

ABSTRACT
Tumor budding occurs at the invasive front of cancer; the tumor cells involved have metastatic and stemness features, indicating a poor prognosis. Tumor budding is partly responsible for cancer metastasis, and its initiation is based on the epithelial-mesenchymal transition (EMT) process. The EMT process involves the conversion of epithelial cells into migratory and invasive cells, and is a profound event in tumorigenesis. The EMT, associated with the formation of cancer stem cells (CSCs) and resistance to therapy, results from a combination of gene mutation, epigenetic regulation, and microenvironmental control. Tumor budding can be taken to represent the EMT in vivo. The EMT process is under the influence of the tumor microenvironment as well as tumor cells themselves. Here, we demonstrate that the tumor microenvironment dominates EMT development and impacts cancer metastasis, as well as promotes CSC formation and mediates drug resistance. In this review, we mainly discuss components of the microenvironment, such as the extracellular matrix (ECM), inflammatory cytokines, metabolic products, and hypoxia, that are involved in and impact on the acquisition of tumor-cell motility and dissemination, the EMT, metastatic tumor-cell formation, tumor budding and CSCs, and cancer metastasis, including subsequent chemo-resistance. From our point of view, the tumor microenvironment now constitutes a promising target for cancer therapy.
KEYWORDS: cancer metastasis, drug resistance, epithelial-mesenchymal transition, microenvironment, tumor budding
The tumor budding concept and its significance
Tumor budding refers to thin, anaplastic cell cords, undifferentiated cancer cells, individual free cells, and is recognized as neoplastic epithelium or aggregates of cancer cells (up to 5 cells) at the invasive front (Fig. 1).1 Tumor budding has recently received much attention, particularly in the setting of colorectal carcinomas.1,2 Tumor budding is identified as a representation of the EMT and the cells display migratory and invasive properties.2,3 From published data, even if the initiation of tumor budding did not equate with the EMT, there are many parallel events between them. For example, tumor budding cells usually have membranous E-cadherin down-regulation and nuclear β-catenin expression, which symbolizes a mesenchymal-like characteristic just as in the EMT process.1,4 Our own data has shown that epithelial biomarkers such as E-cadherin (CDH1) and occludin (OCLN) are down-regulated, and mesenchymal markers such as N-cadherin (CDH2) and fibronectin (FN1), are up-regulated in tumor budding, samples accurately microdissected from patient-derived colorectal cancer specimens, compared to primary tumor cells. Moreover, TGFβ, WNT5A/TCF4, NOTCH3, SNAIL, and TWIST1, which are classic EMT regulators, are also known to be upregulated in tumor budding. Loss or reduction of E-cadherin occurs in most of budding tumor cells. In fact, most colorectal carcinoma with a high degree of budding tumor cells that are detached and migrate a short distance into the adjacent stroma are well or moderately differentiated (Fig. 1). The EMT is needed for the formation of tumor budding.1 When genetic and epigenetic changes occur, cancer cells lose their cell-cell and cell-matrix contacts, remodel the cytoskeleton, and detach from neoplastic glands, then extend lamellipodia or filopodia into the adjacent interstitial component. Membrane-type metalloproteinase and the urokinase plasminogen activator system are needed in the process of pericellular proteolysis.
Figure 1.
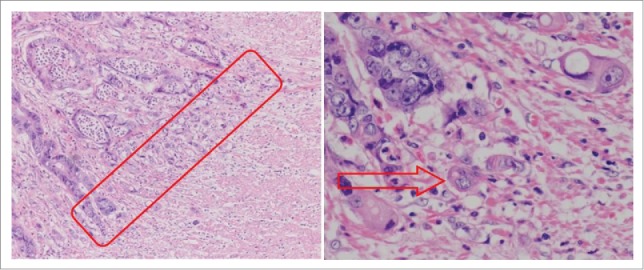
Microscopic finding of colorectal cancer (hematoxylin and eosin staining). Tumor buddings are defined as individual cancer cells or small clusters (up to 5 cancer cells) at the invasive front of cancer.
Tumor budding itself might take part in ECM degradation. This is supported by the overexpression of matrix metalloproteinases MMP-2 and MMP-9 and urokinase plasminogen-activator receptor in high-grade tumor budding specimens, based on immunohistochemical experiments.3 It is important to note that this results in the formation of new extracellular matrix by different components while the old cell-matrix composition is hydrolyzed. It is clear that the tumor microenvironment itself induces the EMT and the initiation of tumor budding. Tumor budding was first described by Imai.5 Evidences showed that the numbers of tumor buddings at the invasive front have significant correlations with lymphatic invasion and lymph-node metastasis.6-10 Others showed that tumor budding in colorectal cancers indicates a poor prognosis and local recurrence.7 Tumor budding is a prognostic biomarker in cancer patients, independent of pathological stage. It has been shown that Dukes' B with tumor budding has a survival rate similar to Dukes' C with no budding in colorectal cancer cases, and it turns out that cases with high-grade tumor budding lead to a worse outcome.7 In sum, tumor budding at the invasive front is strongly associated with lymph-node metastasis, local and distant relapse, and a poor prognosis in advanced colorectal cancer.11 Moreover, tumor budding-associated signal pathways, such as the Wnt pathway, are simultaneously associated with the development of CSCs and the EMT.12
Outline of the epithelial-to-mesenchymal transition
The EMT process is an ubiquitous phenomenon during differentiation and development processes, and is fully functional in embryonic development, wound healing, and neoplastic progress.13,14 For decades, studies have shed light on its critical function in metastasis, CSC formation, and resistance to therapy.15,16 Epithelial cells gradually lose their polarity and transform into mesenchymal phenotypes, accompanied by actin cytoskeleton reprogramming and enhanced motility. At the molecular level, E-cadherin, which is a classical epithelial biomarker, is dramatically downregulated, while some of the mesenchymal biomarkers (N-cadherin, vimentin, and fibronectin) are up-regulated in the EMT process.17 The basic mechanisms of the EMT process involve genetic and epigenetic changes.18 Beyond that, the tumor microenvironment also has significant effect on the EMT program. The tumor microenvironment is composed of cellular and non-cellular components. The former includes lymphocytes, macrophages, cancer-associated fibroblasts (CAFs), and vascular endothelial cells, while the latter includes ECM, pH, oxygen pressure, and various metabolic products. Changes in these influential factors have an impact on the development of the EMT (Fig. 2).19,20 Researches in recent years have shed light on the classic TGF-β, Wnt, Rho, NF-κB, Notch, and STAT signaling pathways and the growth factor pathways, which all play regulatory roles in the EMT process independently or synergistically.21-23 Other transcriptional factors, like Snail, Twist, Smad, Ras, the Zeb family, and GSK-3β, are responsible for the activation of EMT-associated pathways.24 Moreover, inflammatory cytokines, such as tumor necrosis factor α (TNFα) and IL6 can also induce the EMT in vitro. Hypoxia-inducible factor (HIF-1α), which is secreted in an oxygen-deficient environment, can advance the development of the transition program. The EMT is always accompanied by loss of cell polarity. EMT inducers such as Zeb1 and Snail indirectly inhibit the transcription of cell polarity genes like crumbs, and repression of crumbs activates the TGF-β signaling pathway, forming a positive loop to promote the EMT process. The development of the EMT is an integrated network involving transcriptional and post-transcriptional regulation and cytokines from the surrounding environment. Therefore, targeting EMT biomarkers could be an effective therapeutic scheme for
Figure 2.
