

Abstract
To maintain corneal transparency, corneal endothelial cells (CECs) exert a pump function against aqueous inflow. However, human CECs are arrested in the G1-phase and non-proliferative in vivo. Thus, treatment of corneal endothelial decompensation is limited to corneal transplantation, and grafts are vulnerable to immune rejection. Here, we show that ribonuclease (RNase) 5 is more highly expressed in normal human CECs compared to decompensated tissues. Furthermore, RNase 5 up-regulated survival of CECs and accelerated corneal endothelial wound healing in an in vitro wound of human CECs and an in vivo cryo-damaged rabbit model. RNase 5 treatment rapidly induced accumulation of cytoplasmic RNase 5 into the nucleus, and activated PI3-kinase/Akt pathway in human CECs. Moreover, inhibition of nuclear translocation of RNase 5 using neomycin reversed RNase 5-induced Akt activation. As a potential strategy for proliferation enhancement, RNase 5 increased the population of 5-bromo-2′-deoxyuridine (BrdU)-incorporated proliferating CECs with concomitant PI3-kinase/Akt activation, especially in CECs deprived of contact-inhibition. Specifically, RNase 5 suppressed p27 and up-regulated cyclin D1, D3, and E by activating PI3-kinase/Akt in CECs to initiate cell cycle progression. Together, our data indicate that RNase 5 facilitates corneal endothelial wound healing, and identify RNase 5 as a novel target for therapeutic exploitation.
In mammals, the transparent and avascular cornea serves as an optical window that allows entry of light and images for optimal vision. To maintain corneal transparency against the inflow of aqueous humor into the corneal stroma, the cornea utilizes a homeostatic system. Among the three different types of cells comprising the cornea, namely, epithelial, stromal and endothelial cells, corneal endothelial cells (CECs) form the innermost monolayer of the cornea in a well-arranged mosaic pattern that acts as a water barrier due to the presence of ionic pumps1.
Human CECs are arrested in the G1-phase of the cell cycle2 due to several negative regulators suppressing the S-phase entry3,4, and thus do not proliferate in vivo. Instead, in vivo CECs overcome minor corneal endothelial damage caused by ocular trauma, intraocular surgery, diabetes, or glaucoma via the migration of adjacent CECs and cell enlargement rather than mitosis5,6. Unfortunately, if the density of human CECs drops below a critical threshold (400–500 cell/mm2) as a result of serious injury or endothelial dystrophy, the decompensated barrier function of the corneal endothelium results in corneal edema with tearing, recurring pain, and eventual vision loss. Corneal transplantation or keratoplasty is currently the only therapeutic option with a favorable outcome7. Nevertheless, the density of CECs decreases rapidly in the first 6 months postoperative before slowing to a rate that still exceeds the physiological loss rate of 0.6% per year8. Moreover, corneal transplantation may cause severe complications, such as graft failure and allograft rejection, which may require re-grafting9,10. Lastly, there continues to be a worldwide shortage of donor corneas.
A number of studies have reported evidence supporting the proliferative capacity of human CECs in ex vivo corneas11,12. In addition, a subpopulation of CECs with potential proliferative potential at the corneal endothelial periphery was recently reported13,14, suggesting that triggering and controlling the proliferation of CECs could serve as a therapeutic bioengineering approach for treating corneal endothelial dysfunction.
Ribonuclease (RNase) 5, commonly known as angiogenin (ANG), is a 14.4-kDa single-chain protein originally identified as a potent inducer of neovascularization15. ANG translocates to the nucleus, where it binds ribosomal DNA and stimulates the transcription of rRNA16,17,18,19 for diverse functions beyond angiogenesis. Thus, ANG protein is now referred to as RNase 5, and is the 5th member of RNase A family20. Although ANG is up-regulated in a variety of human cancers21,22,23,24,25,26,27,28, there have been recent reports of a neuroprotective role of RNase 5 in neurodegenerative disorders29,30,31,32,33,34,35 and a novel action of RNase 5 for promoting cell survival under stress conditions20,36, suggesting that RNase 5 may be an essential homeostatic factor. In ophthalmology research, in a similar vein, RNase 5 appears to be normally present in human tear fluid and may participate in the host defense system37.
We hypothesized that human CECs may be physiologically armed with self-protective mechanisms against aging and injury, including surgery, trauma, and the lifelong shear stress of aqueous flow in corneal endothelium generated by rapid eye movements during sleep38 and convection flow in the anterior chamber. To explore this hypothesis, we investigated the expression of RNase 5, a candidate homeostatic factor, in normal and decompensated human corneal endothelial tissue. We then investigated potential molecular signals of RNase 5 involved in cellular proliferation, consisting of phosphatidylinositol 3-kinase (PI3-k) activation39 and nuclear translocation of RNase 516.
In the present study, we identified the molecular mechanisms required for G1/S progression which is subsequently provoked by RNase 5 for in vitro proliferation of human CECs. We also investigated the role of RNase 5 in corneal endothelial wound healing in a rabbit model of transcorneal cryogenic endothelial injury. Together, our findings suggest that RNase 5, by virtue of its ability to induce CECs to exit the cell cycle, may be a novel treatment approach for corneal endothelial dysfunction.
Results
RNase 5 is distributed to the cytoplasm of CECs in ex vivo normal human corneal endothelium and, by contrast, is deficient in decompensated corneal endothelial tissues
RNase 5 is a candidate survival factor for CECs based on their embryonic origin from the neural crest40 and its recently discovered neuroprotective role. We compared the expression of RNase 5 in human corneal endothelial tissues from deceased subjects without previous cancer or ocular disorders to RNase 5 expression in excised corneal endothelial tissues from patients with corneal endothelial decompensation caused by pseudophakic bullous keratopathy, infection, or burns obtained during keratoplasty. In normal corneal endothelial tissues, most CECs revealed moderate immunofluorescence of RNase 5 (Fig. 1A). Moreover, the intracellular expression of RNase 5 protein was relatively focused in the cytoplasm. On the contrary, decompensated corneal endothelial tissues contained CECs with decreased immunofluorescence of intracellular RNase 5 protein (Fig. 1B). The overall proportion of RNase 5 protein-defective CECs was significantly higher (p = 0.012) in diseased corneal endothelial tissues (Fig. 1C).
Figure 1. Ex vivo expression patterns of ribonuclease (RNase) 5 protein in normal and decompensated human corneal endothelial tissues.

(A,B) Representative images showing flat-mounted immunofluorescence (IF) staining of RNase 5 protein in normal (A) and decompensated human corneal endothelial tissues (B). Central circular corneal tissues with a diameter of approximately 8 mm (dotted black circles) were punched and excised from donor corneas. Thereafter, RNase 5 protein-expressing corneal endothelial cells (CECs) in the central area of corneas (squares in left column) were microscopically evaluated by IF. Protein expression of RNase 5 was clear in CECs of normal tissue, but defective in CECs of decompensated tissues (white arrows, shown representatively). In the magnified views (small white rectangles in middle column), normal CECs exhibited copious expression of RNase 5 protein in the cytoplasm (yellow arrow, A) between the CEC contour (dotted white circle) and nucleus (white solid circle). On the other hand, the expression of RNase 5 protein was defective by IF stain in CECs in decompensated human corneal endothelial tissues (dotted white circles, B). (B) Photos in the left column are snapshots of a cornea taken prior to surgical excision for corneal transplantation. Scale bar: 50 μm except for those in small white rectangles, which are 10 μm. (C) The proportion of CECs that exhibited defective expression of RNase 5 protein according to IF analysis was significantly higher in corneal tissues from patients with decompensated corneal endothelium. *p = 0.012, vs. normal (t-test). n = 5 corneas in each group. Values represent the mean ± s.e.m.
