDS-PAGE gel stained with Coomassie brilliant blue is shown as a loading control. (E) Comparison of symptoms induced by Cl-90-1 Br2, Cl-No.30, and Cl-RB without GFP at 21 dpi. (Fa and b) Virulence of Cl-90-1 Br2-P3BNo.30, a Cl-90-1 Br2 mutant in which the P3B region of the 90-1 Br2 isolate was replaced with the corresponding region of Cl-No. 30. None of the viruses illustrated had a GFP insertion. The symptoms in a whole plant (a) and an upper uninoculated leaf (b) are shown. Photographs were taken at 19 dpi (a) and 13 dpi (b). (G) Mapping of virulence determinants outside the P3B region. None of the chimeric viruses had a GFP insertion. The photograph was taken at 21 dpi.
The WClMV vectors pWCl/P3N-PIPO-RB, pWCl/P3-RB, pWCl/P3ΔPIPO-RB, and pWCl/GFP were constructed previously (24, 25). In this study, we constructed pWCl/P3-No.30, pWCl/P3N-PIPO-No.30, pWCl/P3ΔPIPO-No.30, pWCl/P3N-PIPO-CS, pWCl/P3NRB-PIPOCS, and pWCl/P3NCS-PIPORB. For the construction of pWCl/P3-No.30, pWCl/P3N-PIPO-No.30, and pWCl/P3ΔPIPO-No.30, pClYVV/C3-S65T (26) was used as a template. The P3 fragment was obtained by PCR with primer set 3406/3412. The P3N-PIPO fragment was amplified by using primer set 3406/3415 from the mixture of two PCR products amplified with primer sets 3406/3653 and 3652/3415. The length of P3N-PIPO carried by Cl-No.30 (647 nucleotides) is shorter than that carried by Cl-RB (692 nucleotides) (7). We cloned P3N-PIPO of Cl-No.30 as a fragment from the 5′ end of P3 to the position corresponding to the stop codon of Cl-RB PIPO (located downstream of the stop codon of the PIPO frame of Cl-No.30). P3N-PIPO has two mutations: (i) a G insertion in the A6 sequence within the G2A6 motif, which is expected to prevent transcriptional slippage or a ribosomal frameshift and translate P3N-PIPO as a zero-frame product, and (ii) a G-to-A substitution that introduces a stop codon in the P3 frame but a silent mutation in the PIPO frame (24). The P3ΔPIPO fragment was amplified by using primer set 3406/3412 from the mixture of two PCR products amplified with primer set 3406/3410 and 3411/3412. P3ΔPIPO has a stop codon in the PIPO frame but a silent mutation in the P3 frame (24). For the construction of pWCl/P3N-PIPO-CS, a cDNA clone constructed from bean yellow mosaic virus (BYMV) isolate CS (BY-CS) (pBY-CS) (27) was used as a template. The P3N-PIPO-CS fragment was amplified by using primer set 3621/3622 from the mixture of two PCR products amplified with primer sets 3621/3669 and 3668/3622. We cloned P3N-PIPO of BY-CS as a fragment from the 5′ end to the 3′ end of the P3 gene carrying the same mutations introduced into P3N-PIPO-RB for enabling P3N-PIPO to be translated as a zero-frame product. For the construction of pWCl/P3NRB-PIPOCS and pWCl/P3NCS-PIPORB, pCl-RB (7) and pBY-CS (27) were used as the templates. The P3NRB-PIPOCS fragment was amplified by using primer set 3945/3625 from the mixture of P3N of Cl-RB and PIPO of BY-CS amplified with primer sets 3945/3885 and 3955/3625, respectively. The P3NCS-PIPORB fragment was amplified by using primer set 3946/3409 from the mixture of P3N of BY-CS and PIPO of Cl-RB amplified with primer sets 3946/3887 and 3954/3409, respectively. For the construction of P3NRB-PIPOCS and P3NCS-PIPORB, we cloned a fragment from the 5′ end of P3 to the stop codon of the PIPO frame. For P3-No.30, P3N-PIPO-No.30, P3ΔPIPO-No.30, and P3N-PIPO-CS, the cDNA fragments were introduced into the pGEM-T Easy plasmid (Promega, Fitchburg, WI), digested with SpeI and XhoI, and inserted into the WClMV vector (25) cut with the same restriction enzymes. For P3NCS-PIPORB, the cDNA fragment introduced into the pGEM-T Easy plasmid was digested with NheI and XhoI and inserted into the WClMV vector cut with the same restriction enzymes. For P3NRB-PIPOCS, the cDNA fragment introduced into the pGEM-T Easy plasmid was cut with SacII, and the 5′ and 3′ ends of the plasmid were blunted by using T4 DNA polymerase (TaKaRa Bio, Kusatsu, Japan). The blunted fragments were digested with XhoI and inserted into the WClMV vector cut with SmaI and XhoI.
The pTA/RB-P3(PIPO:FLAG−1), pTA/No.30-P3(PIPO:FLAG−1), pTA/RB-P3N-PIPO:FLAGmk, and pTA/No.30-P3N-PIPO:FLAGmk constructs were previously described (13).
All viral fragments were amplified by using KOD-plus2 Neo DNA polymerase (Toyobo, Osaka, Japan) according to the manufacturer's instructions, and their nucleotide sequences were confirmed.
Plant growth conditions and viral infection.
Pea (Pisum sativum), broad bean (Vicia faba), and N. benthamiana were cultivated in a growth chamber or growth room at 21°C to 23°C with a 16-h photoperiod. Viral inocula were prepared as described previously (20). For ClYVV, broad bean was inoculated with each infectious cDNA by using particle bombardment. For WClMV, 1 μg of each WClMV plasmid was mechanically inoculated onto a susceptible pea line, PI 250438. The upper symptomatic leaves were harvested and ground in an inoculation buffer (0.1 M phosphate buffer [pH 7.0] and 1% 2-mercaptoethanol). The crude sap was mechanically inoculated onto the second and/or third leaves of 2-week-old pea plants. At the same time, all plants were inoculated with inoculation buffer alone as a negative control (mock inoculation).
Sequence alignment.
P3N-PIPO amino acid sequences (ClYVV, BYMV, and pea seed-borne mosaic virus [PSbMV]) were aligned by using MUSCLE (3.8) (see http://www.ebi.ac.uk/Tools/msa/muscle/) (28). The amino acid sequences of P3N-PIPO were obtained by translating the sequences from the 5′ end of P3 to the stop codon of P3N-PIPO after introducing an A residue into the A6–7 region in the G1–2A6–7 motif of each virus to shift the reading frame.
RNA extraction, reverse transcription, and real-time PCR.
Pea leaves were homogenized in liquid nitrogen, and total RNA was isolated by using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. Each RNA sample was treated with RNase-free DNase I (Roche Diagnostics, Basel, Switzerland), and 1 μg of total RNA was reverse transcribed by using ReverTraAce (Toyobo). The reaction mixture (20 μl) contained 100 U of ReverTraAce, 1 mM deoxynucleoside triphosphate (dNTP), 25 pmol random 9-mers, and 1 to 2 μg total RNA in 1× buffer. Samples were incubated first at 30°C for 10 min, then at 42°C for 30 min, and finally at 99°C for 5 min. Real-time PCR (RT-PCR) was performed by using the DNA Engine Opticon 2 system (Bio-Rad Laboratories, Hercules, CA) as previously described (20). The reaction mixture (25 μl) contained 0.625 U of Ex Taq (TaKaRa), Ex Taq buffer, 0.2 mM dNTP, 0.2 μM (each) forward and reverse primers, SYBR green (30,000× dilution) (Thermo Fisher Scientific), and cDNA obtained by reverse transcribing 12.5 ng of total RNA. Samples were incubated for 5 min at 95°C, followed by 40 cycles of 95°C for 10 s, 53°C for SA-CHI (GenBank accession number L37876) or 55°C for HSR203J (accession number AB026296) for 30 s, and 72°C for 20 s. Transcript levels were normalized to that of the 18S rRNA gene (GenBank accession number U43011), and means and standard deviations were calculated. The primers used for real-time PCR were as follows: SA-CHI-F and SA-CHI-R for SA-CHI, HSR203J-F and HSR203J-R for HSR203J, and 18S rRNA-F and 18S rRNA-R for the 18S rRNA gene.
N. benthamiana leaves were homogenized in liquid nitrogen, and total RNA was isolated by the AGPC (acid guanidinium thiocyanate–phenol-chloroform) extraction method (29), followed by purification with a FARB minicolumn (Favorgen Biotech Corp., Ping-Tung, Taiwan). Total RNA was digested with Turbo DNase (Thermo Fisher Scientific) and reverse transcribed by using PrimeScript reverse transcriptase (TaKaRa) according to the manufacturer's instructions. Real-time PCR was performed by using the StepOnePlus system (Thermo Fisher Scientific). The reaction mixture (10 μl) contained Kapa SYBR Fast quantitative PCR (qPCR) kit master mix (ABI Prism; Kapa Biosystems, Wilmington, MA), 0.3 μM (each) forward and reverse primers, and cDNA obtained by reverse transcribing 50 ng of total RNA. Samples were incubated for 20 s at 95°C, followed by 40 cycles of 95°C for 3 s and 60°C for 30 s. Transcript levels of P3N-PIPO-FLAG were normalized to that of NbEF1α (GenBank accession number AY206004). The primers used were as follows: GGS4-3FL-F and GGS4-3FL-R for P3N-PIPO-FLAG and NbEF1α-F and NbEF1α-R for NbEF1α. Primers were designed in a region of the P3N-PIPO-FLAG genes of Cl-RB and Cl-No.30 with identical nucleotide sequences (linker and FLAG tag coding sequence).
For virus detection by RT-PCR, we used primer set 3735/3736 for Cl-I89-1 and Cl-90-1 Br2 and primer set 3908/3909 for BY-CS. PCR was done by using KOD-FX DNA polymerase (Toyobo) according to the manufacturer's instructions.
Sequences of the primers are available upon request.
Agrobacterium-mediated transient expression.
Agrobacterium-mediated transient expression was conducted as described previously (30). Agrobacterium tumefaciens LBA4404 cells transformed with each construct were suspended in MES buffer [10 mM 2-(N-morpholino)ethanesulfonic acid, 10 mM MgCl2 (pH 5.7)], and the suspensions were adjusted to an optical density at 600 nm (OD600) of 1.0. Acetosyringone was added to the suspensions at a final concentration of 200 μM, followed by incubation at room temperature for 2 to 4 h. Each suspension was infiltrated into N. benthamiana leaves by using needleless syringes. Leaves were sprayed with a 30 μM dexamethasone solution containing 0.01% Tween 20 24 h after agroinfiltration (31). Leaves were collected 24 h after dexamethasone treatment and used for Western blot and real-time PCR analyses.
Western blot analysis.