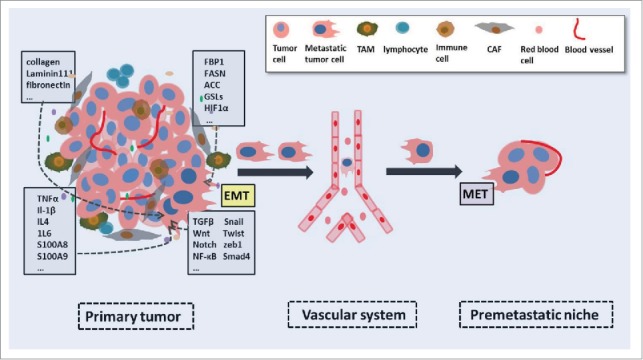
The impact of the tumor microenvironment on the EMT and cancer metastasis. The surrounding environment of tumor cells embraces multiple different type of cells and biomolecules, including CAFs, TAMs, endothelial cells, inflammatory cells, matabolic products. Biomolecules from the tumor microenvironment, such as growth factors, cytotines and glycolipids, influence the arrangment of cytoskeleton and cancer cell behaviors via a varity of signaling pathways or paracrine manners. Regulated cancer cells turn into metastatic ones due to the EMT process. And then metastatic tumor cells swim into the neighboring area, enter into the vascular system and finally settle dowm in their favorite organ.
The ins and outs of the microenvironment and metastasis
Cancer metastasis is a multimodal and multistep progress, and the pathogenesis originates from the internal and external environment. Cancer metastasis resulting in high recurrence and low survival rates urgently needs to be addressed. Traditionally, the metastatic process can be divided into 3 steps. First, cancer cells gain motility and invasive power, likely manipulated by the EMT and the surrounding environment, with loses of cell-cell adhesion and polarity. Second, disseminating neoplastic cells, such as in tumor budding, must penetrate vascular endothelial cells twice, namely intravasation and extravasation, or enter the lymphatic system. Third, some of the circulating tumor cells find a metastatic site in a distant organ for new nodule formation. The tumor microenvironment which includes different effectors of inflammatory system, ECM, hypoxia factors, and metabolic products, is closely involved with all steps of metastasis process.25 While EMT is a likely mechanism for the formation of invasive and metastatic cells. Tumor budding and other disseminating cancer cells may also be responsible for the metastasis.26,27 Budding tumor cells and other disseminating cancer cells are like seeds raised and navigated in their soil microenvironment; this is the “seed and soil” theory, first proposed by Paget.28 The tumor microenvironment at the invasive front of cancer promotes the formation of tumor budding and other disseminating cancer cells.27 While cancer cells with metastatic potential, such as tumor budding, may have specific interactions with their host microenvironment to promote migration, and stimulate proliferation and survival.29 Genetic changes mark the over-proliferation of tumor cells, and the EMT pairs this characteristic with invasive properties leading to the initiation of metastasis, while the dominant regulation of the tumor microenvironment is emphasized in later stages.
Inflammatory response versus EMT and cancer metastasis
The occurrence of the EMT is propelled by the tumor microenvironment which is composed of stromal cells, infiltrating immune cells, and chemical factors.30 In the context of malignance, the inflammatory microenvironment is postulated to participate in angiogenesis, the formation of a hypoxic environment, cancer cell stemness, and changes in microRNA.31 This microenvironment may act in 2 ways. On one hand, genetic changes in cancer cells may result in the over-production of inflammatory mediators. On the other hand, the formation of an inflammatory stroma could be a host-reactive defense against this vicious proliferation, despite the fact that there is no clear evidence to support the existence of a common molecular pathway between those 2 mechanisms.
Inflammatory cytokines such as TNFα and interleukin-1beta (IL-1β), which are usually highly secreted by tumor cells, exist in the tumor microenvironment (Fig. 2).32,33 TNFα is ubiquitously overexpressed in all sorts of cell types, mostly in immune cells that occur in chronic inflammation. TNFα is well known to lead to the EMT by activating the NF-кB pathway.34,35 Aberrant activation of NF-кB is related to cancer cell proliferation, angiogenesis, invasion, and metastasis.36 The inflammatory cytokines S100A8 and S100A9 are reported to mediate metastasis via a TLR4-mediated NF-κB signaling cascade.37 Recent findings have provided evidence that TNFα and IL-1β co-induce tumor cell spreading, the EMT, and invasiveness in breast tumor cells.32 Another study on TNFα and IL-1β finds that cancer cells and macrophages influence each other's behavior.38 As a result, TNFα and IL-1β secreted by macrophages infiltrating the tumor microenvironment promote the expression of TGF-β, which is a classical EMT-inducible factor. TGF-β plays a dual role in tumor development, while TGF-β represses cell proliferation and induces apoptosis in stark contrast to its tumor invasion and metastasis function. This is based on the different context in which TGF-β acts. When a tumor has just started, TGF-β takes on an anti-cancer role in the normal microenvironment with ECM and other antagonistic mediators, while during tumor environment TGF-β is a prominent mediator of tumorigenesis.39-42 TGF-β and TNFα induce the expression of HGF in the tumor stroma. The HGF/Met signaling promotes tumorigenesis and the EMT, and meanwhile, activation of this signaling could activate the MAPK, PI3K/Akt, and NF-кB pathways which are also involved in the EMT and metastasis.43,44
Another inflammatory cytokine with a typical pro-EMT effect is IL-6. Extensive evidence supports the idea that IL-6 promotes the EMT by activating the TGF-β signaling pathway.45 Moreover, IL-6 induces E-cadherin downregulation and vimentin up-regulation through the JAK/STAT3/Snail pathway in head and neck cancer.46 At the location of metastasis, molecules secreted by primary tumor cells contribute to form a metastatic-permissive environment for invasive tumor cells, termed the pre-metastatic niche to stimulate the arrest, adhesion, and the invasion of metastatic tumor cells.