RNase 5 enhances survival of human CECs
RNase 5 has been proposed to mediate both cell survival and growth mechanisms in neurodegenerative disease41. To verify that RNase 5 enhances survival in human CECs, cell viability was measured using the MTT assay in the presence or absence of RNase 5. The number of viable cells was monitored over 48 hours after the start of the culture. Viability of CECs increased over a wide range of RNase 5 concentrations, from 1 to 10 μg/mL, and after culturing for 12, 24 and 48 hours. The effect of cell-survival enhancement by RNase 5 compared to control was noted at all three timepoints (Fig. 2). A concentration of 5 μg/mL, which showed most prominent effect on enhancing CEC viability at all timepoints, was selected for subsequent use in this study.
Figure 2. Dose-dependent cell viability (MTT assay) of human corneal endothelial cells (CECs) treated with ribonuclease (RNase) 5.

Relative CEC viability compared to controls over 48 hours according to the concentration of RNase 5. The number of viable CECs was significantly elevated compared to control by RNase 5, and the lowest concentration of RNase 5 showing maximal CEC viability was 5 μg/mL at all three timepoints (12-, 24- and 48-hour treatments). **p < 0.01, vs. control; ##p < 0.01 and #p < 0.05, vs. 5 μg/mL of RNase 5 (ANOVA followed by Bonferroni’s post-hoc analysis). n = 4 independent experiments at all three timepoints. Values represent the mean ± s.e.m.
RNase 5 promotes wound healing in an in vitro model of human CECs and an in vivo rabbit model
To evaluate the potential effect of RNase 5 on corneal endothelial wound restoration, we administered RNase 5 to an in vitro scrape wound model of cultured human CECs and applied RNase 5 eye drops to an in vivo rabbit corneal endothelial wound model. A time-course treatment with RNase 5 (5 μg/mL) enhanced the migration capacity (wound healing index) of CECs significantly compared with the untreated control (12 hr, p = 2.51e-5; 24 hr, p = 0.002; 36 hr, p = 3.74e-4) (Fig. 3A,B). Although wound healing of CECs pretreated with mitomycin C also tended to be higher in the RNase 5 group, the wound healing index did not differ significantly between both groups (Supplementary Fig. S1), which suggests that RNase 5-induced enhancement of in vitro wound healing may be attributed to the proliferation more than the migration.
Figure 3. Enhancement of corneal endothelial wound healing by ribonuclease (RNase) 5 treatment using in vitro human corneal endothelial cells (CEC) and an in vivo rabbit wound model.

(A,B) In vitro wound healing of cultured CECs with or without RNase 5 (5 μg/mL). (A) The remaining wound area (boundary of yellow line) is shown in both groups. (B) The wound healing index was significantly higher in the RNase 5 group. **p = 2.51e-5 (12 hr); **p = 0.002 (24 hr); **p = 3.74e-4 (36 hr), vs. control (t-test). n = 3 independent experiments. (C) Establishment of a transcorneal cryo-freezing endothelial wound model in rabbits. Scale bar: 40 μm. (D) Representative photos showing the corneal opacity with or without RNase 5 eye drops in rabbits. The corneal opacity (boundary of black line) was more prominent in the control from 72 hours. The iris configuration at pupil margin was completely obscured (grade [Gr] + 4.0) in both groups at 48 hours. Thereafter, the sectorial width of iris contour obscured by corneal opacity (white dotted line) was less at 72 hours (Gr + 3.0) and disappeared from 144 hours (Gr + 0.5) in the RNase 5 group. (E,F) Quantification of corneal opacity and central corneal thickness (CCT) in the control and RNase 5 group. (E) *p = 0.022 (72 hr), vs. control (t-test). (F) **p = 0.003 (144 hr); **p = 0.001 (192 hr), vs. control (t-test). n = 7–10 eyes per group. (G) Representative photos showing the rabbit corneal endothelial wound area and CCT. The remnant wound area by Alizarin red S stain (boundary of yellow dotted line) was 2.696 mm2 at 192 hours in the control, whereas completely healed in the RNase 5 group. The central slit beam (white squares magnified from yellow rectangles) was thicker and the relevant CCT quantified by pachymetry was higher in the control (465 μm). Scale bar: 1 mm. (H) Comparison of corneal endothelial wound area (192 hr) between the control and RNase 5 group. *p = 0.015 (192 hr), vs. control (t-test). n = 4 eyes per group. Values represent the mean ± s.e.m.
To test whether RNase 5 actually enhanced in vivo corneal endothelial wound healing, we used a rabbit model of transcorneal freezing injury of the corneal endothelium, in which the corneal endothelial layer was destroyed by freezing (Fig. 3C). Treatment with RNase 5 eye drops significantly reduced the corneal haziness (Fig. 3D,E) compared to controls at 72 hours after injury (p = 0.022), and the inter-group difference was maintained thereafter, but insignificant. Moreover, RNase 5 significantly lowered central corneal thickness (CCT), which indicates decrease of corneal edema at 144 and 192 hours (p = 0.003 and 0.001, respectively) after injury (Fig. 3F).
To investigate whether RNase 5 could promote corneal endothelial wound healing in an in vivo rabbit model, the wound area in endothelial layer was evaluated by Alizarin red S staining after enucleation at a maximal follow-up period (192 hours). The mean wound area of the RNase 5 group was significantly smaller than that of the control group (p = 0.015) (Fig. 3G–H).
RNase 5 treatment induces nuclear accumulation of RNase 5 in human corneal endothelial tissues and in CECs
It has been reported that RNase 5 exhibits differential subcellular localizations in HeLa cells, being localized to either the nucleus with nucleolar accumulation or the cytoplasm under growth and stress conditions, respectively42. During growth, RNase 5 accumulates in the nucleus where it stimulates rRNA transcription in the vasculature system18,43. To verify this phenomenon in CECs, which express RNase 5 as shown in Fig. 1, we evaluated the immunofluorescence of RNase 5 in ex vivo human corneal endothelial tissues after treatment with exogenous human RNase 5. In both normal and decompensated human corneal endothelial tissues, exogenous RNase 5 treatment (5 μg/mL for 6 hours) induced RNase 5 accumulation in the nucleus (Fig. 4A).
Figure 4. Ribonuclease (RNase) 5 treatment-induced nuclear localization of RNase 5 in human corneal endothelium tissues and cultured human corneal endothelial cells (CECs).

(A) Representative images showing flat-mounted immunofluorescence stain of RNase 5 in normal and decompensated human corneal endothelial tissue with or without RNase 5 treatment. RNase 5 was predominantly expressed in the nucleus after 6-hour treatment with exogenous human RNase 5 (5 μg/mL) in normal and decompensated corneal endothelium (right column). In magnified images of small white rectangles, the contour of CECs (white border lines) and the nuclear expression of RNase 5 after RNase 5 treatment (white dotted circles) are outlined. Scale bar: 50 μm (white) and 10 μm (yellow). n = 3 independent experiments. (B) RNase 5 localization in cultured human CECs with or without RNase 5 treatment. RNase 5 expression was detected at the cytoplasmic perinuclear area in untreated CECs (arrows in the control column). After RNase 5 treatment, intracellular RNase 5 expression became very prominent early (arrows in 1 hr column), and thereafter localized specifically to the nuclei (arrow heads in 3 hr and 6 hr columns). This observation was more apparent with thresholding (upper second row, lightness 30). Scale bar: 100 μm (three upper rows), 20 μm (lowest row). n = 3 independent experiments.
To elucidate the mechanism of this phenomenon, we confirmed that RNase 5-induced nuclear accumulation of RNase 5 in cultured human CECs. Prior to beginning the experiments, the corneal endothelial phenotype of cultured human CECs was identified by the presence and localization of Na+/K+-ATPase and zonula occludens (ZO)-1, which are normally expressed along cell membrane specifically in CECs among cells comprising a cornea, thus are famous markers of CECs (Supplementary Fig. S2).