Western blots were conducted as described previously (30, 32). Proteins were resolved in 12% NuPAGE Bis-Tris gels (Thermo Fisher Scientific) using MES-SDS buffer (FLAG-tagged protein detection) or in 10% SDS-PAGE using Tris-glycine buffer (coat protein [CP] detection [33]), followed by electrotransfer onto a polyvinylidene difluoride (PVDF) membrane. To detect the FLAG-tagged proteins, monoclonal anti-FLAG M2-horseradish peroxidase (HRP) (Sigma-Aldrich Corporation, St. Louis, MO) was used at a 1:5,000 dilution. To detect ClYVV CP, rabbit polyclonal antibody against ClYVV CP was used as the primary antibody, and alkaline phosphatase-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific) was used as the secondary antibody. Chemiluminescence signals were detected with ECL Prime (GE Healthcare, Little Chalfont, United Kingdom) using an LAS-4000 imaging system (GE Healthcare) for FLAG-tagged protein detection or with CDP-Star reagent (New England BioLabs, Ipswich, MA) using an LAS-4000 mini-imaging system (GE Healthcare) for CP detection. As a loading control for the FLAG-tagged protein experiment, membranes after transfer were stained with 0.1% amido black in 45% methanol and 10% acetic acid, followed by destaining in 90% methanol and 2% acetic acid (34).
GFP fluorescence analysis.
Green fluorescent protein (GFP) fluorescence of pea plants infected with GFP-tagged viruses (Cl-No.30/GFP and Cl-RB/GFP) was monitored by using an MVX10 epifluorescence microscope (Olympus Corporation, Tokyo, Japan). The fluorescent area was measured by using the color thresholding tool of ImageJ software (35).
Double-antibody sandwich enzyme-linked immunosorbent assay.
A double-antibody sandwich enzyme-linked immunosorbent assay (DAS-ELISA) was conducted according to methods described in our previous report (32). A single GFP focus derived from the virus was excised from inoculated leaves and used for antigens. We used mouse anti-ClYVV CP IgG as the first antibody and rabbit anti-ClYVV CP as the second antibody. After washing, alkaline phosphatase-conjugated goat anti-rabbit IgG was added, followed by the substrate solution (disodium p-nitrophenyl-phosphate hexahydrate in 10% diethanolamine). The intensity of the signal was measured at an OD405.
Determination of the full-length ORF sequence of Cl-I89-1.
cDNA was synthesized from total RNA isolated from pea leaves infected with Cl-I89-1. The sequence covering the full-length Cl-I89-1 open reading frame (ORF) was amplified with high-fidelity DNA polymerase (KOD-plus2 Neo; Toyobo) into four overlapping fragments by using the following primer sets: 3230/3191, 2978/2493, 2388/2471, and 2470/3229. The four PCR products were directly sequenced by the primer-walking method using primers 157, 2372, 2451, 2464, 2470, 2481, 2491, 2552, 2559, 2621, 3130, 3436, and 3435. Sequences of the primers are available upon request.
Phylogenetic analysis.
Phylogenetic analysis was performed for full-length nucleotide sequences encoding the polyproteins of ClYVV and BYMV. Sequence alignment was conducted by using MUSCLE, and a maximum likelihood tree was inferred by using the MEGA6 package (36). The nucleotide substitution models and rates among sites were general time reversible and gamma distribution. The significance of the nodes was estimated with 1,000 bootstrap replicates.
Nucleotide sequence accession number.
The GenBank/ENA/DDBJ accession number for the full-length ORF sequence of Cl-I89-1 is LC096082.
RESULTS
P3 of Cl-90-1 Br2 is the major virulence determinant in PI 226564.
Cl-90-1 Br2 induced lethal systemic cell death in the pea line PI 226564 (Fig. 1A and Table 1). To identify the virulence determinant of Cl-90-1 Br2 in PI 226564, chimeric viruses were constructed by swapping parts of Cl-90-1 Br2 and Cl-No.30; the latter virus does not induce cell death in PI 226564 (Fig. 1A and Table 1) (20, 21). These chimeric viruses were based on Cl-No.30 infectious cDNA that we previously constructed and developed for use as a gene expression vector (7, 23, 37, 38). The following chimeric viruses tagged with GFP that covered almost all regions of the ClYVV genome were created: Cl-P1HC/GFP, Cl-BB/GFP, Cl-NS/GFP, and Cl-SB/GFP (Fig. 1B) (7). Symptoms indicated that only the BB region of Cl-90-1 Br2 markedly enhanced the virulence of Cl-No.30 (Fig. 1C). Cl-BB/GFP induced cell death in upper uninoculated leaves (Fig. 1C). The P1HC and SB regions of Cl-90-1 Br2 did not enhance Cl-No.30 virulence (Fig. 1C). The NS region slightly enhanced virulence: Cl-NS/GFP occasionally induced cell death associated with yellowing in the upper uninoculated leaves (Fig. 1C). In contrast to Cl-90-1 Br2, Cl-BB/GFP did not kill the plants completely, but a mosaic pattern associated with cell death developed in the upper uninoculated leaves (Fig. 1C).
Further analysis was focused on the virulence enhancement mediated by the Cl-90-1 Br2 BB region, which included ca. 94% of HC-Pro and ca. 79% of P3 from Cl-90-1 Br2. Cl-P1HC/GFP had the full-length HC-Pro gene of Cl-90-1 Br2 but could not enhance virulence (Fig. 1B and C), suggesting that the P3-containing portion of the BB region (designated P3B) (Fig. 1B) was important for high-virulence expression. We constructed Cl-RB/GFP, in which the P3B region of Cl-No.30 was replaced by that of Cl-90-1 Br2 (Fig. 1B). Cl-RB/GFP extensively induced cell death in the upper uninoculated leaves, which was comparable to that induced by Cl-BB/GFP (Fig. 1C).
Like Cl-BB/GFP, Cl-RB/GFP did not kill the plants. To investigate whether the insertion of GFP (which was present in the first set of chimeric constructs tested) attenuated ClYVV virulence, the symptoms induced by Cl-No.30 with GFP (inserted between P1 and HC-Pro or between NIb and CP) were compared with those induced by Cl-No.30 without GFP. The results indicated that the insertion of GFP weakened the symptoms produced by ClYVV (Fig. 1D), although virus accumulation was not visibly different, as measured by Western blotting against CP (Fig. 1D). To investigate whether the weaker virulence of Cl-RB/GFP than of Cl-90-1 Br2 was due to the GFP insertion, we compared the virulence of Cl-RB without GFP with those of Cl-90-1 Br2 (7) and Cl-No.30 without GFP (37). Cl-RB without GFP also induced more severe symptoms than those induced by Cl-No.30 without GFP but did not have the level of virulence of Cl-90-1 Br2 (Fig. 1E). We also examined the reciprocal chimera of Cl-RB, Cl-90-1 Br2-P3BNo.30 without GFP, which contained the P3B region from Cl-No.30 and all other regions from Cl-90-1 Br2 (Fig. 1B). Cl-90-1 Br2-P3BNo.30 without GFP induced yellowing and cell death in the upper uninoculated leaves, but the timing was delayed in comparison with that of Cl-RB without GFP (Fig. 1F). This finding indicated that Cl-90-1 Br2-P3BNo.30 showed less virulence than Cl-RB and more virulence than Cl-No.30. Together, these results indicated that the P3B region was the main determinant of virulence in PI 226564, although regions outside P3B also contributed to virulence expression.
To identify the virulence determinant(s) outside the P3B region, we created chimeric viruses without GFP, each of which has the P1HC, NS, or SB region of Cl-90-1 Br2 in addition to P3B of Cl-90-1 Br2, so that almost all regions of the ClYVV genome were covered (Fig. 1B). All of the chimeric viruses expressed higher virulence than Cl-RB at 21 days postinoculation (dpi) (Fig. 1G); they induced cell death in both inoculated and upper uninoculated leaves. Plants infected with Cl-RB+P1HC, Cl-RB+NS, or Cl-RB+SB were shorter than those infected with Cl-RB; however, none of the chimeric viruses killed the plants completely (Fig. 1G).
Taken together, these data showed that Cl-No.30 carrying Cl-90-1 Br2 regions outside P3B (Cl-P1HC, Cl-NS, Cl-SB, and Cl-90-1 Br2-P3BNo.30) had weaker virulence than Cl-No.30 carrying the P3B region of Cl-90-1 Br2 (Cl-RB) and those in combination with Cl-90-1 Br2 regions (Cl-RB+P1HC, Cl-RB+NS, and Cl-RB+SB). This indicated that the effect of virulence was greatest in exchanging the P3B region, and the effect of virulence enhancement by regions outside P3B was greater in a virus carrying the P3B region of Cl-90-1 Br2 than in a virus carrying the P3B region of Cl-No.30. These observations of symptoms collectively suggested that although regions outside P3 contributed to virulence expression, the P3 gene (P3B region) was the major determinant for inducing lethal systemic cell death in PI 226564.
P3N-PIPO, but not P3, of Cl-RB is responsible for cell death induction in PI 226564.
P3 expresses two mature proteins, P3 and P3N-PIPO (8). To dissect which protein induces cell death, P3, P3N-PIPO, and P3ΔPIPO from Cl-RB were expressed in PI 226564 by WClMV vectors (designated WCl/P3-RB, WCl/P3N-PIPO-RB, and WCl/P3ΔPIPO-RB, respectively) (24). We have previously shown that WClMV can infect PI 226564 but does not induce cell death (25) (Table 1). The P3 construct was expected to produce the P3 protein accompanied by a small amount of the P3N-PIPO protein as a frameshift product (Fig. 2A) (24). P3N-PIPO had mutations enabling it to express P3N-PIPO in the zero frame but not the P3 frame product (Fig. 2A) (24). P3ΔPIPO had a mutation enabling it to produce P3 but not P3N-PIPO (Fig. 2A) (24). We inoculated PI 226564 with the three WClMV vectors and WClMV expressing GFP (WCl/GFP) as a negative control.
FIG 2.
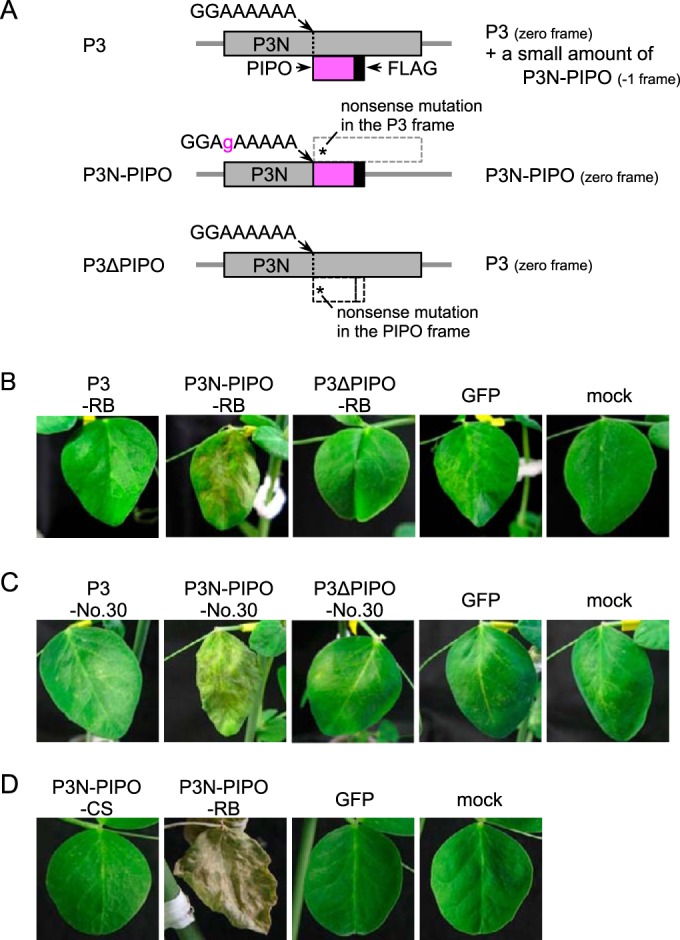
Expression of P3 and/or P3N-PIPO from Cl-RB, Cl-No.30, and BY-CS in PI 226564 plants using a heterologous WClMV vector. (A) Schematic representations of P3, P3N-PIPO, and P3ΔPIPO constructs. The P3 construct was expected to produce P3 accompanied by a small amount of P3N-PIPO as a frameshift product. P3N-PIPO had mutations for expressing P3N-PIPO in the zero frame but no P3. P3ΔPIPO contained a mutation enabling it to produce P3 but not P3N-PIPO. (B and C) P3, P3N-PIPO, and P3ΔPIPO constructs of Cl-RB (B) and Cl-No.30 (C) were expressed by WClMV vectors. The photographs in panels B and C were taken at 5 dpi. (D) P3N-PIPO of BY-CS was expressed by a WClMV vector. The photographs were taken at 14 dpi.