Several molecules and various cell types are involved in metastatic niche formation.47-49 VEGF receptor 1 is reported to be located in the pre-metastasis niche before tumor cells settle down, and is the main molecule of the pre-metastatic niche. Besides, inflammatory factors, such as TGF-β and TNFα, and MMPs together with stromal-derived factor 1 and CXC-chemokine receptor facilitate metastatic niche formation. Bone marrow-derived myeloid cells, CAFs, and endothelial cells are also important parts of metastatic microenvironment. Colony-stimulating factor (CSF-1) and VEGF accelerate the transformation of M1 type macrophages into immunosuppressive M2, as in tumor-associated macrophages (TAMs).50,51
TAMs are associated with a poor prognosis in 80% of clinical studies, especially in prostate, ovarian, breast and cervical cancers.52 They are reported to participate in many steps of tumor metastasis and are known to promote metastasis through the cooperation of signaling pathways.53 They are the main orchestrator of the tumor microenvironment, and directly affect tumor cell growth, angiogenesis, ECM remodeling, and the EMT progress. TAMs at the invasive front have been confirmed to form a feedback loop together with neoplastic cells undergoing the EMT, through the overexpression of CCL18 from TAMs and GM-CSF from EMT-like cells.54 The interaction between these 2 cell types ends with distant metastasis. Silencing of GM-CSF and CCL18 inhibits this positive loop and blocks metastasis. Liu et al. used IL4 to induce M2 polarized TAMs and found that they decrease E-cadherin expression, upregulate mesenchymal biomarkers at the transcription and translation levels, and increase the fibroblastic phenotype in pancreatic cancer models. And TAMs promote the EMT partially via the TLR4/IL10 signaling pathway.55 Recently, Singh and coworkers have shed light on pro-inflammatory cytokines, such as TNFα and IL-6 that are secreted by TAMs; these promote the intracellular accumulation of TGFβ, and then upregulate ROS and Reactive nitrogen species (RNS) leading to oxidative stress in breast cancer cells. This signaling axis would activate CREB, the EMT process, and metastasis.38 Extensive evidence has shown that a series of activatory inflammatory cytokines, growth factors, and chemokines originating from chronic inflammation are capable of advancing carcinogenesis and EMT progress; non-steroidal anti-inflammatory drugs have achieved a good curative effect on cancer.
Extracellular matrix components versus EMT and metastasis
Increasing numbers of studies suggest that the ECM and its receptors are indispensable for the successive development of cancer, from benign lesion to malignant tumor and metastasis.56-58 The ECM is a complicated network that includes several macromolecules, such as laminins, collagens, tenascin, nidogen/entactin, thrombospondin, hyaluronan, chondroitin sulfate, and fibronectins. The ECM is responsible for the architectual support to tumor cells and provides the interactions between tumor cells and the stromal components.59,60 Epithelial-mesenchymal interactions play crucial roles in tumor issue, as disorder of these interactions induces carcinogenesis and tumor cell invasion.61,62 Stromal collagen proliferation in mouse mammary tissues, significantly enhance to the formation of tumors, the invasive phenotype of tumor cells, and distant lung metastasis by approximately three-fold.62 Moreover, tumor cell growth and expansion leads to a responsive proliferation of collagen and other ECM components, inevitably accompanied by matrix reorganization, which involves increased tumor cell motility and the delivery of a series of EMT-related cell signals, such as TGF-β, Wnt, and Rho.
Another study performed by Yasushi Shintani showed that NMuMG breast cancer cells cultured on collagen I become fibroblastic, and E-cadherin expression on the cell-cell border is dramatically down-regulated, with the upregulation of mesenchymal markers, such as N-cadherin and fibronectin. And this typical EMT process on NMuMG breast cancer cells induced by collagen I is mediated by Rac1 and c-Jun NH2-terminal kinase (JNK) signaling pathway.63 Moreover, the morphological phenotype conversion is restored by Rac1 inhibition or JNK signaling inhibition, indicating that the P13K-Rac-JNK signaling pathway might be the critical signaling channel to mediate the collagen-induced EMT process in breast cancer cells.63 The basement membrane protein laminin 111, which is a member of laminin family and presents the best characterized laminin isform, is another ECM component that has an effect on the EMT and metastasis.64,65 It has been noted that laminin 111 blocks the MMP3-mediated EMT by preventing the expression and membrane location of the MMP3 downstream signaling molecule Rac-1b, a highly activated splice variant of small GTPase Rac1. And this EMT-inhibitory effect is achieved via laminin 111 interacting with its special integrin receptor, α6-integrin.66 Conversely, fibronectin restores Rac-1b in the membrane via its integrin receptor α5-integrin and enhances epithelium-to-mesenchyme conversion. Based on the study by Chen, tumor cells cultured in fibronectin-rich not laminin-rich ECM develop a mesenchymal phenotype; while those in the former (fibronectin-rich medium) overexpress classical EMT markers, those in the latter (laminin-rich medium) maintain epithelial characteristics.66 Other ECM molecules, such as hyaluronan, and tenascin, also have been reported to promote EMT process and metastasis.67-69 Taking all these findings together, we are confident in recognizing that the ECM microenvironment as a critical regulator of the EMT and metastasis.
Metabolic products versus EMT and metastasis
Over the past decades, functional studies have implicated carbohydrate metabolism in malignant transformation.70 The development of a tumor consumes considerable amounts of oxygen and ATP, and cancer cells use aerobic glycolysis.71 In general, cancer cells carry out glycolysis in the presence or absence of oxygen in order to satisfy the elevated need for the energy, macromolecular precursors, and NADPH needed for proliferation.