RNase 5 was detected predominantly in the cytoplasmic perinuclear area in untreated CECs (Fig. 4B, arrow in column control). Treatment with RNase 5 (5 μg/mL) induced copious cytoplasmic staining of RNase 5 (Fig. 4B, arrow in column 1 hr) with intranuclear localization after incubation for 1 hour. Thereafter, the nuclear expression of RNase 5 maintained (Fig. 4B, arrow heads in column 3 and 6 hr) with attenuated staining in the cytoplasm after incubation for 3 and 6 hours. These results indicated that CECs may utilize RNase 5-elicited intracellular signaling. Furthermore, these data suggested that the localization of RNase 5 by exogenous application of RNase 5 varies in a time-dependent manner, with localization to the cytoplasm within 1 hour and to the nucleus of CECs thereafter.
RNase 5 activates Akt in CECs and nuclear translocation of RNase 5 is involved in RNase 5-induced Akt activation
The PI3-k/Akt pathway is centrally involved in cell proliferation44. In a previous study, RNase 5 was shown to promote human umbilical vein cell migration and induce angiogenesis in chick chorioallantotic membranes via phosphorylation of Akt, an important downstream effector of PI3-k45. Although a few reports have stressed the involvement of the PI3-k/Akt pathway for human CEC proliferation46,47, there have been no studies regarding RNase 5-induced Akt activation in CECs. In this study, RNase 5 (5 μg/mL) increased Akt phosphorylation in cultured human CECs in a time-dependent manner (Fig. 5A). Notably, Akt, a representative subcellular cytoplasmic signaling molecule, is activated within one hour of RNase 5 treatment, consistent with the timing of intracytoplasmic RNase 5 localization as shown in Fig. 4B.
Figure 5. Ribonuclease (RNase) 5 activates PI3-kinase (PI3-k)/Akt signal pathway in human corneal endothelial cells (CECs) for wound healing enhancement and the nuclear translocation of RNase 5 is associated with RNase 5-induced PI3-k/Akt activation.

(A) Representative immunoblotting of phosphorylated (p-Akt) and total Akt and quantitative analysis of the band density of relative p-Akt in cultured human CECs before (control) and after RNase 5 treatment (5 μg/mL, for 5 minutes to 3 hours). *p = 0.037, vs. control. n = 4 independent experiments. (B) Representative immunoblotting of p-Akt and total Akt and quantitative analysis of the band density of relative p-Akt in cultured human CECs with or without RNase 5 (5 μg/mL, 24 hours), and in the presence or absence of LY294002 (20 μM, 1 hour) or neomycin (1 mM) co-treatment. **p = 0.002, vs. control. ##p = 1.11e-4, RNase 5 treatment with vs. without LY294002. ##p = 2.06e-4, RNase 5 treatment with vs. without neomycin. n = 3 independent experiments. (A,B) β-actin was used as the loading control for Western blots. Full-length gels are presented in Supplementary Fig. S5. (C,D) The in vitro wound healing in a scraped wound model of cultured human CECs with or without RNase 5 (5 μg/mL, 24 hours), and in the presence or absence of LY294002 (20 μM) or neomycin (1 mM) co-treatment. (D) LY294002 or neomycin significantly inhibited RNase 5-induced wound healing enhancement. **p = 1.33e-6, vs. control. ##p = 1.03e-6, RNase 5 treatment with vs. without LY294002. ##p = 3.61e-6, RNase 5 treatment with vs. without neomycin. n = 3 independent experiments. (A,B,D) Statistical analysis was performed with ANOVA followed by Bonferroni’s post-hoc analysis. Values represent the mean ± s.e.m.
It was reported that neomycin inhibited nuclear translocation of RNase 5 in human umbilical vein endothelial cells and RNase 5-induced cell proliferation for angiogenesis48. Likely, the treatment of CECs with neomycin suppressed RNase 5-induced nuclear accumulation of RNase 5 (Supplementary Fig. S3), and reversed RNase 5-induced increase of ratio of nuclear amount of RNase 5 to cytoplasmic amount (N/C expression ratio) (Supplementary Fig. S4). We hypothesized that intranuclear translocation of RNase 5 may be involved in the activation of the PI3-k/Akt pathway in CECs. Co-treatment with neomycin (1 mM) inhibited RNase 5-induced phosphorylation of Akt, like PI3-k inhibitor (LY294002, 20 μM) did (Fig. 5B), indicating that RNase 5-induced Akt activation is regulated by nuclear translocation of RNase 5 in CECs.
PI3-k/Akt activation is associated with RNase 5-induced enhancement of in vitro wound healing of human CECs
In vitro CEC migration was evaluated with or without RNase 5 (5 μg/mL), and in the presence or absence of LY294002 (20 μM) or neomycin (1 mM) co-treatment to verify the effect of RNase 5-induced, and nuclear translocation of RNase 5-related Akt activation (as noted in Fig. 5A,B) on in vitro wound healing. LY294002 or neomycin co-treatment inhibited the effect of CEC migration enhancement induced by RNase 5 treatment to the level of control at 24 hours (Fig. 5C,D), suggesting that PI3-k/Akt activation may play a key role in the signaling of RNase 5-enhanced corneal endothelial wound healing.
RNase 5 up-regulates mitosis-related cell cycle signaling in human CECs
The most desirable strategy for corneal endothelial wound healing is to repair the corneal endothelium by releasing CECs from cell-cycle arrest and replicating residual CECs to replace dead or injured cells. In this context, we explored the possibility of RNase 5-elicited cell cycle signaling for proliferation of human CECs by analyzing Ki-67 expression and 5-bromo-2′-deoxyuridine (BrdU) incorporation into DNA. Subconfluent CECs were treated with RNase 5 (5 μg/mL for 24 hours) and Ki-67+ cells were counted and compared to control CECs to analyze differences in proliferation. The population of Ki-67+ cells after treatment with RNase 5 was significantly increased to approximately 75% among the total population, compared to 18% in the absence of RNase 5 (p = 8.52e-6) (Fig. 6A,B). Moreover, the proliferative effect of RNase 5 was confirmed using an in vitro scrape wound model. Interestingly, RNase 5 elevated Ki-67 expression, which was mainly confined to the initial margin of the wound where migrating CECs were concentrated and not subject to contact-inhibition (Supplementary Fig. S6).
Figure 6. Effect of ribonuclease (RNase) 5 on the proliferation of cultured human corneal endothelial cells (CECs).

(A,B) Representative images of Ki-67 staining of cultured human CECs with or without RNase 5 treatment (5 μg/mL, 24 hours, A) and comparison of proportion of Ki-67+ cells between each group (B). (A) In magnified images of small white rectangles, the Ki-67+ cells with or without RNase 5 treatment are indicated (white arrows). Scale bar: 200 μm (white) and 100 μm (yellow). n = 3 independent experiments. (B) **p = 8.52e-6, vs. control (t-test). n = 6 independent experiments. (C) Relative BrdU incorporation in cultured human CECs with RNase 5 treatment (5 μg/mL, 24 hours) and LY294002 (20 μM) or neomycin (1 mM) co-treatment compared to control. RNase 5 elevated BrdU incorporation in CECs more prominently in cells of the scraping wound model compared with confluent cells. Confluent (ANOVA followed by Bonferroni’s post-hoc analysis): *p = 0.023, vs. control; ##p = 6.04e-6, RNase 5 treatment with vs. without LY294002. Subconfluent (ANOVA followed by Bonferroni’s post-hoc analysis): **p = 3.43e-4, vs. control; ##p = 5.84e-6, RNase 5 treatment with vs. without LY294002; ##p = 7.60e-6, RNase 5 treatment with vs. without neomycin. **p = 0.001, RNase 5 treatment in confluent vs. scrape wound model (t-test). n = 3 independent experiments. (D) Growth curves of CECs with or without RNase 5 treatment (5 μg/mL). *p = 0.043, vs. control (D0); *p = 0.038, vs. control (D4) (t-test). n = 5 independent experiments. Values represent the mean ± s.e.m.