We found that WCl/P3N-PIPO-RB extensively induced cell death along the veins of inoculated leaves at 5 dpi (Fig. 2B and Table 1). In contrast, infection with WCl/P3-RB, WCl/P3ΔPIPO-RB, and WCl/GFP did not induce cell death (Fig. 2B). These results suggested that P3N-PIPO, and not P3, was the factor responsible for inducing cell death in PI 226564. It should be noted that the nucleotide sequence of P3N-PIPO of Cl-RB is the same as that of Cl-90-1 Br2.
P3N-PIPO of Cl-No.30 also induces cell death in PI 226564.
Cl-No.30 does not induce cell death in PI 226564, which suggested that P3N-PIPO of Cl-No.30 would not induce cell death either (20). We inoculated PI 226564 with WClMV carrying P3N-PIPO, P3, or P3ΔPIPO of Cl-No.30 (designated WCl/P3N-PIPO-No.30, WCl/P3-No.30, and WCl/P3ΔPIPO-No.30, respectively). Unexpectedly, WCl/P3N-PIPO-No.30 extensively induced cell death along the veins on the inoculated leaves of PI 226564 plants (Fig. 2C and Table 1). This symptom was comparable to that induced by WCl/P3N-PIPO-RB (Fig. 2B). WCl/P3-No.30, WCl/P3ΔPIPO-No.30, and WCl/GFP did not induce cell death at 5 dpi in PI 226564 (Fig. 2C).
We constructed a WClMV vector that expresses P3N-PIPO from the BYMV CS strain (BY-CS), designated WCl/P3N-PIPO-CS, in order to rule out the possibility that cell death was nonspecifically caused by the overexpression of P3N-PIPO. We expected that P3N-PIPO of BY-CS would not induce cell death because BY-CS has never been reported to induce cell death in pea, including PI 226564 (20, 21). Like ClYVV, however, BY-CS is a member of the genus Potyvirus, and the two viruses are closely related (39). The nucleotide sequence identity of the P3N-PIPO genes between BY-CS and Cl-90-1 Br2 or Cl-No.30 is 64.2% or 61.9%, respectively (see Fig. S1 in the supplemental material). The amino acid sequence identity (similarity) of the P3N-PIPO proteins between BY-CS and Cl-90-1 Br2 or Cl-No.30 is 56.5% (73.8%) or 54.9% (71.3%), respectively (Fig. 3). In PI 226564 plants infected with WCl/P3N-PIPO-CS, cell death was not induced in either inoculated or upper uninoculated leaves, even at 14 dpi (Fig. 2D and Table 1), thus ruling out the possibility that the overexpression of P3N-PIPO nonspecifically induced cell death.
FIG 3.

Multiple-sequence alignment of amino acid sequences of P3N-PIPO. Alignment was performed by using the program MUSCLE (3.8) (http://www.ebi.ac.uk/Tools/msa/muscle/). GenBank accession numbers are NC_001671 for PSbMV-DPD1, AJ252242 for PSbMV-L1, AJ311841 for PSbMV-NEP1, X89997 for PSbMV-NY, LC096082 for ClYVV-I89-1, AB011819 for ClYVV-No.30, HG970870 for ClYVV-CYVV, KF975894 for ClYVV-Gm, AB732962 for ClYVV-90-1 Br2, AB373203 for BYMV-CS, AB439731 for BYMV-90-2, AB439732 for BYMV-92-1, AY192568 for BYMV-GDD, and JN692500 for BYMV-Vfaba2. The amino acid (aa) sequences of P3N-PIPO were obtained by translating the sequences from the 5′ end of P3 to the stop codon of P3N-PIPO after the introduction of an A residue into the A6–7 region in the G1–2A6–7 motif of each virus to shift the reading frame.
P3N-PIPO of Cl-No.30 and Cl-RB, but not that of BY-CS, activates the SA signaling pathway in PI 226564.
One of the possible mechanisms that induces cell death by P3N-PIPO is high-level activation of the SA signaling pathway. We previously showed that the activation of the SA signaling pathway contributes to the induction of systemic cell death by ClYVV in the susceptible pea lines PI 118501 and PI 226564 (20). Therefore, we hypothesized that the expression of Cl-90-1 Br2 P3N-PIPO activated the SA signaling pathway, which led to the induction of systemic cell death in PI 226564.
To test this hypothesis, we analyzed the expression of the SA-responsive chitinase gene (SA-CHI) and an HR-related gene homologous to tobacco HSR203J by real-time PCR. We conducted an expression analysis in leaves of PI 226564 plants inoculated with Cl-90-1 Br2, Cl-No.30, or BY-CS. The expression level of SA-CHI was significantly higher in leaves inoculated with Cl-90-1 Br2 than in leaves inoculated with either Cl-No.30 or BY-CS (Fig. 4A). There were no significant differences among mock, Cl-No.30, and BY-CS inoculations (Fig. 4A). The expression level of HSR203J was also significantly higher in leaves inoculated with Cl-90-1 Br2 than in mock-inoculated leaves (Fig. 4B). These results indicated that Cl-90-1 Br2 infection activated SA and HR-like signaling pathways in PI 226564.
FIG 4.
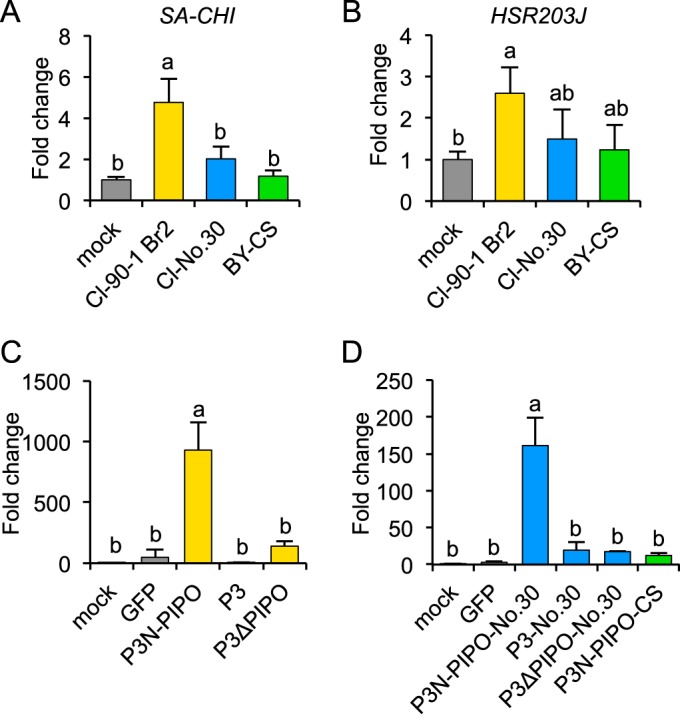
Real-time PCR analysis of defense-associated gene expression levels in susceptible pea cultivar PI 226564. (A and B) SA-responsive (SA-CHI) and HR-associated (HSR203J) gene expression levels in response to Cl-90-1 Br2, Cl-No.30, and BY-CS infections. Total RNA was extracted from leaves (n = 3) inoculated with Cl-90-1 Br2, Cl-No.30, and BY-CS at 6 dpi. cDNA was synthesized from total RNA and used for real-time PCR analysis. (C and D) SA-CHI expression in response to P3N-PIPO expression via WClMV. WClMV vectors carrying P3N-PIPO, P3, or P3ΔPIPO of Cl-RB (C), the corresponding genes from Cl-No.30 (D), P3N-PIPO from BY-CS (D), and GFP (C and D) as a negative control were inoculated onto PI 226564 plants. Total RNA was extracted from the inoculated leaves (n = 3) at 4 dpi and used to synthesize cDNA for real-time PCR analysis. Expression levels of SA-CHI and HSR203J were normalized to that of the 18S rRNA gene. Fold changes from mock infection are shown. Error bars indicate standard deviations. Statistical analyses were conducted by using the Tukey-Kramer method. Different letters above bars indicate statistically significant differences (P < 0.05 [A and B] and P < 0.01 [C and D]).
We carried out an expression analysis of SA-CHI in leaves of PI 226564 plants inoculated with WCl/P3N-PIPO, WCl/P3, or WCl/P3ΔPIPO from Cl-RB; the corresponding set of sequences from Cl-No.30; or WCl/GFP. At 4 dpi, SA-CHI was significantly induced only in leaves inoculated with WCl/P3N-PIPO-RB or WCl/P3N-PIPO-No.30 (Fig. 4C and D). WCl/P3 and WCl/P3ΔPIPO (from both Cl-RB and Cl-No.30), WCl/GFP, and mock inoculations did not significantly induce SA-CHI (Fig. 4C and D). We also investigated the expression level of SA-CHI in leaves inoculated with WCl/P3N-PIPO-CS (from BY-CS) and confirmed that WCl/P3N-PIPO-CS infection did not induce SA-CHI expression (Fig. 4D). The amplitudes of SA-CHI upregulation in plants infected with WCl/P3N-PIPO of Cl-RB and Cl-No.30 (Fig. 4C and D) were several orders of magnitude higher than those in plants infected with Cl-RB (Fig. 4A), possibly because Cl-RB produced lower levels of P3N-PIPO (produced by transcriptional slippage) than did WClMV carrying P3N-PIPO (produced in the zero frame).
The lower level of accumulation of Cl-No.30 P3N-PIPO than of Cl-RB P3N-PIPO is presumably the reason for the lower virulence of Cl-No.30.
We found that heterologous expression of Cl-No.30 P3N-PIPO could induce cell death in PI 226564 (Fig. 2C and Table 1), which was seemingly inconsistent with the fact that Cl-No.30 does not induce cell death in this cultivar (Fig. 1A and Table 1) (20, 21). We previously showed that P3N-PIPO was detected in Cl-RB-infected plants but was below the level of detection in Cl-No.30-infected plants of a susceptible pea cultivar, PI 250438, indicating that the level of P3N-PIPO from Cl-No.30 is significantly lower than that of P3N-PIPO from Cl-90-1 Br2 (7). The same difference was observed when P3N-PIPO was produced from the P3 cistron of each virus by using an in vitro translation system with an A. thaliana cell-free system and wheat germ extract (7, 13). In this study, we compared the accumulation of Cl-No.30 P3N-PIPO with that of Cl-RB P3N-PIPO in a transient-expression system, agroinfiltration in N. benthamiana leaf tissues.