Glucose homeostasis in vivo depends on catabolic glycolysis/oxidative phosphorylation and gluconeogenesis. Fructose-1, 6-biophosphase (FBP1) is a critical enzyme in catabolic oxidative phosphorylation. Silencing of the FBP1 promoter by DNA methylation has been reported in liver, gastric, and colon cancers.72,73 Dong showed that the FBP1 promoter embraces 9 reduplicative E-box (CAGGTG) binding sites for Snail. Coincidently, silencing of both the FBP1 promoter and E-cadherin promoter is mediated by the Snail-G9a-Dnmt1 complex, suggesting that FBP1 might be required for Snail-mediated EMT (Fig. 2). Dong and colleagues found that FBP1, which directly inhibits glucose uptake, directly binds Snail and restores Snail-mediated E-cadherin down-regulation, finally inhibiting the basal-like phenotype transition in luminal breast tumor cells. They also found that FBP1 expression significantly restrained the percentage of CD44+/CD24-/EpCAM+ population in BLBC cell lines, suggesting that FBP1 is probably associated with induced pluripotent stem cell reprogramming.74 Collectively, these results suggest that epigenetic silencing of FBP1 is of great importance in basal-like breast cancer cell EMT and metastasis. Given these results, they speculated that targeting this Snail-G9a-Dnmt1-FBP1 chromatin modification complex would be an efficient therapy for metastatic breast cancer.74,75
In addition to glucose metabolism, cancer cell proliferation needs lipogenesis and membrane production. Several lipogenic enzymes are required for tumor cell proliferation, such as fatty acid synthase (FASN) and acetyl-CoA carboxylase. FASN is associated with EMT and cancer metastasis.76 Jiang et al. found that stable knockdown of FASN is enough to induce the EMT, advance cancer cell extravasation in vitro, and promote lung metastasis in vivo. Meanwhile, they found that sterol regulatory element binding protein, carbohydrate-responsive-element binding protein, and transcriptional factors of lipogenesis, undergo a sharp decline in the TGFβ-induced EMT process. Glycosphingolipids (GSLs), located at the cell membrane and functionally united with cell growth factors and signal transducers, have been reported to participate in several cellular activities, including cytoplasmic signal transduction, cell adhesion, and motility. Guan and colleagues showed that complete in vitro GSL knockdown by EtDO-P4, which is a GSLs synthase inhibitor, causes a phenotype reversion from epithelium to mesenchyme, accompanied by downregulation of epithelial markers and upregulation of mesenchymal markers.77 Another report from the same laboratory showed that GSLs, particularly ganglioside, are of great importance in the regulation of HGF-induced cell motility and invasiveness through Met tyrosine kinase.78 Collectively, current studies indicate that metabolism is not simply a consequence but rather plays a crucial role in dictating the phenotype conversions and cell dissemination exhibited by cancer cells.
Hypoxia-mediated EMT and cancer metastasis
Another factor impacting the EMT and tumor metastasis is hypoxia.79 There is a balance between tissue oxygen consumption in vivo. When cancer cells enter into an infinite proliferation cycle, the oxygen consumption sharply increases, causing a relatively hypoxic environment around neoplastic cells.80 Increasing evidence shows that hypoxia participates in every step of cancer progression.81,82 Hypoxia increases angiogenesis, cancer cell survival, and metastasis in various cancer types, including breast, colorectum, head, and neck.83,84
Using a computerized polarographic electrode system to test 52 patients with cervical cancer, Hockel et al. found that hypoxic tumors have a worse survival rate than non-hypoxic tumors.85 Actually, hypoxia plays a dual role in regulating human cancer progression.86,87 Hypoxia predominantly induces the overexpression of HIF-1 (Fig. 2).88 Microarray profiling has identified the target genes of HIF-1 that execute different functions in cancer progression, including proliferation and survival (IGF2, TNFα, IGFBP1/2/3), apoptosis (NIX, NIP3), motility (c-MET, AMF/GPI, TNFα ), cytoskeletal structure (KIR14/18/19, VIM), angiogenesis (EG-VEGF, TGF-β3, VEGF), ECM metabolism (FN1, MMP2, collagen type V), and drug resistance.79 It is well established that HIF-1 can directly induce the EMT, elevating the protein levels of EMT-related transcription factors and mesenchymal biomarkers (VIM, FN1, CDH2) and downregulating the expression of epithelial characteristics (CDH1, TJP1 ). It has been reported that HIF-1α reduces caveolin-1 (Cav-1), which is controlled by heat shock protein 90 (HSP90) under hypoxic conditions in gastric carcinogenesis. The downregulation of Cav-1 is associated with the expression level of epidermal growth factor receptor (EGFR) which stably activates its downstream effector STAT3. Overexpression of STAT3 in the nucleus leads to decreasing expression of epithelial biomarkers, along with the overexpression of mesenchymal molecules. Meanwhile, crosstalk between EGFR and the TGF-β signaling pathway associated with Wnt signaling also elevates the EMT progression and cancer cell invasion.89 Discoidin domain receptor (DDR2), being recognized as a unique subfamily of tyrosine kinases, is reported to be closely related to the expression level of HIF-1α under hypoxic conditions in breast cancer cells.90 DDR2 is usually secreted by mesenchymal cells or fibroblasts. Ren and coworkers found that DDR2 participates in hypoxia-mediated cell migration, invasion, and the EMT, which probably requires the regulation of transcription factor Snail. They also confirmed that DDR2 silencing interrupts the hypoxia-activated ERK/MAPK signaling pathway, suggesting that DDR2 is a reliable therapeutic target in breast cancer therapy. In addition, HIF-1α cooperating with the transcription factors ROS, STAT3, and TWIST1 promote the EGF-mediated prostate cancer cell EMT through the ROS/STAT3/HIF-1α/TWIST1/N-cadherin signaling cascade.91
The occurrence and development of malignant tumors benefit from the complicated network of the tumor itself and the crosstalk between tumor cells and the microenvironment. Ample evidence enables us to reasonably conclude that tumor cell hypoxia is good for the EMT and metastasis. Hepatocyte growth factor (HGF) is overexpressed by CAFs under hypoxia, following up with the up-regulation of Met tyrosine kinase. Co-expression of HGF and Met are beneficial to cancer cell motility and EMT progress.92 In this context, we conclude that hypoxia advances the process of malignant progression intrinsically and externally. More and more evidences have shed light on the influence of hypoxia on tumor procession. Hypoxia probably participates in angiogenesis, cell invasion, the EMT, and metastasis. Thus, targeting hypoxia-regulated genes may be used to cancer therapy.
Stem-like characteristics in the EMT Program and microenvironment
Studies in recent years have shown that the EMT process contributes to building the stemness of cancer cells, even directly generating cancer stem cells.93,94 CSCs, which usually express a CD44high/CD24low antigen phenotype, are capable of self-renewal and differentiating into adult cancer cells.95 Xenografts of CSCs in immune-deficient mice promote the mammosphere-forming efficiency of cancer.93 It has been reported that using Snail, Twist, or TGF-β1 to successfully induce the EMT in both non-tumorigenic and immortalized human mammary epithelial cells results in the reshaping of cancer cells into mesenchymal-like cells and they simultaneously acquire the CD44high/CD24low marker profile.93,94,96 It is further known that E-cadherin is a regulator of pluripotency; it is downregulated in stem-like cells and N-cadherin is conversely upregulated.97 Cancer cell stemness is also deeply influenced by the microenvironment. Myofibroblast-secreted factors, which are stromal activators of Wnt signaling pathway, are available for the expression of CSC markers and restoration of the CSC phenotype of differentiated cancer cells.98 The EMT process has long been associated with the formation of cancer cell stemness. Moreover, it is known that crosstalk between signaling pathways regulates the EMT and stem cell formation. The tight relationships among CSCs, the EMT, and the microenvironment is confirmed by the above evidences.93,94,96,97
EMT-related therapeutic resistance in the tumor microenvironment
Resistance to cancer therapy is responsible for a poor outcome. Environment-mediated drug resistance (EMDR) is inherent in tumors, unlike acquired resistance which is a consequence of adaptive genetic and epigenetic mutation due to chemotherapy and radiotherapy.99 Generally, the tumor microenvironment contains many factors including soluble factors such as interleukins, extracellular matrix components such as fibronectin and collagen, and stromal cells that contribute to weakening drug activity and result in minimal residual disease. EMDR is instantaneous and less complicated than acquired resistance. EMDR is regulated by integrated signaling pathways between the tumor cell and its surrounding environment.99 Hazlehurst et al. used myeloma cells in vitro to compare gene expression differences between EMDR and acquired resistance when myeloma cells were treated with melphalan.100 They concluded that there were 69 gene expression changes associated with EMDR, contrasting with 1479 for acquired resistance. It is deduced from their data that targeting microenvironment will be more effective to cure cancer.