Although it is still not fully understood what mediates the inhibition of CEC proliferation in mature corneal endothelia, contact-inhibition is a strong candidate49. Thus, we treated cultured CECs with RNase 5 under two different conditions: 1) fully confluent and 2) scrape-wound subconfluent plated CECs. Proliferation, assessed by BrdU incorporation, increased with RNase 5 (5 μg/mL for 24 hours) in confluent and subconfluent conditions by approximately 10% and 60%, respectively (Fig. 6C). Moreover, blockade of either PI3-k/Akt signaling by LY294002 or nuclear translocation of RNase 5 by neomycin, significantly inhibited RNase5-induced proliferation in subconfluent CECs under scrape wound condition (p = 5.84e-6 and 7.60e-6, respectively). The increase of BrdU incorporation by RNase 5 was significantly higher in subconfluent CECs (p = 0.001) compared to fully confluent cells. Based on these results, we hypothesized that RNase 5 may have a sufficient mitotic effect on CECs under wound conditions where there is a loss of contact-inhibition.
The velocity of CEC growth either in the absence or in the presence of RNase 5 was subsequently verified to evaluate the effect of RNase 5 on the eventual proliferation of CECs. According to daily growth curves of CECs, RNase 5 enhanced CEC growth compared to control with significance at initial growth (day 1) and at close to being confluent (day 4) (Fig. 6D).
RNase 5 mediates p27Kip1 phosphorylation and upregulation of cyclin D1, D3, and E via activation of PI3-k/Akt pathway
P27Kip1 protein binds to and prevents the activation of cyclin E-CDK2 or cyclin D-CDK4 complexes, thus inhibiting the S phase progression. Activation of cyclin D in early G1 and cyclin E in late G1 promote S phase entry and progression, respectively, as positive G1 regulators11. The abundance of p27 is regulated by its phosphorylation and subsequent ubiquitin-proteasome-mediated degradation47. Thus, phosphorylation of p27 is a prerequisite for degradation of p27 and a major mechanism for G1/S cell cycle progression50. Importantly, PI3-k/Akt signaling is involved in the removal of p27, furthering the proliferation of CECs51,52. To reveal the mechanism for CEC proliferation driven by RNase 5 as shown in Fig. 6, we investigated the alteration of positive and negative G1 regulators in cultured human CECs treated with RNase 5. In cultured human CECs, RNase 5 up-regulated phosphorylation of p27Kip1 and induced the expression of cyclins D1, D3, and E. These effects were reversed by blockade of PI3-k signaling using LY294002, or by inhibition of nuclear translocation of RNase 5 using neomycin (Fig. 7). The enhancement of cyclin D1 and D3 by RNase 5 (Fig. 7B,C) was less prominent than that of cyclin E (Fig. 7D). In addition, neomycin did not suppress RNase 5-induced cyclin D1 (Fig. 7B) unlike cyclin D3 and E, suggesting that activation of cyclin D1 by RNase 5 occurs through PI3-k/Akt signaling itself rather than nuclear localization of RNase 5 in CECs.
Figure 7. The effect of ribonuclease (RNase) 5 on G1 to S cell cycle progression via PI3-kinase/Akt activation in cultured human corneal endothelial cells (CECs).
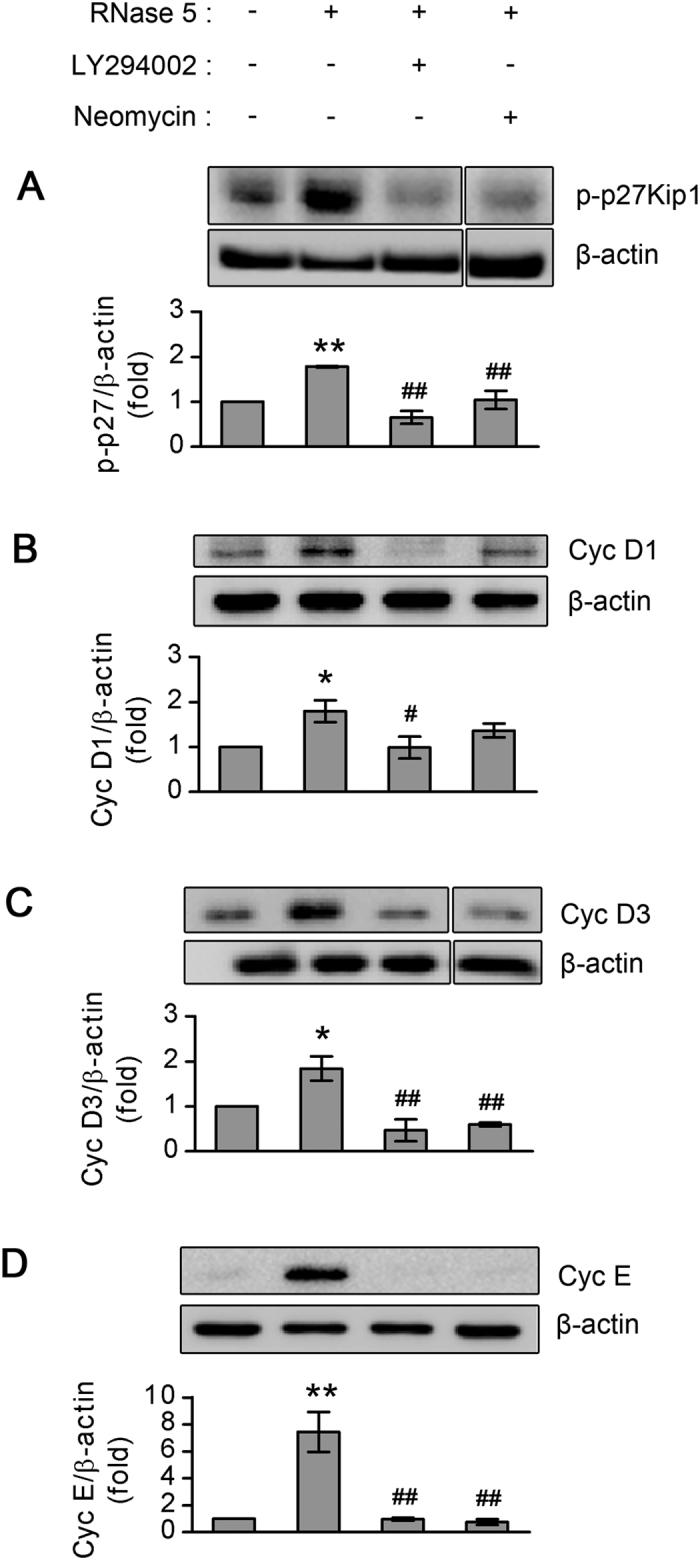
(A–D) Representative immunoblotting and quantitative analysis of band density of phosphorylated p27Kip1 (p-p27Kip1, A), cyclin (Cyc) D1 (B), cyc D3 (C) and cyc E (D) in cultured human CECs with or without RNase 5 (5 μg/mL, 24 hours), and in the presence or absence of LY294002 (20 μM) or neomycin (1 mM) co-treatment. P27Kip1 (A) **p = 0.005, vs. control; ##p = 1.86e-4, RNase 5 treatment with vs. without LY294002; ##p = 0.007, RNase 5 treatment with or without neomycin. Cyc D1 (B) *p = 0.046, vs. control; #p = 0.042, RNase 5 treatment with vs. without LY294002. Cyc D3 (C) *p = 0.042, vs. control; ##p = 0.001, RNase 5 treatment with vs. without LY294002; ##p = 0.003, RNase 5 treatment with vs. without neomycin. Cyc E (D) **p = 3.63e-4, vs. control; ##p = 3.45e-4, RNase 5 treatment with vs. without LY294002; ##p = 2.53e-4, RNase 5 treatment with or without neomycin. Statistical analysis was performed with ANOVA followed by Bonferroni’s post-hoc analysis. n = 4 independent experiments. β-actin was used as a loading control for Western blotting, and the small gaps indicate skipped lanes from the same membrane. Values represent the mean ± s.e.m. Full-length gels are presented in Supplementary Fig. S7.
Based on these results, we speculated that RNase 5 activates PI3-k/Akt signaling in CECs via its ability to undergo nuclear localization, ultimately leading to cell cycle progression and cellular proliferation (Fig. 8).