We prepared the construct P3(PIPO:FLAG−1), in which a FLAG epitope tag sequence was inserted in front of the stop codon of the PIPO coding sequence, as a means to detect PIPO frame products (Fig. 5A) (13). P3(PIPO:FLAG−1) expresses the P3N-PIPO protein fused to FLAG as a −1 frame product. P3(PIPO:FLAG−1) constructs of Cl-RB and Cl-No.30 were transiently expressed in the same N. benthamiana leaf, and the levels of production of protein from the PIPO frame and mRNA of P3(PIPO:FLAG−1) were compared by measuring the protein/mRNA ratio. Western blotting using a FLAG antibody indicated that the level of accumulation of PIPO frame products of Cl-RB was higher than that of Cl-No.30 in three independent plants (Fig. 5B). Similar results were observed in at least three independent experiments. In contrast, real-time PCR analysis indicated that the mRNA level of Cl-No.30 P3(PIPO:FLAG−1) was higher than that of Cl-RB (Fig. 5C). Thus, the protein/mRNA ratio of Cl-No.30 was significantly lower than that of Cl-RB (Fig. 5D). As a control experiment, we compared the protein/mRNA ratios of P3N-PIPO:FLAGmk, which has mutations that enable the production of P3N-PIPO in the zero frame but does not produce P3 frame product, between Cl-RB and Cl-No.30 (Fig. 5A) (13). Western blot analysis indicated that there were no visible differences in protein accumulation between Cl-RB and Cl-No.30 (Fig. 5E). Real-time PCR analysis indicated that the mRNA level of Cl-No.30 P3N-PIPO:FLAGmk tended to be higher than that of Cl-RB (Fig. 5F). Thus, the protein/mRNA ratio of Cl-No.30 was lower than that of Cl-RB (Fig. 5G). These results showed that level of P3N-PIPO production from the P3 cistron of Cl-No.30 was lower than that from the P3 cistron of Cl-RB in a transient-expression system in N. benthamiana leaf tissues.
FIG 5.

Comparison of the amounts of P3N-PIPO produced from the P3 cistron between Cl-RB and Cl-No.30 using an agroinfiltration assay in N. benthamiana leaf tissues. P3(PIPO:FLAG−1) or P3N-PIPO:FLAGmk of Cl-RB and Cl-No.30 was transiently expressed in the same leaf of N. benthamiana, and the levels of production of protein from the PIPO frame were compared. (A) Schematic diagrams of plasmids for analysis of P3N-PIPO expression. To detect P3N-PIPO produced via a frameshift to the −1 reading frame, we prepared the P3(PIPO:FLAG−1) construct, in which a FLAG epitope tag sequence was inserted in front of the stop codon of the sequence encoding PIPO. P3N-PIPO:FLAGmk has mutations that enable the expression of P3N-PIPO tagged with FLAG in the zero frame. These modified P3 cistrons were inserted in a binary vector between a dexamethasone (DEX)-inducible promoter and a poly(A) addition signal (pAs). (B) P3N-PIPO-FLAG accumulation was detected by Western blotting using an antibody against FLAG. The numbers below the top panel are relative band intensities of Cl-No.30 compared to those of Cl-RB in each plant. Arrowheads indicate bands corresponding to P3N-PIPO. Yellow fluorescent protein was expressed as a negative control. (C) The levels of P3(PIPO:FLAG−1) mRNA of Cl-RB and Cl-No.30 were compared by real-time PCR analysis. The mRNA levels of P3(PIPO:FLAG−1) were normalized to those of EF1α. The relative value for the P3N-PIPO-FLAG transcript of Cl-No.30 compared to that of Cl-RB is indicated for each plant. (D) Protein/mRNA ratios were calculated by dividing the relative level of protein expression (B) by that of mRNA (C) for each plant. (E to G) P3N-PIPO-FLAG proteins of Cl-RB and Cl-No.30 were expressed in the zero frame (P3N-PIPO:FLAGmk of Cl-RB and Cl-No.30). The results in panels E to G are presented similarly to those shown in panels B to D, respectively. Welch's t test was applied to the data in panels D and G. * and ** indicate P values of <0.05 and <0.01, respectively.
We also obtained data supporting the possibility that the increased levels of P3N-PIPO produced by Cl-RB enhance virus accumulation in infected plants. We compared the accumulation of Cl-RB with that of Cl-No.30 by using GFP-tagged versions of each virus. P3N-PIPO is an essential factor for potyviruses to move from cell to cell in infected leaves (40). Therefore, we anticipated that Cl-RB would accumulate more efficiently than Cl-No.30 in PI 226564 if the level of Cl-RB P3N-PIPO was higher than that of Cl-No.30 P3N-PIPO. Virus accumulation levels of Cl-No.30/GFP and Cl-RB/GFP in PI 226564 were compared. We excised infection foci (GFP-expressing areas) from inoculated leaves using an epifluorescence microscope (Fig. 6A) and measured the amount of CP of each virus by a DAS-ELISA (Fig. 6B). The results showed that Cl-RB/GFP accumulated to higher levels than did Cl-No.30/GFP at 5 dpi. By measuring the GFP-fluorescent area, we also found that Cl-RB/GFP spread more rapidly than Cl-No.30/GFP at 5 dpi (Fig. 6C). These results suggested that the higher level of production of P3N-PIPO enhanced the ability of Cl-RB to accumulate in infected peas, perhaps synergistically increasing the difference in the levels of accumulation of P3N-PIPO between pea plants infected with Cl-RB and those infected with Cl-No.30.
FIG 6.
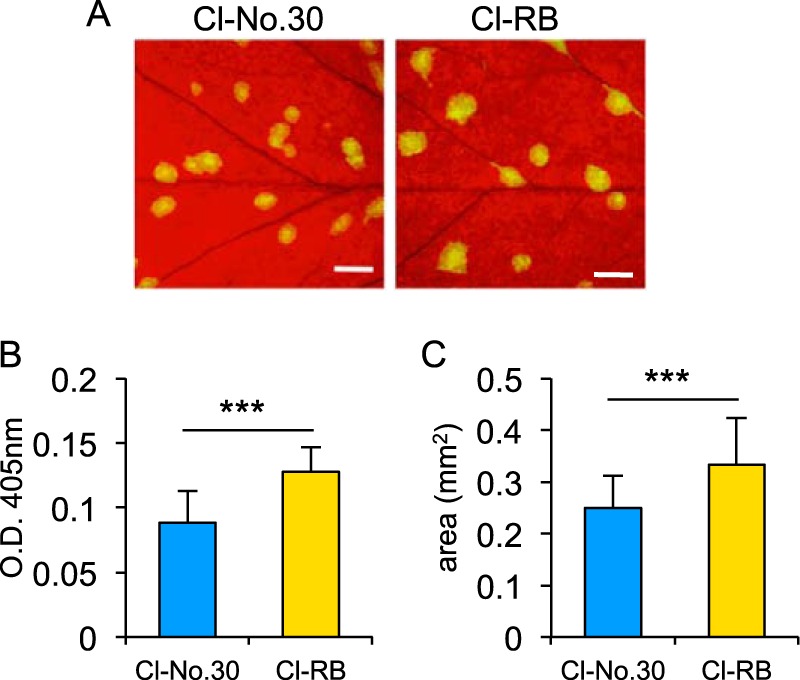
Comparison of virus accumulation between Cl-No.30 and Cl-RB by using GFP-tagged viruses. (A) Pea cultivar PI 226564 was inoculated with Cl-No.30/GFP and Cl-RB/GFP, and photographs of GFP fluorescence were taken at 5 dpi. Bar = 1 mm. (B) Comparison of virus accumulation per single infection focus at 5 dpi. Each sample was collected from 10 spots from five plants (2 spots/plant) and used for a DAS-ELISA. Error bars indicate standard deviations. Welch's t test was applied to the data. *** indicates a P value of <0.001. (C) Comparison of virus cell-to-cell movement. GFP-fluorescent areas in inoculated leaves at 5 dpi were measured by using ImageJ software. The areas of 50 spots from five plants (10 spots/plant) were measured for each virus. Error bars indicate standard deviations. Welch's t test was applied to the data. *** indicates a P value of <0.001.
These results collectively suggest that the lower level of Cl-No.30 P3N-PIPO enabled Cl-No.30 to avoid activating the SA signaling pathway, resulting in the loss of cell death induction in PI 226564.
The increased virulence of Cl-90-1 Br2 relative to that of Cl-90-1 appears to be caused by a single amino acid difference in P3N-PIPO(P3).
Cl-90-1 Br2, a mutant isolate that originated from Cl-90-1, expressed higher virulence than did Cl-90-1, which induced cell death in upper uninoculated leaves of PI 226564 plants but did not kill the plants (Fig. 7 and Table 1). We anticipated that the P3B region was responsible for the virulence enhancement of Cl-90-1 Br2 relative to Cl-90-1. We created a chimeric virus based on Cl-RB/GFP with the P3B region (from Cl-90-1 Br2) replaced by that of Cl-90-1 (Fig. 7A). There is a single nonsynonymous difference between the two sequences (T [Cl-90-1 Br2] versus G [Cl-90-1]) that causes a replacement of methionine (Cl-90-1 Br2) with arginine (Cl-90-1) at amino acid position 28 of P3 (Fig. 7B); thus, the chimeric virus was designated Cl-RBM28R/GFP. As expected, the symptoms induced by Cl-RBM28R/GFP were weaker than those induced by Cl-RB/GFP (Fig. 7C and D). Cl-RBM28R/GFP induced yellowing and cell death in upper uninoculated leaves, but the timing was delayed in comparison with Cl-RB/GFP (Fig. 7D). The plants infected with Cl-RBM28R/GFP were reproducibly taller than those infected with Cl-RB/GFP (Fig. 7C). The substitution at amino acid position 28 of P3N-PIPO(P3) is in the N-terminal region, distant from the PIPO coding region (Fig. 3), suggesting that the substitution did not affect PIPO frame translation and qualitatively affected virulence through some other mechanism. It should be noted that Cl-RB/GFP can break cyv1 resistance, whereas Cl-RBM28R/GFP cannot, indicating that the same single-amino-acid substitution affected both the breaking of cyv1 recessive resistance and symptom severity in susceptible cultivar PI 226564 (7). The substitution at amino acid position 28 is also close to the position important for PSbMV virulence in pea lines carrying sbm-2 recessive resistance (Fig. 3) (41).
FIG 7.

Mapping of the virulence determinant of Cl-90-1 in PI 226564. (A) Schematic representations of chimeric viruses constructed from Cl-No.30 (blue) and either Cl-90-1 Br2 (yellow) or Cl-90-1 (yellow; the sequence is not revealed in the shaded yellow area). Black triangles indicate the positions of the GFP insertion. Cl-RBM28R has the P3B region of Cl-90-1, which contains an amino acid substitution (methionine to arginine) at position 28 of Cl-RB P3 (asterisk). The severity of symptoms in the susceptible pea line PI 226564 is indicated by the number of + symbols. We also show the infection profile of PI 429853 carrying the recessive gene cyv1, which was reported previously (7). (B) Alignment of amino acid sequences surrounding amino acid position 28 in P3. (C and D) Symptoms induced by Cl-RBM28R were compared with those induced by Cl-RB. The photographs were taken at 14 dpi (C) and 12 dpi (D).
P3N-PIPO also induces cell death in two other pea lines, PI 118501 and PI 429853.