Damiano et al. used drug-sensitive 8266 human myeloma cells to demonstrate the mechanism of cell adhesion-mediated resistance. Human myeloma cells stably express VLA-4(α4β1) integrin fibronectin receptors, and β1 integrin stably upregulates BCL-2 which is a drug resistance-related biomarker. Using RNase assays to guarantee the transcriptional level of BCL-2 and BCL-XL were stable, the author and his colleague investigated whether VLA-4(α4β1) integrin fibronectin acceptor acts against the apoptotic effects induced by doxorubicin and melphalan when pre-adhered to fibronectin in comparison to cells in suspended culture.101 Actually, β1 integrin may confer multi-drug resistance. Another study in the same laboratory showed that β1 integrin also regulates Bim degradation, a member of the BCL-2 family known to contribute to drug-resistance as mentioned above, to eliminate the efficacy of chemotherapy through mediating the adhesion of tumor cells to fibronectin.102 To sum up, cell adhesion-mediated drug resistance provides an index for environmental factors when accessing the efficacy of chemo-therapeutic drugs.
The tumor microenvironment is inundated with soluble factors, such as cytokines and chemokines derived from either autocrine or paracrine sources, and they play an essential role in chemotherapy-resistance. IL-6 has been implicated in various stages of malignancy by mediating several signaling pathways.103,104 Robyn Catlett-Falcone and his colleagues studied IL-6-related tumor cell apoptosis in the in vitro human myeloma cell line U226, which is intrinsically resistant to Fas-mediated apoptosis and expresses the anti-apoptotic protein BCL-XL.105 They found that down-regulated expression of IL-6 effectively blocks BCL-XL expression and induces tumor cell apoptosis. What is more, IL-6 was confirmed to enhance myeloma cell survival through the STAT signaling pathway, demonstrating that IL-6 and the STAT pathway might be efficient therapeutic targets for cancer.
Tumorigenesis is a comprehensive result of genetic and epigenetic changes accompanied by crosstalk with the tumor microenvironment. That is why cancer therapy remains a challenge. Acquired and de novo resistance are the 2 mechanisms of chemotherapeutic resistance. The EMT is another pathway to acquired drug resistance. In a study by Zhang and colleagues, >50% resistant non-small cell lung cancer (NSCLC) were shown to harbor an EGFR mutation. Using an EGFR-targeted tyrosine kinase inhibitor such as erlotinib is effective in NSCLC, but does not last, since resistance is acquired.106 The authors found that genetic or pharmacological alteration of receptor tyrosine kinase AXL affects the sensitivity of tumor cells to erlotinib in NSCLC models. They also found that overexpression of AXL is closely linked to some of the EMT biomarkers, such as vimentin. They showed that knockdown of vimentin by siRNA partially inhibits AXL expression and restores erlotinib sensitivity in HCC827 cells compared to parental HCC827 cells. Taken together, this study shows that the EMT marked by vimentin overexpression plays a significant role in acquired drug resistance to erlotinib regulated by AXL in human EGFR-mutant NSCLC. As the tumor microenvironment is responsible for the EMT, we can presumably say that the tumor microenvironment may support a possible role in acquired tumor resistance.
Snail at the crossroads of different cancer programs
The tumor microenvironment plays critical roles in stem-like cell formation, such as tumor budding, and EMT-mediated metastasis. But among all these EMT-related inducers, which is the “hub gene” that cross-regulates EMT-related CSC formation, metastasis, and drug resistance? We collected evidence and found that Snail took part in all EMT-related aspects, including tumor cell stemness, metastasis, and drug resistance (Fig. 3). Sufficient evidence shows that overexpression of Snail is sufficient to induce the EMT in vivo by directly binding to the E-cadherin promoter.21,107,108 Moreover, compared to other transcriptional factors involved in the EMT process, Snail might be the initiator.109 Zhu et al. found that Snail induces the EMT in oral squamous cell carcinoma (OSCC) tumor cells, and facilitates the formation of OSCC tumor cell stemness.110 It has also been demonstrated that breast cancer cells obtain stemness characteristics when Snail is overexpressed.93 Another study shows that upregulation of Snail is significantly associated with tumor budding and lymphatic metastasis.27 Spatiotemporal regulation of the EMT is critical to efficient tumor cell metastasis in vivo. Tumor cells undergoing the EMT in primary tumors are capable of non-proliferation and mobility, since EMT-related transcriptional factors, such as Snail, inhibit cell proliferation.111,112 When tumor cells reach distant sites, they need to revert to an epithelial identity to form metastases.109 Transcriptional EMT regulators upregulated in the primary tumor are blocked in distant metastatic niches to activate tumor cell proliferation and metastasis formation. Especially the down-regulation of Snail activates the mesenchymal epithelial transition (MET) process which favors the distant metastasis formation (Fig. 3).113 The ATP binding cassette (ABC transporters) is associated with multi-drug resistance. Tumor cells that overexpress ABC transporters are usually more invasive and chemoresistant.114-116 Snail can directly bind to the promoter of ABC transporters and reduce the chemo-sensitivity of tumor cells in breast cancer.117 We speculate that targeting Snail is an effective means of invasive cancer therapy. The EMT process and cancer metastasis involve multiple steps that are influenced by many chemokines and cytokines. We have confidence in the speculation that more potential “hub genes” await experimental certification.
Figure 3.