Figure 8. Proposed mechanism of mitotic pathways in human corneal endothelial cells induced by ribonuclease (RNase) 5-elicited PI3-kinase/Akt activation and by nuclear translocation of RNase 5.

Discussion
Finding a therapeutic pharmaceutical to restore the function of damaged corneal endothelia has long been a goal of ophthalmologists due to the invasiveness and risk of corneal transplantation, which is the only current surgical treatment option. However, no definite regenerative zone has been ascertained in the human corneal endothelium. Therefore, the ability to maintain CEC viability and promote proliferation is in high demand as an alternative to waiting for passive recovery by a homeostatic mechanism that may not exist in the corneal endothelia. In the present study, we identified a role of RNase 5 not previously implicated in human CECs. In brief, treatment of CECs with exogenous human RNase 5 led to strong localization of RNase 5 in the cytoplasm and nucleus in cultured human CECs. Moreover, application of RNase 5 enhanced corneal endothelial wound healing both in vitro and in vivo corneal endothelial wound models. Furthermore, we demonstrated that these actions of RNase 5 were likely due to enhanced PI3-k/Akt signaling-mediated cell cycle progression in CECs.
Unlike general wound healing mechanisms, the wound repair process observed in the human corneal endothelium is accomplished not by cell proliferation, but instead by migration of neighboring CECs53. Thus, the abundant cell population on the flattened monolayer of the corneal endothelium is not protected against injury or disease54. Moreover, the excessive cellularity of the corneal endothelium would rather induce refractive alteration or blurring that threatens the vision as seen in the retrocorneal membrane, a representative vision-threatening corneal endothelial pathologic condition. We hypothesized that the unsatisfactory proliferative capacity of human CECs may be evolutionarily linked to several necessary mechanisms for anti-proliferation to maintain corneal endothelium as one definite layer, including the low level of growth factors in aqueous humor, lack of autocrine growth factors produced by CECs, and blocked G1/S phase transition in CECs2,11,55. However, there may be some potential proliferative capacity in CECs when exposed to extraordinary conditions, such as trauma or invasive eye surgery. In support of this idea, recent reports have demonstrated the possible presence of stem cell niches in the extreme periphery of the human corneal endothelium13. In addition, rho-kinase inhibitors have been investigated as factors that stimulate the proliferation of human and monkey CECs56. In the present study, we postulated two separate mechanisms responsible for the homeostasis of the human corneal endothelium: 1) anti-proliferation of CECs in ordinary healthy conditions and 2) unlocking mitosis of CECs under wound conditions.
RNase 5 has been studied with respect to tumor growth and the pathogenesis of several cancer types. Moreover, the PI3-k/Akt pathway, which was activated by RNase 5 in this study, is known significantly involved in regulating growth, but is responsible for tumorigenesis. In this context, it is notable that treatment with RNase 5 increased the number of BrdU-incorporating CECs to a significantly greater degree in wound conditions than under confluency (Fig. 6C). Although RNase 5 significantly elevated proliferative capacity in confluent CECs, the increase in the level of BrdU incorporation was not distinct compared to controls. In addition, after RNase 5 treatment, Ki-67-expressing CECs were mainly confined to the migrating cells adjacent to the wound scrape (Supplementary Fig. S6). Together, these results suggest that the growth effect of RNase 5 on CECs follows the contact-inhibition theory, and that RNase 5 treatment is unlikely to induce tumor formation through the uninhibited and persistent proliferation of CECs, especially when the corneal endothelium is nearly healed or normal.
A clear difference following RNase 5 treatment in ex vivo corneal endothelial tissues as well as cultured CECs was the intracellular differential localization of RNase 5. Prior to treatment, the intracellular expression of RNase 5 was confined to the cytoplasm (Fig. 1A), while RNase 5 expression became dominant in the nucleus with accumulation in the putative nucleolus after RNase 5 treatment. This observation was consistent with reports that RNase 5 undergoes nuclear translocation and accumulates in the nucleolus, where it is involved in rRNA transcription during proliferative growth in vascular endothelial16, cancer19 and neuronal cells31. We postulated PI3-k/Akt signaling as one of the key pathways for corneal endothelial wound healing which, importantly, occurs via nuclear localization of RNase 5 in CECs.
In vivo human CECs express p27Kip13,4, and the action of p27Kip1 differs depending on its subcellular localization57. P27Kip1 controls cells in the G1 phase by binding to cyclin D or E complexes in the nucleus, but can no longer inhibit cell proliferation as it is phosphorylated and degraded upon being exported from the nucleus into the cytoplasm. Blocking the nuclear translocation of RNase 5 inhibited the p27Kip1 phosphorylation in human CECs (Fig. 7A). This result indicated that RNase 5, in the nucleus, may affect the nuclear export of p27, thereby activating cyclin D and E in CECs. Notably, ex vivo expression of RNase 5 protein are predominantly in the cytoplasm of normal human corneal endothelial tissues (Fig. 1A). We suggest that resident endogenous RNase 5 in CECs exists mainly in the cytoplasm in the stable, unthreatened corneal endothelium to suppress cell cycle progression by keeping the action of negative regulators, including p27Kip1, thereby maintaining contact-inhibition.
Decompensated corneal endothelial tissues included many CECs with defective overall intracellular expression of RNase 5 protein (Fig. 1B). Based on copious cytoplasmic existence of RNase 5 in CECs from normal tissues (Fig. 1A), our results suggest that the varied degrees of deficiency of RNase 5 in CECs from decompensated tissues may have been due to consumption of pre-existing intracellular RNase 5 promoting survival or proliferation of CECs. However, the correlation between intracellular shortage of RNase 5 and limitation of proliferation of CECs should be confirmed in future in vivo studies.
This is the first study to demonstrate an effect of RNase 5 on the mitosis of human CECs. In addition, this is the first report to show RNase 5-induced activation of mitosis-related factors including cyclin D1, D3, and E. Although it is unclear why RNase 5 activated cyclin E much more prominently than cyclin D1 and D3, one possibility is that cyclin D and E control different events in addition to having an additive effect on shortening the G1 interval as previously reported58.
For the purpose of exploring RNase 5-elicited pathways other than PI3-k/Akt in CECs, we investigated signaling pathways known to enhance cell proliferation, including mTOR, Erk, and inhibition of rho-kinase56 (Supplementary Fig. S8). Unexpectedly, RNase 5 treatment did not affect the phosphorylation of Erk and P70s6k (the downstream target of mTOR) and only moderately inhibited myosin phosphatase target subunit 1 (MYPT1, the downstream target of rho-kinase). This result was inconsistent with the role of RNase 5 in human trabecular meshwork cells reported in our previous study59. Thus, it seems that RNase 5-elicited signals differ according to the type of ocular cell. We expect that future genome sequencing or microarray analysis with RNase 5 may aid in discovering other promising target specific for proliferation of CECs.
One limitation of this study was that we only used LY294002 to inhibit PI3-k specifically and we applied LY294002 at a fixed concentration (20 μM). Several previous reports46,47,56,60 have used LY294002 at the known optimal concentration of 20 μM61 to inhibit PI3-k in CECs. Furthermore, LY294002 does not inhibit other kinases at higher concentrations (50 μM) according to its product data sheet. Nevertheless, specific inhibition of PI3-k in our study should have been confirmed using other PI3-k inhibitors to reduce concerns that LY294002 may have inhibited other targets such as mTOR to trigger unexpected feedback mechanisms involving Akt activation.
In future studies, RNase 5-induced enhancement of wound healing should be reproduced in the eyes of primate mammals, because rabbit CECs are known to be capable of undergoing proliferation. However, we identified that RNase 5 is expressed in the normal human corneal endothelium and is capable of unlocking the halted mitosis of CECs via its nuclear accumulation and activation of PI3-k/Akt signaling. Thus, our results contribute to an increased understanding of a long-standing problem in the management of corneal endothelial diseases. In our opinion, co-treatment of RNase 5 in conjunction with future CEC transplantation or pre-established endothelial keratoplasty may result in higher rates of successful treatment.