To investigate whether the P3N-PIPO proteins of Cl-90-1 Br2, Cl-No.30, and BY-CS had the ability to induce cell death in PI 118501 (Cyn1) (21) and PI 429853 (cyv1) (7), these P3N-PIPO proteins were expressed by WClMV vectors in these two lines. P3N-PIPO proteins of Cl-90-1 Br2 and Cl-No.30 extensively induced cell death at 5 dpi in PI 118501, whereas P3N-PIPO of BY-CS did not induce cell death until 8 dpi (Fig. 8A and Table 1). The same pattern was observed for infection of PI 429853 plants (Fig. 8B and C and Table 1). These results indicated that each P3N-PIPO protein had the ability to induce cell death in both PI 118501 and PI 429853. The results were inconsistent with symptoms in the context of virus infection (Table 1), suggesting that cell death induction was determined by other factors in addition to P3N-PIPO.
FIG 8.

P3N-PIPO expression by WClMV in PI 118501 and PI 429853. P3N-PIPO-RB, P3N-PIPO-No.30, and P3N-PIPO-CS were expressed by WClMV in PI 118501 (A) and PI 429853 (B and C). The photographs were taken at 5 or 8 dpi, as indicated.
Cell death induction is not determined solely by the PIPO region.
In PI 226564, P3N-PIPO of Cl-RB induced cell death, but P3 did not (Fig. 2B). As P3N-PIPO has the same N-terminal region (P3N) as P3, PIPO is the only region distinguishing P3N-PIPO from P3. Therefore, we inferred that the PIPO domain of Cl-RB was responsible for cell death induction in PI 226564. To test this possibility, we created chimeric P3N-PIPO genes that had either P3N of Cl-RB and PIPO of BY-CS (P3NRB-PIPOCS) or P3N of BY-CS and PIPO of Cl-RB (P3NCS-PIPORB); these combinations were chosen because P3N-PIPO of BY-CS did not induce cell death in PI 226564 (Fig. 2D). P3NRB-PIPOCS and P3NCS-PIPORB were expressed by WClMV vectors in PI 226564. P3NCS-PIPORB expression induced cell death in the inoculated leaves, but it was markedly weaker than that induced by P3N-PIPO of Cl-RB (Fig. 9A). P3NRB-PIPOCS expression did not induce cell death (Fig. 9A).
FIG 9.

Mapping of the region determining virulence in P3N-PIPO in PI 226564 and PI 118501. P3NCS-PIPORB, P3NRB-PIPOCS, P3N-PIPO-RB, P3N-PIPO-CS, and GFP were expressed by WClMV vectors in PI 226564 (A) and PI 118501 (B). P3NCS-PIPORB has the P3N region of BY-CS and PIPO of Cl-RB; P3NRB-PIPOCS has the P3N region of Cl-RB and PIPO of BY-CS. Mock indicates treatment with inoculation buffer only. The photographs of the inoculated leaves were taken at 8 dpi (A) and 5 and 18 dpi (B). The bottom left panel of panel A shows a magnified picture of the area indicated in the top panel. Arrowheads indicate regions in which cell death was induced.
We also expressed the chimeric P3N-PIPO proteins in PI 118501 by using the same WClMV vectors. When we inoculated PI 118501 plants with WCl/P3NRB-PIPOCS or WCl/P3NCS-PIPORB, neither one induced cell death at 5 dpi or even at 18 dpi in the inoculated leaves, although both WCl/P3N-PIPO-RB and WCl/P3N-PIPO-CS induced severe cell death at 18 dpi (Fig. 9B).
A consistent gradation of virulence is observed among ClYVV isolates in cyv1-carrying and susceptible peas.
Previous studies using PI 429853 (cyv1) indicated that Cl-No.30 never infects this line, Cl-90-1 can produce resistance-breaking mutants, and Cl-90-1 Br2 and Cl-I89-1 can systemically infect this line (Table 1) (7). In this study, we compared the symptoms induced by Cl-90-1 Br2 and Cl-I89-1. Cl-90-1 Br2 induced no symptoms in PI 429853 (cyv1), whereas Cl-I89-1 induced chlorosis and cell death systemically at 25 dpi (Fig. 10A). RT-PCR analysis of upper uninoculated leaves confirmed Cl-I89-1 infection of all three plants and confirmed Cl-90-1 Br2 infection of two out of three plants (Fig. 10A and B). Plants inoculated with BY-CS did not show any symptoms at 25 dpi in PI 429853 (cyv1), and RT-PCR analysis indicated that no plants were infected with BY-CS (Fig. 10C). Therefore, the order of virulence in PI 429853 (cyv1) was Cl-I89-1 > Cl-90-1 Br2 > Cl-90-1 > Cl-No.30 and BY-CS (Table 1). Similarly, the order of symptom severity in the susceptible pea line PI 226564 was Cl-90-1 Br2 > Cl-90-1 > Cl-No.30 (Fig. 7 and Table 1) (20, 42). We compared the severities of symptoms between Cl-I89-1 and Cl-90-1 Br2 and between Cl-No.30 and BY-CS. The symptoms induced by Cl-I89-1 were more severe than those induced by Cl-90-1 Br2 at 10 dpi in PI 226564 (Fig. 10D). The symptoms induced by Cl-No.30 were more severe than those induced by BY-CS at 10 dpi in PI 226564 (Fig. 10E) These results indicated a consistent gradation of virulence among these viruses in both the PI 429853 (cyv1) and susceptible PI 226564 lines (Table 1).
FIG 10.

Gradation of virulence among ClYVVs and BYMV in susceptible and cyv1 (recessive resistance)-carrying peas. (A) Symptoms of Cl-90-1 Br2 and Cl-I89-1 in PI 429853 (cyv1) at 25 dpi were compared. The presence or absence of virus infection (determined by RT-PCR, as shown in panels B and C) is indicated below the photograph. (B) Cl-I89-1 and Cl-90-1 Br2 infections in upper uninoculated leaves (shown in panel A) were confirmed by RT-PCR at 25 dpi in PI 429853 (cyv1). (C) BY-CS infection in upper uninoculated leaves of PI 429853 (cyv1) at 25 dpi was confirmed by RT-PCR. For the positive control (p.c.), RT-PCR was done by using an upper uninoculated leaf of PI 118501 inoculated with BY-CS. (D) The symptoms induced by Cl-I89-1 and Cl-90-1 Br2 in PI 226564 at 10 dpi were compared. (E) The symptoms induced by Cl-No.30 (without GFP) and BY-CS in PI 226564 at 10 dpi were compared. (F to H) Cl-I89-1, Cl-90-1 Br2, Cl-90-1, Cl-No.30, and BY-CS were inoculated onto PI 118501 plants, and their symptoms were compared at 12 dpi. Upper symptomatic leaves of plants inoculated with BY-CS (G) or mock-inoculated plants (H) are shown. (I) Gradation of virulence among ClYVVs and BYMV in recessive resistant (PI 429853 [cyv1 Cyn1]) (top) and susceptible (PI 226564 [weak Cyn1] and PI 118501 [Cyn1]) (bottom) cultivars. The graphs indicate the consistent gradation observed in this study: the more efficiently a ClYVV isolate broke the resistance conferred by cyv1 (top), the more it expressed virulence in susceptible peas (bottom). (J) Molecular phylogenetic analysis of full-length nucleotide sequences encoding polyproteins of ClYVV and BYMV. The sequences were aligned by using MUSCLE, and the maximum likelihood tree was inferred by using the MEGA6 package (36). The tree is drawn to scale, with branch lengths measured as the number of substitutions per site. TuMV-Tu-2R1 was set as an outgroup. The significance of the nodes was estimated with 1,000 bootstrap replicates. The GenBank accession number for each virus is listed in the legend of Fig. 3, except for TuMV-Tu-2R1 (accession number AB105135).
Next, we investigated whether the same gradation was observed for another susceptible cultivar, PI 118501, which shows lethal systemic cell death following Cl-No.30 infection but not following BY-CS infection (20, 21). The inoculation test indicated that Cl-I89-1, Cl-90-1 Br2, Cl-90-1, and Cl-No.30 similarly induced lethal systemic cell death (Fig. 10F), whereas BY-CS induced only mosaic symptoms in the upper uninoculated leaves at 12 dpi (Fig. 10G and H). Cell death induction by Cl-No.30 was slightly slower than induction by Cl-I89-1, Cl-90-1 Br2, and Cl-90-1. These results indicated that the order of symptom severity in PI 118501 was Cl-I89-1, Cl-90-1 Br2, and Cl-90-1 > Cl-No.30 > BY-CS (Table 1). Taken together, these results indicated that there is a consistent gradation in virulence among ClYVVs and BYMV in PI 429853 (cyv1 recessive resistance) and in PI 226564 and PI 118501 (susceptible) (Table 1 and Fig. 10I).
Phylogenetic analysis using full-length ORF sequences encoding polyproteins suggested that high-virulence isolates (Cl-90-1, Cl-90-1 Br2, and Cl-I89-1) form a group distinct from low-virulence isolate Cl-No.30 (Fig. 10J).
DISCUSSION
We revealed that the main determinant of lethal systemic cell death induction by ClYVV was P3N-PIPO. This was determined by analyses using chimeric viruses and transient expression from WClMV vectors in a susceptible pea cultivar, PI 226564. SMV strain G7 induces lethal systemic cell death in soybean carrying the dominant resistance gene Rsv1 (14). In this virus, P3, and not P3N-PIPO, determines virulence (40). TuMV induces lethal systemic cell death in A. thaliana Ler carrying the TuNI gene (15). TuMV P3 expression alone is sufficient for the induction of cell death at the single-cell level, and the region required for cell death induction is upstream of the PIPO coding sequence (43). Our study is the first report to suggest that induction of lethal systemic cell death could be attributed to P3N-PIPO.
As mentioned above, it was unexpected to see that the expression of P3N-PIPO from Cl-No.30 by a WClMV vector induced cell death in PI 226564 (Fig. 2C) because Cl-No.30 itself does not induce cell death in this cultivar (Fig. 1A) (20, 21). In N. benthamiana, Plantago asiatica mosaic virus (PlAMV) isolate Li1 induces systemic necrosis that has HR-like characteristics (44, 45). Transient expression of PlAMV strain Li1 RNA-dependent RNA polymerase (RdRp) (helicase domain) by agroinfiltration induces cell death in N. benthamiana (46). These results suggest that N. benthamiana recognizes PlAMV RdRp and induces an HR-like response systemically, resulting in systemic necrosis. Interestingly, transient expression of RdRp encoded by asymptomatic PlAMV isolate Li6 also induces cell death in N. benthamiana (46). The level of RdRp accumulation is higher in areas infiltrated with Agrobacterium carrying infectious cDNA of the Li1 isolate than when infectious cDNA of the Li6 isolate is used (46). These results suggest that N. benthamiana has the ability to recognize both Li1 and Li6 RdRp proteins, but the difference in their protein accumulation levels determines the induction of systemic necrosis in the context of virus infection. The results reported here can be explained in a similar fashion. Previously, we reported that in the susceptible pea line PI 250438 infected with Cl-90-1 Br2 or Cl-No.30, P3N-PIPO of Cl-90-1 Br2 could be detected, but the amount of P3N-PIPO of Cl-No.30 was below the level of detection when tested by using an antibody against the PIPO peptide (7). Furthermore, we showed that the amount of P3N-PIPO produced from the P3 cistron of Cl-No.30 is significantly smaller than that of Cl-RB in an in vitro translation system using MM2 dL (an extract derived from A. thaliana MM2d cells) and wheat germ extract (7, 13). In this study, we showed evidence that the P3 cistron of Cl-No.30 produced less P3N-PIPO protein than did Cl-RB in vivo in agroinfiltrated N. benthamiana leaves (Fig. 5B). The protein/mRNA ratio of Cl-No.30 was lower than that of Cl-RB (Fig. 5D). These results suggest that the level of P3N-PIPO production, or transcriptional slippage efficiency, from Cl-No.30 P3(PIPO:FLAG−1) (which tagged P3N-PIPO produced by a frameshift) is lower than that of Cl-RB. On the other hand, the protein/mRNA ratio of Cl-No.30 P3N-PIPO:FLAGmk (which produced tagged P3N-PIPO as a zero-frame product) was also lower than that of Cl-RB (Fig. 5G). The protein/mRNA ratio of P3(PIPO:FLAG−1) tended to be lower than that of P3N-PIPO:FLAGmk in repeated experiments, although there was not a statistically significant difference. These findings suggest that the difference in P3N-PIPO accumulation between Cl-No.30 and Cl-RB was determined by a combination of transcriptional slippage efficiency and other factors such as protein stability or translation efficiency. Taken together, these results support the hypothesis that in the susceptible pea line PI 226564, Cl-90-1 Br2 can produce P3N-PIPO protein levels sufficient for the induction of cell death, but Cl-No.30 cannot, even though host cells are able to recognize P3N-PIPO proteins from both isolates.