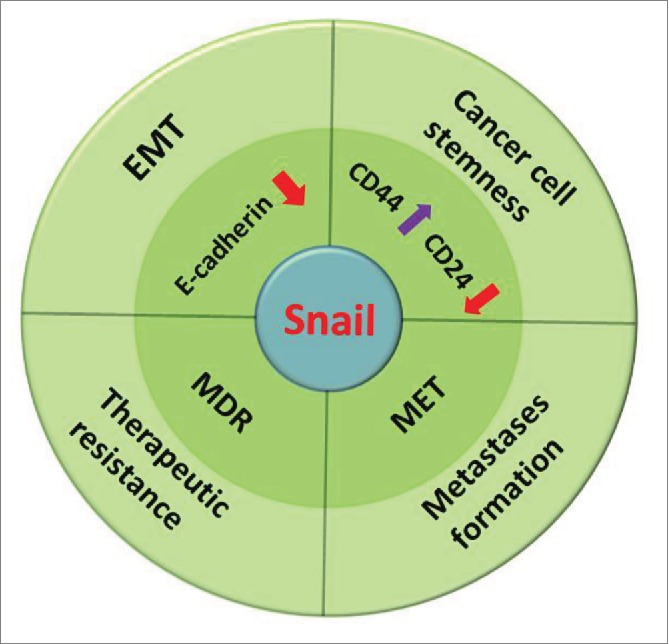
Snail at the crossroads of different cancer programs.
Summary
Surrounding stromal cells that are recruited by tumor cells allow the latter to escape from senescence or apoptosis, enter the circulation, and seed in distant sites. The local microenvironment is of great importance to the final fate of metastatic tumor cells. Metastatic tumor cells, such as budding cells, have preferred organs for settlement. For example, colorectal cancer prefers to metastasize to liver and brain; breast cancer to bone, liver, and brain. It is well-documented that the preferred organ sites for metastases are encoded by genetic alteration of specific tumor cells themselves118,119 and the signal communication between tumor cells and their constantly-changing microenvironment which partially derives from the products secreted by metastatic tumor cells.47,120,121 Evidence indicates that the local microenvironment of tumor cells critically influences their ability for stemness formation, migration, intravasation and extravasation, and secondary tumor formation. Although the mechanisms involved in these processes have been well characterized in both in vivo and in vitro models, crosstalk between tumor cells and the microenvironment is still poorly understood. Far more attention should be paid to clarify the dynamic interactions between tumor cells and the microenvironment.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by National Natural Science Foundation of China (81090420/81090421), Key Science & Technology special project of Zhejiang Province (2012C13014-3), 111 Project (B13026), National Natural Science Foundation of China (81572716).
References
- [1].Prall F. Tumour budding in colorectal carcinoma. Histopathol 2007; 50:151-62; http://dx.doi.org/ 10.1111/j.1365-2559.2006.02551.x [DOI] [PubMed] [Google Scholar]
- [2].Mitrovic B, Schaeffer DF, Riddell RH, Kirsch R. Tumor budding in colorectal carcinoma: time to take notice. Modern Pathol Inc 2012; 25:1315-25; http://dx.doi.org/ 10.1038/modpathol.2012.94 [DOI] [PubMed] [Google Scholar]
- [3].Zlobec I, Lugli A. Epithelial mesenchymal transition and tumor budding in aggressive colorectal cancer: tumor budding as oncotarget. Oncotarget 2010; 1:651-61; PMID:21317460; http://dx.doi.org/ 10.18632/oncotarget.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T. Variable β-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A 2001; 98:10356-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Imai T. The growth of human carcinoma: A morphological analysis. Fukuoka Igaku Zasshi 1954; 45:72-102 [Google Scholar]
- [6].Morodomi T, Isomoto H, Shirouzu K, Kakegawa K, Irie K, Morimatsu M. An index for estimating the probability of lymph node metastasis in rectal cancers. Lymph node metastasis and the histopathology of actively invasive regions of cancer. Cancer 1989; 63:539-43; PMID:2912530; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- [7].Hase K, Shatney C, Johnson D, Trollope M, Vierra M. Prognostic value of tumor “budding” in patients with colorectal cancer. Dis Colon Rectum 1993; 36:627-35; PMID:8348847; http://dx.doi.org/ 10.1007/BF02238588 [DOI] [PubMed] [Google Scholar]
- [8].Ono M, Sakamoto M, Ino Y, Moriya Y, Sugihara K, Muto T, Hirohashi S. Cancer cell morphology at the invasive front and expression of cell adhesion-related carbohydrate in the primary lesion of patients with colorectal carcinoma with liver metastasis. Cancer 1996; 78:1179-86; PMID:8826938; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- [9].Goldstein NS, Hart J. Histologic features associated with lymph node metastasis in stage T1 and superficial T2 rectal adenocarcinomas in abdominoperineal resection specimens. Identifying a subset of patients for whom treatment with adjuvant therapy or completion abdominoperineal resection should be considered after local excision. Am J Clin Pathol 1999; 111:51-8; PMID:9894454 [DOI] [PubMed] [Google Scholar]
- [10].Okuyama T, Oya M, Ishikawa H. Budding as a risk factor for lymph node metastasis in pT1 or pT2 well-differentiated colorectal adenocarcinoma. Dis Colon Rectum 2002; 45:628-34; PMID:12004212; http://dx.doi.org/ 10.1007/s10350-004-6259-0 [DOI] [PubMed] [Google Scholar]
- [11].Xu FY, Dong JK, Zhu YM, Qu MJ, Wang FJ, Jin YS, Ren GP, Lai MD. Study on independent factors on the prognosis of colorectal carcinoma: TNM stage, tumor budding, perineural invasion, peritumoral-lymphocytic infiltration and urine glucose. Zhonghua liu xing bing xue za zhi 2005; 26:366-9; PMID:16053766 [PubMed] [Google Scholar]
- [12].Katoh M. Network of WNT and other regulatory signaling cascades in pluripotent stem cells and cancer stem cells. Curr Pharmaceutical Biotechnol 2011; 12:160-70; http://dx.doi.org/ 10.2174/138920111794295710 [DOI] [PubMed] [Google Scholar]
- [13].Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest 2009; 119:1438-49; PMID:19487820; http://dx.doi.org/ 10.1172/JCI38019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139:871-90; PMID:19945376; http://dx.doi.org/ 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- [15].Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012; 22:725-36; PMID:23201165; http://dx.doi.org/ 10.1016/j.ccr.2012.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Krantz SB, Shields MA, Dangi-Garimella S, Munshi HG, Bentrem DJ. Contribution of epithelial-to-mesenchymal transition and cancer stem cells to pancreatic cancer progression. J Surg Res 2012; 173:105-12; PMID:22099597; http://dx.doi.org/ 10.1016/j.jss.2011.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell 2004; 118:277-9; PMID:15294153; http://dx.doi.org/ 10.1016/j.cell.2004.07.011 [DOI] [PubMed] [Google Scholar]
- [18].Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, Meyer-Schaller N, Schübeler D, van Nimwegen E, Christofori G. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 2013; 23:768-83; PMID:23764001; http://dx.doi.org/ 10.1016/j.ccr.2013.04.020 [DOI] [PubMed] [Google Scholar]
- [19].Jing Y, Han Z, Zhang S, Liu Y, Wei L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci 2011; 1:29; PMID:21880137; http://dx.doi.org/ 10.1186/2045-3701-1-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sounni NE, Noel A. Targeting the tumor microenvironment for cancer therapy. Clin Chem 2013; 59:85-93; PMID:23193058; http://dx.doi.org/ 10.1373/clinchem.2012.185363 [DOI] [PubMed] [Google Scholar]