In conclusion, with respect to RNase 5 treatment-elicited enhancement of wound healing in both in vitro and in vivo corneal endothelial wound models, RNase 5 may provide a mechanism to restore the injured corneal endothelium and is thus a useful target for therapeutic exploitation.
Methods
Materials, reagents and antibodies
Detailed information of product numbers and commercial suppliers of materials, reagents and antibodies used in this study is listed in the Supplementary Methods.
Whole mount staining of cadaveric corneal tissue
Human corneal tissues from the control group (normal) and patient group with decompensated corneal endothelium, were obtained for research and central 8 mm round corneal tissues excised were stored at 4 °C in storage medium (Optisol-GS, Bausch & Lomb, Rochester, NY, USA). When treating with RNase 5, the corneal tissues were stored in Endothelial Growth Medium (EGM)-2MV BulletKitTM (Lonza, Walkersville, MD, USA) medium with human RNase 5 (5 μg/mL) for 6 hours at 37 °C in an incubator. Next, cornea tissues were fixed in 4% paraformaldehyde at 4 °C overnight and washed 3 times with phosphate-buffered saline (PBS) at room temperature (RT). Corneal endothelium was then permeabilized by incubation for 20 minutes at RT with PBS supplemented with 0.5% Triton X-100 and washed by rocking 3 times in PBS. Subsequent steps are described in the Supplementary Methods.
The expression of RNase 5 in CECs of human corneal endothelial tissues was determined based on standard photos of CECs expressing RNase 5 (small white rectangle in Fig. 1A) or defective intracellular RNase 5 (small white rectangles in Fig. 1B).
Animals
New Zealand White rabbits (total 24 eyes) weighing 2.0 to 3.0 kg and raised at the Clinic Research Center, Chung-Ang University, College of Medicine, were used and in this study. Animals were assigned to one of three groups: control group, RNase 5 treatment group, and a LY294002 + RNase 5 treatment group.
Preparation of cryo-damage of corneal endothelium in rabbit model
A combination of tiletimine and zolazepam-mixed agent (ZoletilTM, Virbac, Fort Worth, TX, USA) and xylazine (RompunTM, Bayer, Leverkusen, Germany) was injected intramuscularly for anesthesia (12.5 mg/kg). The corneal endothelium of rabbits was cryo-damaged according to the established protocol by Okumura et al.62. In brief, a stainless steel probe with a diameter of 7 mm was immersed in liquid nitrogen tank for 3 minutes to achieve a stabilized temperature of approximately −196 °C. Thereafter, corneal endothelium was damaged by transcorneal freezing by gently touching the central corneal surface with the probe for 15 seconds as noted in Fig. 3C. The ocular surface was then irrigated using 10 mL of normal saline. Topical levofloxacin eye drops (CravitTM, Santen, Osaka, Japan) were applied 4 times a day for 3 days to prevent infection.
Instillation of eye drops in rabbit model
RNase 5 (200 μg/mL) eye drops and PBS eye drops as a control were applied 6 times (50 μL in each application) a day for 48 hours, and then 4 times a day until 192 hours after injury. RNase 5 was diluted in PBS.
Analysis of corneal edema and central corneal thickness in rabbit model
Corneal edema was evaluated and central corneal thickness was measured at 48, 72, 144 and 192 hours after cryo-injury in the rabbit model. Corneal edema was evaluated under slit-lamp biomicroscopic examination. The severity of corneal edema was quantified from 0 to +4.0 according to the well-known grading system of corneal opacity (0: completely clear cornea; +0.5: faint haze, only detectable by careful oblique illumination; +1.0: minimal haze, seen with difficulty with direct illumination; +2.0: mild haze, easily visible with direct focal slit illumination; +3.0: moderately dense haze that partially obscured iris details; +4.0: severe dense haze that obscured completely iris details) established by Fantes et al.63. Central corneal thickness was measured using an ultrasound corneal pachymeter (POCKET-II, Quantel medical, Clemont-Ferrand, France; Measureable up to maximal 1,000 μm). The average of three repeated measurements was recorded for analysis.
Analysis of corneal endothelial wound area in rabbit model
Rabbit eyes were enucleated at 192 hours and corneal button tissues were excised from the eyeball. The excised corneal tissues were then immersed in 1% Alizarin red S solution (Lab Chem, Pittsburgh, PA, USA) for 2 minutes and washed with normal saline. Next, flat-mounts were placed on glass slides with the endothelial layer facing up and photo-documented under inverted optical microscopic examination (IX71, Olympus, Tokyo, Japan). The corneal endothelial wound area was measured using Image J software ver. 1.46 (National Institutes of Health (NIH); http://rsbweb.nih.gov/ij/).
Culture of human CECs
Human corneal tissues for corneal transplantation without previous ocular diseases were obtained and the peripheral corneal tissues after excision of central 8 mm round cornea were used for the culture of human CECs. Corneal tissues were washed 3 times in a 5× antibiotics (penicillin/streptomycin) solution followed by 6 times in PBS. Cells on the corneal endothelial layer were isolated using a peel-and-digest approach. Subsequent steps are described in the Supplementary Methods.
Cell viability (MTT) assay
Human CECs were cultured in 96-well culture plates for 16 hours. Cell viability was assessed using the MTT assay method. Briefly, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was dissolved in PBS at a concentration of 5 mg/mL. MTT was then added to each well (10 μL per 100 μL medium) and plates were incubated at 37 °C for 2 hours in the absence of light. The medium was replaced with 100 μL dimethyl sulfoxide (DMSO), and the absorbance for each well was measured at 570 nm using a SpectramaxTM 340PC384 microplate photometer (Molecular Devices, Sunnyvale, CA, USA).
Immunocytochemistry of RNase 5 and Ki-67
Human CECs were seeded at a density of 1.6 × 104 cells/cm2 on FNC-coated 12 mm coverslips (Deckglasser, Mülheim, Germany). CECs were incubated in the presence or absence of human RNase 5 treatment (5 μg/mL) 24 hours after seeding. For RNase 5 treatment, cells were treated for 1, 3, 6 (immunocytochemistry of RNase 5), or 24 hours (immunocytochemistry of Ki-67) as indicated. Scraping using a 200 μL pipette (yellow tip) was done in some of slides to detect Ki-67-positive CECs before addition of RNase 5 to analyze the effect of RNase 5 as an in vitro subconfluent wound model. Subsequent steps are described in the Supplementary Methods.
Immunocytochemistry of Na+/K+ATPase and ZO-1
Human CECs were seeded at a density of 2.0 × 105 cells/cm2 on FNC-coated 8-well Lab-Tek II chamber glass slides (Nalge Nunc Inc., Naperville, IL, USA). CECs cultured for 2 days were washed twice with PBS and fixed with 4% paraformaldehyde for 20 minutes. Subsequent steps are described in the Supplementary Methods.
Wound healing assay of cultured CECs
Human CECs were grown in 6-well plates to create 80 to 90% confluent monolayer. To create scratch wounds, a straight line in the cell monolayer was made by scraping with 200 μL pipette (yellow tip) in a straight line. Next, for RNase 5 treatments, CECs were treated with RNase 5 (5 μg/mL) until 36 hours. When treating with inhibitors, CECs were pre-incubated with LY294002 (20 μM) or neomycin (1 mM) for 1 hour before and during exposure to RNase 5 (5 μg/mL for 24 hours). Images of migrated cells were documented by inverted optical microscopic examination (IX71, Olympus, Tokyo, Japan) and wound areas were analyzed using Image J software. The extent of wound healing was determined by the areas of migration into the denuded area and presented as a wound healing index (%): (initial wound area-remaining wound area)/initial wound area. For the wound healing assay using mitomycin C, CECs were treated with 5 μg/mL mitomycin C for 2 hours prior to wounding in order to eliminate the influence of cell proliferation.