We showed that each of the tested regions outside P3 are accessorily involved in the virulence enhancement in PI 226564 (Fig. 1G). In previous studies, we revealed that the HC-Pro gene is indirectly involved in the induction of lethal systemic cell death in PI 118501 (20, 22, 32). We found that a Cl-No.30 mutant with a D-to-Y substitution at amino acid position 193 of HC-Pro (Cl-D193Y) loses the ability to induce cell death in PI 118501 (20). Potyvirus HC-Pro is an RNA-silencing suppressor required for efficient virus accumulation in host plants (47). The D193Y mutation in HC-Pro markedly reduces its ability to suppress RNA silencing, and the level of accumulation of Cl-D193Y is significantly lower than that of wild-type Cl-No.30 in PI 118501 (20, 22). Heterologous expression of viral suppressors of RNA silencing (tomato bushy stunt virus P19 or cucumber mosaic virus 2b) can complement the virulence of Cl-D193Y in PI 118501 (32). These results suggest that HC-Pro itself is not an elicitor but indirectly affects cell death induction via its RNA-silencing suppressor activity in PI 118501: the reduced accumulation of ClYVV leads to the reduced accumulation of the elicitor molecules that induce cell death. In this study, we found that the elicitor molecule active against PI 118501 was P3N-PIPO (Fig. 8A). Thus, it is likely that Cl-D193Y is not lethal in PI 118501 because it cannot produce enough P3N-PIPO to induce cell death. Taken together, these results indicate that lethal systemic cell death in PI 118501 was induced by P3N-PIPO, the production of which was indirectly regulated by the RNA-silencing suppressor activity of HC-Pro. As observed for HC-Pro, viral genes other than P3 might increase the accumulation of P3N-PIPO, enhancing virulence in PI 226564 (Fig. 1G).
We found that the expression of P3N-PIPO from Cl-90-1 Br2, Cl-No.30, and BY-CS by WClMV vectors induced cell death in PI 226564, PI 118501, and PI 429853, with the exception of BY-CS P3N-PIPO in PI 226564 (Table 1), suggesting that many peas have a common factor that recognizes P3N-PIPO proteins of two different Potyvirus species (ClYVV and BYMV). The Rx gene in potato encodes an NB-LRR-type resistance gene that confers genetically dominant resistance against potato virus X (PVX) (48). Rx specifically recognizes CP of avirulent PVX strains and induces extreme resistance (epistatic to an HR) (49). N. benthamiana expressing Rx also shows resistance to PVX (50). Interestingly, Rx is able to recognize CP of three other species of the genus Potexvirus (narcissus mosaic virus, WClMV, and cymbidium mosaic virus) and to induce an HR in N. benthamiana expressing Rx (51). The product of the L4 resistance gene (NB-LRR) isolated from pepper is able to recognize CP of several distant species of the genus Tobamovirus, including tomato mosaic virus (TMV), paprika mild mottle virus, and pepper mild mottle virus, and to induce cell death accompanied by an HR when both CP and L4 are transiently expressed in N. benthamiana by agroinfiltration (34). Similarly, the product of N′ (NB-LRR) isolated from Nicotiana sylvestris is able to recognize CP of tomato mosaic virus, paprika mild mottle virus, and pepper mild mottle virus and induces cell death accompanied by an HR when both CP and N′ are transiently expressed in N. benthamiana by agroinfiltration (34). These studies suggest that a single resistance protein has the potential to recognize a wide range of elicitor molecules, at least within the same genus.
In pea, one of the candidate factors to recognize P3N-PIPO is the product of the Cyn1 gene (21). Genetic analysis indicated that lethal systemic cell death induced by Cl-No.30 in PI 118501 is controlled by the dominant gene Cyn1 (21). Cyn1 is located on linkage group 3 (LG3), where many R gene analogs were suggested to be clustered by a previous study of synteny between pea and Medicago truncatula (21). In this study, we showed that the expression of P3N-PIPO of Cl-No.30 in a WClMV vector induced cell death in PI 118501 (Fig. 8A), suggesting that Cyn1 recognized P3N-PIPO and induced lethal systemic cell death. We previously showed the possibility that PI 226564 also has a Cyn1 allele that weakly recognizes Cl-No.30 (20). Cl-No.30 does not induce cell death in PI 226564 (Fig. 1A) (20, 21); however, the activation of the SA signaling pathway by the application of an SA analog, benzo(1,2,3)thiadiazole-7-carbothioic acid S-methyl ester (BTH), induces systemic cell death in PI 226564 plants infected with Cl-No.30 (20). In this study, we obtained the supporting result that the expression of Cl-No.30 P3N-PIPO by a WClMV vector induced cell death in PI 226564 (Fig. 2C). In contrast, BTH treatment does not induce cell death in PI 226564 plants infected with BY-CS (20). Consistent with this result, the expression of BY-CS P3N-PIPO by WClMV did not induce cell death in PI 226564 (Fig. 2D). These results suggest that PI 226564 has a Cyn1 allele whose product has the potential to specifically recognize Cl-No.30 but not BY-CS. Cyn1 of PI 226564 might be able to recognize P3N-PIPO effectively when P3N-PIPO accumulates to high levels, e.g., in situations such as overexpression by a WClMV vector or Cl-RB infection, but not when it is at low levels, e.g., in a situation such as Cl-No.30 infection (20). In PI 429853 (cyv1), the P3N-PIPO protein could be recognized when expressed by a WClMV vector, suggesting that PI 429853 has a Cyn1 allele whose product recognizes ClYVV. This was inconsistent with symptoms in the context of virus infection (Table 1). cyv1-mediated resistance would inhibit or reduce the accumulation of ClYVV and, thus, the recognition of P3N-PIPO under natural infection conditions (Table 1). In contrast to its lack of an effect on PI 226564, P3N-PIPO of BY-CS could induce cell death in PI 118501 and PI 429853 although more slowly than P3N-PIPO of either Cl-90-1 Br2 or Cl-No.30 (Fig. 2 and 8); these data suggest that Cyn1 of PI 118501 and PI 429853 can recognize BY-CS although less efficiently than Cl-90-1 Br2 and Cl-No.30. Note that we could not detect P3N-PIPO expressed via a WClMV vector by Western blot analysis and thus could not compare the levels of accumulation of P3N-PIPO among the three tested pea cultivars. Taken together, these results suggest that the product of Cyn1 recognizes matching P3N-PIPO proteins of ClYVV and BYMV and activates the SA-mediated defense pathway, resulting in systemic cell death induction in many pea lines (20).
We showed that the expression of P3N-PIPO by WClMV induced cell death in PI 226564 but that the expression of P3 did not (Fig. 2). One possible explanation is that a pea protein (e.g., the Cyn1 product) recognizes the PIPO peptide, which is part of P3N-PIPO but not P3. However, the PIPO domain of Cl-P3N-PIPO alone did not appear to induce cell death, as shown by an experiment using chimeric P3N-PIPO (constructed from P3N-PIPOs of Cl-RB and BY-CS) in PI 226564 (Fig. 9). Although the PIPO domain contributed to recognition by peas, the overall structure of P3N-PIPO may be important for the full activation of the signaling pathway to induce cell death. A second possible explanation is that a pea protein (e.g., the Cyn1 product) recognizes a P3N-PIPO-targeted host factor(s) (the guard/decoy model) (52). Recently, it was found that PCaP1 of A. thaliana and its homolog NbDREPP of N. benthamiana interact with P3N-PIPO (53, 54). PCaP1 interacts with P3N-PIPO via the PIPO domain and does not interact with P3, indicating that PCaP1 is specifically targeted by P3N-PIPO (53). Therefore, a pea protein may monitor a target of P3N-PIPO such as PCaP1 and induce cell death. A third explanation is that pea recognizes P3N-PIPO in a localization-dependent manner. Previous studies reported that P3 localizes to the endoplasmic reticulum (ER)-Golgi membrane interface and that P3N-PIPO localizes to plasmodesmata (9, 55). Therefore, P3 may not be recognized due to its localization.
Our results using five virus isolates (four ClYVV isolates and one BYMV isolate) and three pea genotypes showed that the more efficiently a particular ClYVV isolate broke cyv1 recessive resistance, the more it expressed virulence in susceptible peas carrying Cyn1 (Fig. 10I and Table 1). In particular, we observed adaptive evolution from Cl-90-1 to Cl-90-1 Br2, which overcame cyv1 resistance through attaining a point mutation in the P3N domain (7). This mutation also resulted in Cl-90-1 Br2 gaining higher virulence than Cl-90-1 in the susceptible pea line PI 226564 (Fig. 7). Many studies have suggested that trade-offs are observed for plant virus infection across hosts, and antagonistic pleiotropy (when mutations beneficial for infection of one host are deleterious for infection of another one) explains such trade-offs well (56, 57). For example, tobacco etch virus (TEV) infects several solanaceous plants, such as Nicotiana tabacum, in nature, and some nonsolanaceous plants (e.g., Helianthus annuus and Spinacia oleracea) are also susceptible under experimental conditions. Analysis using a TEV mutant series indicated that fitness trade-offs due to antagonistic pleiotropy are observed between N. tabacum and nonsolanaceous plants (58). Several studies suggest that viruses pay a fitness cost when they overcome dominant resistance due to adaptive mutations with antagonistic pleiotropic effects. In pepper, tobamoviruses that can overcome dominant resistance conferred by L genes are less able to accumulate in susceptible plants, and virus particles of resistance-breaking isolates in the soil are less stable than those of the wild-type virus (59, 60). In Brassica napus, TuMV isolates CZE1 and CDN1 are able to overcome dominant resistance conferred by TuRB01 but are outcompeted by avirulent isolate UK1 in susceptible plants (61). Soybeans carrying the dominant Rsv1 or Rsv4 allele are resistant to SMV-N (62) or three strains (SMV-N, SMV-G7, and SMV-G7d) (63), respectively. SMV-N HC-Pro mutants or SMV P3 mutants of the three strains can overcome these respective resistances, and the accumulation of these mutants is reduced in susceptible cultivars (62, 63). Those studies indicated that their fitness trade-offs are caused by antagonistic pleiotropy of the viral genes that overcome dominant resistance. Similar observations have also been reported for viruses that overcome recessive resistance. Potato virus Y VPg double mutants show more virulence than VPg single mutants in pepper carrying a pvr23 recessive resistance gene (64). In contrast, potato virus Y VPg double mutants are less virulent than single mutants in susceptible pepper (64). In rice, rice yellow mottle virus mutants that can overcome rymv1-2 recessive resistance are less virulent than the wild-type virus in susceptible cultivars (65).