- [21].Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal 2014; 7:re8; PMID:25249658; http://dx.doi.org/ 10.1126/scisignal.2005189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype?. Nature Rev Cancer 2007; 7:415-28; http://dx.doi.org/ 10.1038/nrc2131 [DOI] [PubMed] [Google Scholar]
- [23].Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res 2011; 71:245-54; PMID:21199805; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Ann Rev Cell Dev Biol 2011; 27:347-76; http://dx.doi.org/ 10.1146/annurev-cellbio-092910-154036 [DOI] [PubMed] [Google Scholar]
- [25].Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem 2007; 101:816-29; PMID:17243120; http://dx.doi.org/ 10.1002/jcb.21215 [DOI] [PubMed] [Google Scholar]
- [26].Kazama S, Watanabe T, Ajioka Y, Kanazawa T, Nagawa H. Tumour budding at the deepest invasive margin correlates with lymph node metastasis in submucosal colorectal cancer detected by anticytokeratin antibody CAM5.2. Br J Cancer 2006; 94:293-8; PMID:16404429; http://dx.doi.org/ 10.1038/sj.bjc.6602927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yusra Semba S, Yokozaki H. Biological significance of tumor budding at the invasive front of human colorectal carcinoma cells. Int J Oncol 2012; 41:201-10; PMID:22569829 [DOI] [PubMed] [Google Scholar]
- [28].Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev 1989; 8:98-101; PMID:2673568 [PubMed] [Google Scholar]
- [29].Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature 2001; 411:375-9; PMID:11357145; http://dx.doi.org/ 10.1038/35077241 [DOI] [PubMed] [Google Scholar]
- [30].Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014; 2014:149185; PMID:24901008; http://dx.doi.org/ 10.1155/2014/149185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gao F, Liang B, Reddy ST, Farias-Eisner R, Su X. Role of inflammation-associated microenvironment in tumorigenesis and metastasis. Curr Cancer Drug Targets 2014; 14:30-45; PMID:24200082; http://dx.doi.org/ 10.2174/15680096113136660107 [DOI] [PubMed] [Google Scholar]
- [32].Leibovich-Rivkin T, Liubomirski Y, Bernstein B, Meshel T, Ben-Baruch A. Inflammatory factors of the tumor microenvironment induce plasticity in nontransformed breast epithelial cells: EMT, invasion, and collapse of normally organized breast textures. Neoplasia 2013; 15:1330-46; PMID:24403855; http://dx.doi.org/ 10.1593/neo.131688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, Weitzenfeld P, Meshel T, Shabtai E, Gutman M, et al.. Inflammatory mediators in breast cancer: coordinated expression of TNFalpha & IL-1beta with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011; 11:130; PMID:21486440; http://dx.doi.org/ 10.1186/1471-2407-11-130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene 2007; 26:711-24; PMID:16862183; http://dx.doi.org/ 10.1038/sj.onc.1209808 [DOI] [PubMed] [Google Scholar]
- [35].Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 2003; 3:745-56; PMID:12949498; http://dx.doi.org/ 10.1038/nri1184 [DOI] [PubMed] [Google Scholar]
- [36].Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005; 5:749-59; PMID:16175180; http://dx.doi.org/ 10.1038/nri1703 [DOI] [PubMed] [Google Scholar]
- [37].Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat Rev Cancer 2015; 15:96-109; PMID:25614008; http://dx.doi.org/ 10.1038/nrc3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Singh R, Shankar BS, Sainis KB. TGF-beta1-ROS-ATM-CREB signaling axis in macrophage mediated migration of human breast cancer MCF7 cells. Cell Signal 2014; 26:1604-15; PMID:24705025; http://dx.doi.org/ 10.1016/j.cellsig.2014.03.028 [DOI] [PubMed] [Google Scholar]
- [39].Achyut BR, Yang L. Transforming growth factor-β in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology 2011; 141:1167-78; PMID:21839702; http://dx.doi.org/ 10.1053/j.gastro.2011.07.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fuxe J, Vincent T, Garcia de Herreros A. Transcriptional crosstalk between TGF-β and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell cycle 2010; 9:2363-74; PMID:20519943; http://dx.doi.org/ 10.4161/cc.9.12.12050 [DOI] [PubMed] [Google Scholar]
- [41].Shin JA, Hong OK, Lee HJ, Jeon SY, Kim JW, Lee SH, Cho JH, Lee JM, Choi YH, Chang SA, et al.. Transforming growth factor-β induces epithelial to mesenchymal transition and suppresses the proliferation and transdifferentiation of cultured human pancreatic duct cells. J Cell Biochem 2011; 112:179-88; PMID:21069735; http://dx.doi.org/ 10.1002/jcb.22929 [DOI] [PubMed] [Google Scholar]
- [42].Zhu L, Qin H, Li PY, Xu SN, Pang HF, Zhao HZ, Li DM, Zhao Q. Response gene to complement-32 enhances metastatic phenotype by mediating transforming growth factor β-induced epithelial-mesenchymal transition in human pancreatic cancer cell line BxPC-3. J Exp Clin Cancer Res 2012; 31:29; PMID:22458379; http://dx.doi.org/ 10.1186/1756-9966-31-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4:915-25; PMID:14685170; http://dx.doi.org/ 10.1038/nrm1261 [DOI] [PubMed] [Google Scholar]
- [44].Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol 2010; 11:834-48; PMID:21102609; http://dx.doi.org/ 10.1038/nrm3012 [DOI] [PubMed] [Google Scholar]
- [45].Abulaiti A, Shintani Y, Funaki S, Nakagiri T, Inoue M, Sawabata N, Minami M, Okumura M. Interaction between non-small-cell lung cancer cells and fibroblasts via enhancement of TGF-β signaling by IL-6. Lung Cancer 2013; 82:204-13; PMID:24011634; http://dx.doi.org/ 10.1016/j.lungcan.2013.08.008 [DOI] [PubMed] [Google Scholar]
- [46].Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res 2011; 9:1658-67; PMID:21976712; http://dx.doi.org/ 10.1158/1541-7786.MCR-11-0271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al.. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005; 438:820-7; PMID:16341007; http://dx.doi.org/ 10.1038/nature04186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nature Rev Cancer 2009; 9:285-93; http://dx.doi.org/ 10.1038/nrc2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sceneay J, Smyth MJ, Moller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev 2013; 32:449-64; PMID:23636348; http://dx.doi.org/ 10.1007/s10555-013-9420-1 [DOI] [PubMed] [Google Scholar]