BrdU cell proliferation assay
Cell proliferation was measured using the BrdU Cell Proliferation Kit (Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. Human CECs were seeded at a density of 7,500 cells/well in FNC-coated 8-well Lab-Tek II chamber glass slides (Nunc, Naperville, IL, USA). To establish the scraping wound model, cells were scratched with a 200 μL pipette in the middle of wells 24 hours after seeding. For inhibitor treatments, human CECs were pre-incubated with either LY294002 (20 μM) or neomycin (1 mM) for 1 hour before and during exposure to RNase 5 (5 μg/mL for 24 hours). Subsequent steps are described in the Supplementary Methods.
Cell growth analysis
CECs were seeded at a concentration of 1 × 105 cells/mL in a 35 mm plate (SPL life sciences, Pocheon, Gyeonggi-do, Republic of Korea) and treated with or without RNase 5 (5 ug/mL). Following 24, 48, 72, 96 and 120 hours (day 5), CECs were harvested, diluted by trypan blue working solution and counted using hemocytometer.
Western blot
When verifying the time-dependent effect of RNase 5 on Akt expression, cultured human CECs were treated with RNase 5 (5 μg/mL) for various times (5 minutes to 3 hours). When CECs were treated with inhibitors, cells were pre-incubated with either LY294002 (20 μM) or neomycin (1 mM) for 1 hour before and during exposure to RNase 5 (5 μg/mL for 24 hours). The protocol for Western blot analysis performed in this study is described in the Supplementary Methods.
Statistics
Two-tailed Student’s t-test and analysis of variance (ANOVA) followed by post-hoc analysis were used for the statistical analysis between two groups and among three or more groups, respectively. p values < 0.05 were considered significant.
Study approval
All experiments other than animal studies were approved by the Institutional Review Boards at the Chung-Ang University Hospital, and the methods were carried out in accordance with the approved guidelines. Informed consent was obtained for the use of human cadaveric ocular tissues in accordance with the Declaration of Helsinki. Animal procedures in this study were performed in accordance with ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and were approved by the Chung-Ang University Ethics Committee for Animal Experiment.
Additional Information
How to cite this article: Kim, K. W. et al. Ribonuclease 5 facilitates corneal endothelial wound healing via activation of PI3-kinase/Akt pathway. Sci. Rep. 6, 31162; doi: 10.1038/srep31162 (2016).
Supplementary Material
Acknowledgments
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (NRF-2015R1A2A2A01004643). We wish to respect the memory of the late Prof. S.I. Chang (College of Biochemistry, Chungbuk National University, Cheongju, Chungbuk, Republic of Korea) who passed away at 2014 and devoted his life to the research of angiogenin (RNase 5).
Footnotes
Author Contributions K.W.K. and J.C.K. designed all the experiments. In vitro experiments were performed by S.H.P. and S.J.L. Animal experiments were performed by K.W.K. and S.H.P. The data was analyzed by K.W.K., S.H.P. and S.J.L. K.W.K. wrote the manuscript and J.C.K. revised the manuscript.
References
- Hodson S. The endothelial pump of the cornea. Invest Ophthalmol Vis Sci 16, 589–591 (1977). [PubMed] [Google Scholar]
- Joyce N. C. Proliferative capacity of the corneal endothelium. Prog Retin Eye Res 22, 359–389 (2003). [DOI] [PubMed] [Google Scholar]
- Joyce N. C., Meklir B., Joyce S. J. & Zieske J. D. Cell cycle protein expression and proliferative status in human corneal cells. Invest Ophthalmol Vis Sci 37, 645–655 (1996). [PubMed] [Google Scholar]
- Joyce N. C., Navon S. E., Roy S. & Zieske J. D. Expression of cell cycle-associated proteins in human and rabbit corneal endothelium in situ. Invest Ophthalmol Vis Sci 37, 1566–1575 (1996). [PubMed] [Google Scholar]
- Matsuda M. et al. Cellular migration and morphology in corneal endothelial wound repair. Invest Ophthalmol Vis Sci 26, 443–449 (1985). [PubMed] [Google Scholar]
- Sherrard E. S. The corneal endothelium in vivo: its response to mild trauma. Exp Eye Res 22, 347–357 (1976). [DOI] [PubMed] [Google Scholar]
- Tan D. T., Dart J. K., Holland E. J. & Kinoshita S. Corneal transplantation. Lancet 379, 1749–1761 (2012). [DOI] [PubMed] [Google Scholar]
- Bourne W. M., Nelson L. R. & Hodge D. O. Central corneal endothelial cell changes over a ten-year period. Invest Ophthalmol Vis Sci 38, 779–782 (1997). [PubMed] [Google Scholar]
- Rahman I. et al. The influence of donor and recipient factors in allograft rejection of the human cornea. Eye (Lond) 24, 334–339 (2010). [DOI] [PubMed] [Google Scholar]
- Terry M. A. et al. Endothelial keratoplasty: the influence of preoperative donor endothelial cell densities on dislocation, primary graft failure, and 1-year cell counts. Cornea 27, 1131–1137 (2008). [DOI] [PubMed] [Google Scholar]
- Joyce N. C. Proliferative capacity of corneal endothelial cells. Exp Eye Res 95, 16–23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoo T. & Joyce N. C. Cell cycle kinetics in corneal endothelium from old and young donors. Invest Ophthalmol Vis Sci 41, 660–667 (2000). [PubMed] [Google Scholar]
- He Z. et al. Revisited microanatomy of the corneal endothelial periphery: new evidence for continuous centripetal migration of endothelial cells in humans. Stem cells 30, 2523–2534 (2012). [DOI] [PubMed] [Google Scholar]
- Espana E. M., Sun M. & Birk D. E. Existence of Corneal Endothelial Slow-Cycling Cells. Invest Ophthalmol Vis Sci 56, 3827–3837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fett J. W. et al. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry 24, 5480–5486 (1985). [DOI] [PubMed] [Google Scholar]
- Moroianu J. & Riordan J. F. Nuclear translocation of angiogenin in proliferating endothelial cells is essential to its angiogenic activity. Proc Natl Acad Sci USA 91, 1677–1681 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. P., Tsuji T., Riordan J. F. & Hu G. F. Identification and characterization of an angiogenin-binding DNA sequence that stimulates luciferase reporter gene expression. Biochemistry 42, 121–128 (2003). [DOI] [PubMed] [Google Scholar]
- Xu Z. P., Tsuji T., Riordan J. F. & Hu G. F. The nuclear function of angiogenin in endothelial cells is related to rRNA production. Biochem Biophys Res Commun 294, 287–292 (2002). [DOI] [PubMed] [Google Scholar]
- Tsuji T. et al. Angiogenin is translocated to the nucleus of HeLa cells and is involved in ribosomal RNA transcription and cell proliferation. Cancer Res 65, 1352–1360 (2005). [DOI] [PubMed] [Google Scholar]
- Li S. & Hu G. F. Emerging role of angiogenin in stress response and cell survival under adverse conditions. J Cell Physiol 227, 2822–2826 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka N., Wang L., Kishimoto K., Tsuji T. & Hu G. F. A therapeutic target for prostate cancer based on angiogenin-stimulated angiogenesis and cancer cell proliferation. Proc Natl Acad Sci USA 103, 14519–14524 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozman A., Silar M. & Kosnik M. Angiogenin and vascular endothelial growth factor expression in lungs of lung cancer patients. Radiol Oncol 46, 354–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto K. et al. Hypoxia-induced up-regulation of angiogenin, besides VEGF, is related to progression of oral cancer. Oral Oncol 48, 1120–1127 (2012). [DOI] [PubMed] [Google Scholar]
- Bodner-Adler B. et al. Serum levels of angiogenin (ANG) in invasive cervical cancer and in cervical intraepithelial neoplasia (CIN). Anticancer Res 21, 809–812 (2001). [PubMed] [Google Scholar]
- Shimoyama S. & Kaminishi M. Increased angiogenin expression in gastric cancer correlated with cancer progression. J Cancer Res Clin Oncol 126, 468–474 (2000). [DOI] [PubMed] [Google Scholar]