These studies collectively support the hypothesis that many viruses pay fitness costs to adapt to new hosts or to overcome resistances and that these across-host trade-offs are caused by adaptive mutations with antagonistic pleiotropic effects. In the case of ClYVV, our results suggest that many susceptible peas carry Cyn1, whose product recognizes ClYVVs (or their P3N-PIPO proteins, in particular) that break cyv1 resistance. In Cyn1-carrying peas, these ClYVVs induce an HR-like response associated with systemic cell death, resulting in plant death. As noted above, the more efficiently a particular ClYVV isolate broke cyv1 recessive resistance, the more it induced systemic cell death, resulting in a loss of tissue to support virus accumulation and leading to a reduction of fitness in susceptible peas carrying Cyn1. This observation suggests that there are fitness trade-offs between overcoming cyv1 resistance and reducing recognition by Cyn1 via the antagonistic pleiotropy of P3N-PIPO. The trade-offs shown in previous studies (described above) involve adaptation to a nonhost or overcoming a resistant cultivar, resulting in a reduction of virus viability or virulence in a susceptible host. This study is unique in terms of showing trade-offs in a virus overcoming two independent defense systems in a single plant species.
We hypothesize the following model of coevolution between ClYVV and pea, driven by the antagonistic pleiotropy of P3N-PIPO: (i) ClYVV cannot infect peas carrying the cyv1 recessive resistance gene; (ii) selection favors mutations in the P3 gene of ClYVV that enable P3N-PIPO to accumulate to higher levels and/or alter its protein structure, enabling the virus to overcome cyv1 resistance; (iii) after overcoming resistance, the virus can infect and accumulate effectively, but Cyn1 now recognizes the more abundant (Cl-No.30 versus Cl-90-1 Br2) and/or adapted (Cl-90-1 versus Cl-90-1 Br2) P3N-PIPO protein directly or indirectly and induces cell death systemically; (iv) systemic cell death leads to a loss of host viability, which is also unfavorable for the virus; and (v) selection favors mutations in ClYVV that reduce the accumulation of P3N-PIPO and/or change the amino acids of P3N-PIPO required for its recognition or function, resulting in a loss of the ability to overcome cyv1 recessive resistance. Based on the proposed model, Cyn1 may have evolved to recognize ClYVVs (or their P3N-PIPO proteins, in particular) that break cyv1 resistance in susceptible peas. Although Cyn1-mediated activation of the SA defense pathway does not appear to inhibit ClYVV infection efficiently, as observed for authentic HRs (20), systemic cell death may oppose adaptive evolution to overcome cyv1 resistance because the induction of systemic cell death leads to a loss of host viability. We assume that two independent defense mechanisms (recessive resistance and the SA defense pathway) in pea impose antagonistic pleiotropy on P3N-PIPO. Such a trade-off for the virus in overcoming paired defense mechanisms may sustain the durability of resistance against fast-evolving viruses (66).
Supplementary Material
ACKNOWLEDGMENTS
We thank Kazue Obara and Kami Murakami for technical assistance. We also thank Takashi Aoyama and Nam-Hai Chua for the use of binary vector pTA7001, and we thank Kappei Kobayashi and Kentaro Yoshida for critical readings of the manuscript and useful discussions.
G.A., K.S.N., and I.U. designed the research. G.A., H.S., Y.M., S.H.C., Y.H., S.R., R.S., E.J.J., J.A., and K.S.N conducted the experiments. G.A., K.S.N., and I.U. discussed the results and wrote the manuscript.
We declare that we have no conflicts of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00190-16.
REFERENCES
- 1.Mandadi KK, Scholthof KBG. 2013. Plant immune responses against viruses: how does a virus cause disease? Plant Cell 25:1489–1505. doi: 10.1105/tpc.113.111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diaz-Pendon JA, Truniger V, Nieto C, Garcia-Mas J, Bendahmane A, Aranda MA. 2004. Advances in understanding recessive resistance to plant viruses. Mol Plant Pathol 5:223–233. doi: 10.1111/j.1364-3703.2004.00223.x. [DOI] [PubMed] [Google Scholar]
- 3.Gibbs AJ, Ohshima K, Phillips MJ, Gibbs MJ. 2008. The prehistory of potyviruses: their initial radiation was during the dawn of agriculture. PLoS One 3:e2523. doi: 10.1371/journal.pone.0002523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robaglia C, Caranta C. 2006. Translation initiation factors: a weak link in plant RNA virus infection. Trends Plant Sci 11:40–45. doi: 10.1016/j.tplants.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Andrade M, Abe Y, Nakahara KS, Uyeda I. 2009. The cyv-2 resistance to Clover yellow vein virus in pea is controlled by the eukaryotic initiation factor 4E. J Gen Plant Pathol 75:241–249. doi: 10.1007/s10327-009-0163-3. [DOI] [Google Scholar]
- 6.Provvidenti R, Hampton RO. 1991. Chromosomal distribution of genes for resistance to seven potyviruses in Pisum sativum. Pisum Genet 23:26–28. [Google Scholar]
- 7.Choi SH, Hagiwara-Komoda Y, Nakahara KS, Atsumi G, Shimada R, Hisa Y, Naito S, Uyeda I. 2013. Quantitative and qualitative involvement of P3N-PIPO in overcoming recessive resistance against Clover yellow vein virus in pea carrying the cyv1 gene. J Virol 87:7326–7337. doi: 10.1128/JVI.00065-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung BYW, Miller WA, Atkins JF, Firth AE. 2008. An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci U S A 105:5897–5902. doi: 10.1073/pnas.0800468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei T, Zhang C, Hong J, Xiong R, Kasschau KD, Zhou X, Carrington JC, Wang A. 2010. Formation of complexes at plasmodesmata for potyvirus intercellular movement is mediated by the viral protein P3N-PIPO. PLoS Pathog 6:e1000962. doi: 10.1371/journal.ppat.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen RH, Hajimorad MR. 2010. Mutational analysis of the putative pipo of soybean mosaic virus suggests disruption of PIPO protein impedes movement. Virology 400:1–7. doi: 10.1016/j.virol.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 11.Rodamilans B, Valli A, Mingot A, San León D, Baulcombe D, López-Moya JJ, García JA. 2015. RNA polymerase slippage as a mechanism for the production of frameshift gene products in plant viruses of the Potyviridae family. J Virol 89:6965–6967. doi: 10.1128/JVI.00337-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olspert A, Chung BYW, Atkins JF, Carr JP, Firth AE. 2015. Transcriptional slippage in the positive-sense RNA virus family Potyviridae. EMBO Rep 16:995–1004. doi: 10.15252/embr.201540509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hagiwara-Komoda Y, Choi SH, Sato M, Atsumi G, Abe J, Fukuda J, Honjo MN, Nakahara KS, Nagano AJ, Komoda K, Uyeda I, Naito S. 2016. Truncated yet functional viral protein produced via RNA polymerase slippage implies underestimated coding capacity of RNA viruses. Sci Rep 6:21411. doi: 10.1038/srep21411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jayaram C, Hill JH, Miller WA. 1992. Complete nucleotide sequences of two soybean mosaic virus strains differentiated by response of soybean containing the Rsv resistance gene. J Gen Virol 73:2067–2077. doi: 10.1099/0022-1317-73-8-2067. [DOI] [PubMed] [Google Scholar]
- 15.Kaneko YH, Inukai T, Suehiro N, Natsuaki T, Masuta C. 2004. Fine genetic mapping of the TuNI locus causing systemic veinal necrosis by turnip mosaic virus infection in Arabidopsis thaliana. Theor Appl Genet 110:33–40. doi: 10.1007/s00122-004-1824-4. [DOI] [PubMed] [Google Scholar]
- 16.Hajimorad MR, Eggenberger AL, Hill JH. 2003. Evolution of Soybean mosaic virus-G7 molecularly cloned genome in Rsv1-genotype soybean results in emergence of a mutant capable of evading Rsv1-mediated recognition. Virology 314:497–509. doi: 10.1016/S0042-6822(03)00456-2. [DOI] [PubMed] [Google Scholar]
- 17.Kim B, Masuta C, Matsuura H, Takahashi H, Inukai T. 2008. Veinal necrosis induced by Turnip mosaic virus infection in Arabidopsis is a form of defense response accompanying HR-like cell death. Mol Plant Microbe Interact 21:260–268. doi: 10.1094/MPMI-21-2-0260. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Kim BM, Kaneko Y, Inukai T, Masuta C. 2015. Identification of the TuNI gene causing systemic necrosis in Arabidopsis ecotype Ler infected with Turnip mosaic virus and characterization of its expression. J Gen Plant Pathol 81:180–191. doi: 10.1007/s10327-015-0583-1. [DOI] [Google Scholar]
- 19.Wen RH, Khatabi B, Ashfield T, Saghai Maroof MA, Hajimorad MR. 2013. The HC-Pro and P3 cistrons of an avirulent Soybean mosaic virus are recognized by different resistance genes at the complex Rsv1 locus. Mol Plant Microbe Interact 26:203–215. doi: 10.1094/MPMI-06-12-0156-R. [DOI] [PubMed] [Google Scholar]
- 20.Atsumi G, Kagaya U, Kitazawa H, Nakahara KS, Uyeda I. 2009. Activation of the salicylic acid signaling pathway enhances Clover yellow vein virus virulence in susceptible pea cultivars. Mol Plant Microbe Interact 22:166–175. doi: 10.1094/MPMI-22-2-0166. [DOI] [PubMed] [Google Scholar]
- 21.Ravelo G, Kagaya U, Inukai T, Sato M, Uyeda I. 2007. Genetic analysis of lethal tip necrosis induced by Clover yellow vein virus infection in pea. J Gen Plant Pathol 73:59–65. doi: 10.1007/s10327-006-0324-6. [DOI] [Google Scholar]
- 22.Yambao MLM, Yagihashi H, Sekiguchi H, Sekiguchi T, Sasaki T, Sato M, Atsumi G, Tacahashi Y, Nakahara KS, Uyeda I. 2008. Point mutations in helper component protease of clover yellow vein virus are associated with the attenuation of RNA-silencing suppression activity and symptom expression in broad bean. Arch Virol 153:105–115. doi: 10.1007/s00705-007-1073-3. [DOI] [PubMed] [Google Scholar]
- 23.Masuta C, Yamana T, Tacahashi Y, Uyeda I, Sato M, Ueda S, Matsumura T. 2000. Development of clover yellow vein virus as an efficient, stable gene-expression system for legume species. Plant J 23:539–546. doi: 10.1046/j.1365-313x.2000.00795.x. [DOI] [PubMed] [Google Scholar]