- Hisai H. et al. Increased expression of angiogenin in hepatocellular carcinoma in correlation with tumor vascularity. Clin Cancer Res 9, 4852–4859 (2003). [PubMed] [Google Scholar]
- Verstovsek S. et al. Significance of angiogenin plasma concentrations in patients with acute myeloid leukaemia and advanced myelodysplastic syndrome. Br J Haematol 114, 290–295 (2001). [DOI] [PubMed] [Google Scholar]
- Raghu H. et al. Suppression of uPA and uPAR attenuates angiogenin mediated angiogenesis in endothelial and glioblastoma cell lines. PLoS One 5, e12458 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D. et al. Angiogenin loss-of-function mutations in amyotrophic lateral sclerosis. Ann Neurol 62, 609–617 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es M. A. et al. Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann Neurol 70, 964–973 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiyagarajan N., Ferguson R., Subramanian V. & Acharya K. R. Structural and molecular insights into the mechanism of action of human angiogenin-ALS variants in neurons. Nat Commun 3, 1121 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidinger T. U., Standaert D. G. & Yacoubian T. A. A neuroprotective role for angiogenin in models of Parkinson’s disease. J Neurochem 116, 334–341 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. N. & Kim do H. Decreased serum angiogenin level in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry 38, 116–120 (2012). [DOI] [PubMed] [Google Scholar]
- Kieran D. et al. Control of motoneuron survival by angiogenin. J Neurosci 28, 14056–14061 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenway M. J. et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 38, 411–413 (2006). [DOI] [PubMed] [Google Scholar]
- Czech A., Wende S., Morl M., Pan T. & Ignatova Z. Reversible and rapid transfer-RNA deactivation as a mechanism of translational repression in stress. PLoS Genet 9, e1003767 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack R. A. et al. Membrane array characterization of 80 chemokines, cytokines, and growth factors in open- and closed-eye tears: angiogenin and other defense system constituents. Invest Ophthalmol Vis Sci 46, 1228–1238 (2005). [DOI] [PubMed] [Google Scholar]
- Modarreszadeh S., Abouali O., Ghaffarieh A. & Ahmadi G. Physiology of aqueous humor dynamic in the anterior chamber due to rapid eye movement. Physiol Behav 135, 112–118 (2014). [DOI] [PubMed] [Google Scholar]
- Trouillon R., Kang D. K., Park H., Chang S. I. & O’Hare D. Angiogenin induces nitric oxide synthesis in endothelial cells through PI-3 and Akt kinases. Biochemistry 49, 3282–3288 (2010). [DOI] [PubMed] [Google Scholar]
- Bahn C. F. et al. Classification of corneal endothelial disorders based on neural crest origin. Ophthalmology 91, 558–563 (1984). [DOI] [PubMed] [Google Scholar]
- Li S. & Hu G. F. Angiogenin-mediated rRNA transcription in cancer and neurodegeneration. Int J Biochem Mol Biol 1, 26–35 (2010). [PMC free article] [PubMed] [Google Scholar]
- Pizzo E. et al. Ribonuclease/angiogenin inhibitor 1 regulates stress-induced subcellular localization of angiogenin to control growth and survival. J Cell Sci 126, 4308–4319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Monti D. M. & Hu G. Angiogenin activates human umbilical artery smooth muscle cells. Biochem Biophys Res Commun 285, 909–914 (2001). [DOI] [PubMed] [Google Scholar]
- Liang J. & Slingerland J. M. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2, 339–345 (2003). [PubMed] [Google Scholar]
- Kim H. M., Kang D. K., Kim H. Y., Kang S. S. & Chang S. I. Angiogenin-induced protein kinase B/Akt activation is necessary for angiogenesis but is independent of nuclear translocation of angiogenin in HUVE cells. Biochem Biophys Res Commun 352, 509–513 (2007). [DOI] [PubMed] [Google Scholar]
- Lee J. G., Song J. S., Smith R. E. & Kay E. P. Human corneal endothelial cells employ phosphorylation of p27(Kip1) at both Ser10 and Thr187 sites for FGF-2-mediated cell proliferation via PI 3-kinase. Invest Ophthalmol Vis Sci 52, 8216–8223 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. G. & Kay E. P. PI 3-kinase/Rac1 and ERK1/2 regulate FGF-2-mediated cell proliferation through phosphorylation of p27 at Ser10 by KIS and at Thr187 by Cdc25A/Cdk2. Invest Ophthalmol Vis Sci 52, 417–426 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G. F. Neomycin inhibits angiogenin-induced angiogenesis. Proc Natl Acad Sci USA 95, 9791–9795 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce N. C., Harris D. L. & Zieske J. D. Mitotic inhibition of corneal endothelium in neonatal rats. Invest Ophthalmol Vis Sci 39, 2572–2583 (1998). [PubMed] [Google Scholar]
- Kikuchi M., Harris D. L., Obara Y., Senoo T. & Joyce N. C. p27kip1 Antisense-induced proliferative activity of rat corneal endothelial cells. Invest Ophthalmol Vis Sci 45, 1763–1770 (2004). [DOI] [PubMed] [Google Scholar]
- Lee H. T. & Kay E. P. Regulatory role of PI 3-kinase on expression of Cdk4 and p27, nuclear localization of Cdk4, and phosphorylation of p27 in corneal endothelial cells. Invest Ophthalmol Vis Sci 44, 1521–1528 (2003). [DOI] [PubMed] [Google Scholar]
- Lee J. G. & Kay E. P. Two populations of p27 use differential kinetics to phosphorylate Ser-10 and Thr-187 via phosphatidylinositol 3-Kinase in response to fibroblast growth factor-2 stimulation. J Biol Chem 282, 6444–6454 (2007). [DOI] [PubMed] [Google Scholar]
- Honda H., Ogita Y., Higuchi S. & Kani K. Cell movements in a living mammalian tissue: long-term observation of individual cells in wounded corneal endothelia of cats. J Morphol 174, 25–39 (1982). [DOI] [PubMed] [Google Scholar]
- Bloomfield S. E., Jakobiec F. A. & Iwamoto T. Fibrous ingrowth with retrocorneal membrane. Ophthalmology 88, 459–465 (1981). [DOI] [PubMed] [Google Scholar]
- Sheerin A. N. et al. Characterization of cellular senescence mechanisms in human corneal endothelial cells. Aging Cell 11, 234–240 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N. et al. Involvement of cyclin D and p27 in cell proliferation mediated by ROCK inhibitors Y-27632 and Y-39983 during corneal endothelium wound healing. Invest Ophthalmol Vis Sci 55, 318–329 (2014). [DOI] [PubMed] [Google Scholar]
- le Sage C., Nagel R. & Agami R. Diverse ways to control p27Kip1 function: miRNAs come into play. Cell Cycle 6, 2742–2749 (2007). [DOI] [PubMed] [Google Scholar]
- Resnitzky D. & Reed S. I. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol Cell Biol 15, 3463–3469 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. W. et al. Ribonuclease 5 coordinates signals for the regulation of intraocular pressure and inhibits neural apoptosis as a novel multi-functional anti-glaucomatous strategy. Biochim Biophys Acta 1862, 145–154 (2016). [DOI] [PubMed] [Google Scholar]
- Lee J. G. & Kay E. P. NF-kappaB is the transcription factor for FGF-2 that causes endothelial mesenchymal transformation in cornea. Invest Ophthalmol Vis Sci 53, 1530–1538 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. T. & Kay E. P. FGF-2 induced reorganization and disruption of actin cytoskeleton through PI 3-kinase, Rho, and Cdc42 in corneal endothelial cells. Mol Vis 9, 624–634 (2003). [PubMed] [Google Scholar]
- Okumura N. et al. Enhancement of corneal endothelium wound healing by Rho-associated kinase (ROCK) inhibitor eye drops. Br J Ophthalmol 95, 1006–1009 (2011). [DOI] [PubMed] [Google Scholar]
- Fantes F. E. et al. Wound healing after excimer laser keratomileusis (photorefractive keratectomy) in monkeys. Arch Ophthalmol 108, 665–675 (1990). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.