- 24.Hisa Y, Suzuki H, Atsumi G, Choi SH, Nakahara KS, Uyeda I. 2014. P3N-PIPO of Clover yellow vein virus exacerbates symptoms in pea infected with White clover mosaic virus and is implicated in viral synergism. Virology 449:200–206. doi: 10.1016/j.virol.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 25.Ido Y, Nakahara KS, Uyeda I. 2012. White clover mosaic virus-induced gene silencing in pea. J Gen Plant Pathol 78:127–132. doi: 10.1007/s10327-012-0360-3. [DOI] [Google Scholar]
- 26.Sato M, Masuta C, Uyeda I. 2003. Natural resistance to Clover yellow vein virus in beans controlled by a single recessive locus. Mol Plant Microbe Interact 16:994–1002. doi: 10.1094/MPMI.2003.16.11.994. [DOI] [PubMed] [Google Scholar]
- 27.Nakahara KS, Nishino K, Uyeda I. 2015. Construction of infectious cDNA clones derived from the potyviruses Clover yellow vein virus and Bean yellow mosaic virus. Methods Mol Biol 1236:219–227. doi: 10.1007/978-1-4939-1743-3_16. [DOI] [PubMed] [Google Scholar]
- 28.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chomczynski P, Sacchi N. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159. doi: 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]
- 30.Atsumi G, Tomita R, Yamashita T, Sekine KT. 2015. A novel virus transmitted through pollination causes ring-spot disease on gentian (Gentiana triflora) ovaries. J Gen Virol 96:431–439. doi: 10.1099/vir.0.071498-0. [DOI] [PubMed] [Google Scholar]
- 31.Aoyama T, Chua NH. 1997. A glucocorticoid-mediated transcriptional induction system in transgenic plants. Plant J 11:605–612. doi: 10.1046/j.1365-313X.1997.11030605.x. [DOI] [PubMed] [Google Scholar]
- 32.Atsumi G, Nakahara KS, Wada TS, Choi SH, Masuta C, Uyeda I. 2012. Heterologous expression of viral suppressors of RNA silencing complements virulence of the HC-Pro mutant of clover yellow vein virus in pea. Arch Virol 157:1019–1028. doi: 10.1007/s00705-012-1281-3. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Sekine KT, Tomita R, Takeuchi S, Atsumi G, Saitoh H, Mizumoto H, Kiba A, Yamaoka N, Nishiguchi M, Hikichi Y, Kobayashi K. 2012. Functional differentiation in the leucine-rich repeat domains of closely related plant virus-resistance proteins that recognize common Avr proteins. Mol Plant Microbe Interact 25:1219–1229. doi: 10.1094/MPMI-11-11-0289. [DOI] [PubMed] [Google Scholar]
- 35.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi Y, Takahashi T, Uyeda I. 1997. A cDNA clone to clover yellow vein potyvirus genome is highly infectious. Virus Genes 14:235–243. doi: 10.1023/A:1007940028058. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Ueda S, Uyeda I, Yagihashi H, Sekiguchi H, Tacahashi Y, Sato M, Ohya K, Sugimoto C, Matsumura T. 2003. Positional effect of gene insertion on genetic stability of a clover yellow vein virus-based expression vector. J Gen Plant Pathol 69:327–334. doi: 10.1007/s10327-003-0057-8. [DOI] [Google Scholar]
- 39.Nakazono-Nagaoka E, Takahashi T, Shimizu T, Kosaka Y, Natsuaki T, Omura T, Sasaya T. 2009. Cross-protection against Bean yellow mosaic virus (BYMV) and Clover yellow vein virus by attenuated BYMV isolate M11. Phytopathology 99:251–257. doi: 10.1094/PHYTO-99-3-0251. [DOI] [PubMed] [Google Scholar]
- 40.Wen RH, Maroof MAS, Hajimorad MR. 2011. Amino acid changes in P3, and not the overlapping pipo-encoded protein, determine virulence of Soybean mosaic virus on functionally immune Rsv1-genotype soybean. Mol Plant Pathol 12:799–807. doi: 10.1111/j.1364-3703.2011.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hjulsager CK, Olsen BS, Jensen DMK, Cordea MI, Krath BN, Johansen IE, Lund OS. 2006. Multiple determinants in the coding region of Pea seed-borne mosaic virus P3 are involved in virulence against sbm-2 resistance. Virology 355:52–61. doi: 10.1016/j.virol.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 42.Andrade M, Sato M, Uyeda I. 2007. Two resistance modes to Clover yellow vein virus in pea characterized by a green fluorescent protein-tagged virus. Phytopathology 97:544–550. doi: 10.1094/PHYTO-97-5-0544. [DOI] [PubMed] [Google Scholar]
- 43.Kim BM, Suehiro N, Natsuaki T, Inukai T, Masuta C. 2010. The P3 protein of Turnip mosaic virus can alone induce hypersensitive response-like cell death in Arabidopsis thaliana carrying TuNI. Mol Plant Microbe Interact 23:144–152. doi: 10.1094/MPMI-23-2-0144. [DOI] [PubMed] [Google Scholar]
- 44.Ozeki J, Takahashi S, Komatsu K, Kagiwada S, Yamashita K, Mori T, Hirata H, Yamaji Y, Ugaki M, Namba S. 2006. A single amino acid in the RNA-dependent RNA polymerase of Plantago asiatica mosaic virus contributes to systemic necrosis. Arch Virol 151:2067–2075. doi: 10.1007/s00705-006-0766-3. [DOI] [PubMed] [Google Scholar]
- 45.Komatsu K, Hashimoto M, Ozeki J, Yamaji Y, Maejima K, Senshu H, Himeno M, Okano Y, Kagiwada S, Namba S. 2010. Viral-induced systemic necrosis in plants involves both programmed cell death and the inhibition of viral multiplication, which are regulated by independent pathways. Mol Plant Microbe Interact 23:283–293. doi: 10.1094/MPMI-23-3-0283. [DOI] [PubMed] [Google Scholar]
- 46.Komatsu K, Hashimoto M, Maejima K, Shiraishi T, Neriya Y, Miura C, Minato N, Okano Y, Sugawara K, Yamaji Y, Namba S. 2011. A necrosis-inducing elicitor domain encoded by both symptomatic and asymptomatic Plantago asiatica mosaic virus isolates, whose expression is modulated by virus replication. Mol Plant Microbe Interact 24:408–420. doi: 10.1094/MPMI-12-10-0279. [DOI] [PubMed] [Google Scholar]
- 47.Anandalakshmi R, Pruss GJ, Ge X, Marathe R, Mallory AC, Smith TH, Vance VB. 1998. A viral suppressor of gene silencing in plants. Proc Natl Acad Sci U S A 95:13079–13084. doi: 10.1073/pnas.95.22.13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cockerham G. 1970. Genetical studies on resistance to potato virus X and Y. Heredity 25:309–348. [Google Scholar]
- 49.Bendahmane A, Köhn BA, Dedi C, Baulcombe DC. 1995. The coat protein of potato virus X is a strain-specific elicitor of Rx1-mediated virus resistance in potato. Plant J 8:933–941. doi: 10.1046/j.1365-313X.1995.8060933.x. [DOI] [PubMed] [Google Scholar]
- 50.Bendahmane A, Kanyuka K, Baulcombe DC. 1999. The Rx gene from potato controls separate virus resistance and cell death responses. Plant Cell 11:781–792. doi: 10.1105/tpc.11.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baurès I, Candresse T, Leveau A, Bendahmane A, Sturbois B. 2008. The Rx gene confers resistance to a range of potexviruses in transgenic Nicotiana plants. Mol Plant Microbe Interact 21:1154–1164. doi: 10.1094/MPMI-21-9-1154. [DOI] [PubMed] [Google Scholar]
- 52.van der Hoorn RAL, Kamoun S. 2008. From guard to decoy: a new model for perception of plant pathogen effectors. Plant Cell 20:2009–2017. doi: 10.1105/tpc.108.060194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vijayapalani P, Maeshima M, Nagasaki-Takekuchi N, Miller WA. 2012. Interaction of the trans-frame potyvirus protein P3N-PIPO with host protein PCaP1 facilitates potyvirus movement. PLoS Pathog 8:e1002639. doi: 10.1371/journal.ppat.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geng C, Cong QQ, Li XD, Mou AL, Gao R, Liu JL, Tian YP. 2015. Developmentally regulated plasma membrane protein of Nicotiana benthamiana contributes to potyvirus movement and transports to plasmodesmata via the early secretory pathway and the actomyosin system. Plant Physiol 167:394–410. doi: 10.1104/pp.114.252734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cui X, Wei T, Chowda-Reddy RV, Sun G, Wang A. 2010. The Tobacco etch virus P3 protein forms mobile inclusions via the early secretory pathway and traffics along actin microfilaments. Virology 397:56–63. doi: 10.1016/j.virol.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 56.García-Arenal F, Fraile A. 2013. Trade-offs in host range evolution of plant viruses. Plant Pathol 62:2–9. doi: 10.1111/ppa.12104. [DOI] [Google Scholar]
- 57.Elena SF, Fraile A, García-Arenal F. 2014. Evolution and emergence of plant viruses. Adv Virus Res 88:161–191. doi: 10.1016/B978-0-12-800098-4.00003-9. [DOI] [PubMed] [Google Scholar]
- 58.Lalić J, Cuevas JM, Elena SF. 2011. Effect of host species on the distribution of mutational fitness effects for an RNA virus. PLoS Genet 7:e1002378. doi: 10.1371/journal.pgen.1002378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fraile A, Pagán I, Anastasio G, Sáez E, García-Arenal F. 2011. Rapid genetic diversification and high fitness penalties associated with pathogenicity evolution in a plant virus. Mol Biol Evol 28:1425–1437. doi: 10.1093/molbev/msq327. [DOI] [PubMed] [Google Scholar]
- 60.Fraile A, Hily JM, Pagán I, Pacios LF, García-Arenal F. 2014. Host resistance selects for traits unrelated to resistance-breaking that affect fitness in a plant virus. Mol Biol Evol 31:928–939. doi: 10.1093/molbev/msu045. [DOI] [PubMed] [Google Scholar]
- 61.Jenner CE, Wang X, Ponz F, Walsh JA. 2002. A fitness cost for Turnip mosaic virus to overcome host resistance. Virus Res 86:1–6. doi: 10.1016/S0168-1702(02)00031-X. [DOI] [PubMed] [Google Scholar]
- 62.Khatabi B, Wen RH, Hajimorad MR. 2013. Fitness penalty in susceptible host is associated with virulence of Soybean mosaic virus on Rsv1-genotype soybean: a consequence of perturbation of HC-Pro and not P3. Mol Plant Pathol 14:885–897. doi: 10.1111/mpp.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y, Hajimorad MR. 13 December 2015. Gain of virulence by Soybean mosaic virus on Rsv4-genotype soybeans is associated with a relative fitness loss in a susceptible host. Mol Plant Pathol doi: 10.1111/mpp.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quenouille J, Montarry J, Palloix A, Moury B. 2013. Farther, slower, stronger: how the plant genetic background protects a major resistance gene from breakdown. Mol Plant Pathol 14:109–118. doi: 10.1111/j.1364-3703.2012.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Poulicard N, Pinel-Galzi A, Hébrard E, Fargette D. 2010. Why Rice yellow mottle virus, a rapidly evolving RNA plant virus, is not efficient at breaking rymv1-2 resistance. Mol Plant Pathol 11:145–154. doi: 10.1111/j.1364-3703.2009.00582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miyashita Y, Atsumi G, Nakahara KS. 13 June 2016. Trade-offs for viruses in overcoming innate immunities in plants. Mol Plant Microbe Interact. doi: 10.1094/MPMI-05-16-0103-CR. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.