- [50].Ohm JE, Carbone DP. VEGF as a mediator of tumor-associated immunodeficiency. Immunologic Res 2001; 23:263-72; http://dx.doi.org/ 10.1385/IR:23:2-3:263 [DOI] [PubMed] [Google Scholar]
- [51].Wang HW, Joyce JA. Alternative activation of tumor-associated macrophages by IL-4: priming for protumoral functions. Cell Cycle 2010; 9:4824-35; PMID:21150330; http://dx.doi.org/ 10.4161/cc.9.24.14322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, Zhao YW, Wei YQ. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PloS One 2012; 7:e50946; PMID:23284651; http://dx.doi.org/ 10.1371/journal.pone.0050946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010; 141:39-51; PMID:20371344; http://dx.doi.org/ 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Su S, Liu Q, Chen J, Chen J, Chen F, He C, Huang D, Wu W, Lin L, Huang W, et al.. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014; 25:605-20; PMID:24823638; http://dx.doi.org/ 10.1016/j.ccr.2014.03.021 [DOI] [PubMed] [Google Scholar]
- [55].Liu CY, Xu JY, Shi XY, Huang W, Ruan TY, Xie P, Ding JL. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Investi J Tech Methods Pathol 2013; 93:844-54; http://dx.doi.org/ 10.1038/labinvest.2013.69 [DOI] [PubMed] [Google Scholar]
- [56].Ou J, Peng Y, Deng J, Miao H, Zhou J, Zha L, Zhou R, Yu L, Shi H, Liang H. Endothelial cell-derived fibronectin extra domain A promotes colorectal cancer metastasis via inducing epithelial-mesenchymal transition. Carcinogenesis 2014; 35:1661-70; PMID:24743511; http://dx.doi.org/ 10.1093/carcin/bgu090 [DOI] [PubMed] [Google Scholar]
- [57].Raglow Z, Thomas SM. Tumor matrix protein collagen XIalpha1 in cancer. Cancer Letters 2015; 357:448-53; PMID:25511741; http://dx.doi.org/ 10.1016/j.canlet.2014.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Stanisavljevic J, Loubat-Casanovas J, Herrera M, Luque T, Pena R, Lluch A, Albanell J, Bonilla F, Rovira A, Peña C, et al.. Snail1-expressing fibroblasts in the tumor microenvironment display mechanical properties that support metastasis. Cancer Res 2015; 75:284-95; PMID:25488750; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-1903 [DOI] [PubMed] [Google Scholar]
- [59].Lochter A, Bissell MJ. Involvement of extracellular matrix constituents in breast cancer. Seminars Cancer Biol 1995; 6:165-73; http://dx.doi.org/ 10.1006/scbi.1995.0017 [DOI] [PubMed] [Google Scholar]
- [60].Radisky D, Muschler J, Bissell MJ. Order and disorder: the role of extracellular matrix in epithelial cancer. Cancer Invest 2002; 20:139-53; PMID:11852996; http://dx.doi.org/ 10.1081/CNV-120000374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Niu YN, Xia SJ. Stroma-epithelium crosstalk in prostate cancer. Asian J Androl 2009; 11:28-35; PMID:19098934; http://dx.doi.org/ 10.1038/aja.2008.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med 2006; 4:38; PMID:17190588; http://dx.doi.org/ 10.1186/1741-7015-4-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Shintani Y, Wheelock MJ, Johnson KR. Phosphoinositide-3 kinase-Rac1-c-Jun NH2-terminal kinase signaling mediates collagen I-induced cell scattering and upregulation of N-cadherin expression in mouse mammary epithelial cells. Mol Biol Cell 2006; 17:2963-75; PMID:16624865; http://dx.doi.org/ 10.1091/mbc.E05-12-1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Aumailley M. The laminin family. Cell Adhesion Migration 2013; 7:48-55; PMID:23263632; http://dx.doi.org/ 10.4161/cam.22826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Horejs CM, Serio A, Purvis A, Gormley AJ, Bertazzo S, Poliniewicz A, Wang AJ, DiMaggio P, Hohenester E, Stevens MM. Biologically-active laminin-111 fragment that modulates the epithelial-to-mesenchymal transition in embryonic stem cells. Proc Natl Acad Sci U S A 2014; 111:5908-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chen QK, Lee K, Radisky DC, Nelson CM. Extracellular matrix proteins regulate epithelial-mesenchymal transition in mammary epithelial cells. Differ Res Biol Diversity 2013; 86:126-32; PMID:23660532; http://dx.doi.org/ 10.1016/j.diff.2013.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Turley EA. Hyaluronan and cell locomotion. Cancer Metastasis Rev 1992; 11:21-30; PMID:1380898; http://dx.doi.org/ 10.1007/BF00047600 [DOI] [PubMed] [Google Scholar]
- [68].Takahashi Y, Sawada G, Kurashige J, Matsumura T, Uchi R, Ueo H, Ishibashi M, Takano Y, Akiyoshi S, Iwaya T, et al.. Tumor-derived tenascin-C promotes the epithelial-mesenchymal transition in colorectal cancer cells. Anticancer Res 2013; 33:1927-34; PMID:23645740 [PubMed] [Google Scholar]
- [69].El-Haibi CP, Bell GW, Zhang J, Collmann AY, Wood D, Scherber CM, Csizmadia E, Mariani O, Zhu C, Campagne A, et al.. Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci U S A 2012; 109:17460-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Vaupel P. Metabolic microenvironment of tumor cells: a key factor in malignant progression. Exp Oncol 2010; 32:125-7; PMID:21403604 [PubMed] [Google Scholar]
- [71].Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 2011; 11:325-37; PMID:21508971; http://dx.doi.org/ 10.1038/nrc3038 [DOI] [PubMed] [Google Scholar]
- [72].Chen M, Zhang J, Li N, Qian Z, Zhu M, Li Q, Zheng J, Wang X, Shi G. Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PloS One 2011; 6:e25564; PMID:22039417; http://dx.doi.org/ 10.1371/journal.pone.0025564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Liu X, Wang X, Zhang J, Lam EK, Shin VY, Cheng AS, Yu J, Chan FK, Sung JJ, Jin HC. Warburg effect revisited: an epigenetic link between glycolysis and gastric carcinogenesis. Oncogene 2010; 29:442-50; PMID:19881551; http://dx.doi.org/ 10.1038/onc.2009.332 [DOI] [PubMed] [Google Scholar]
- [74].Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T, et al.. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013; 23:316-31; PMID:23453623; http://dx.doi.org/ 10.1016/j.ccr.2013.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Schieber MS, Chandel NS. ROS links glucose metabolism to breast cancer stem cell and EMT phenotype. Cancer Cell 2013; 23:265-7; PMID:23518342; http://dx.doi.org/ 10.1016/j.ccr.2013.02.021 [DOI] [PubMed] [Google Scholar]
- [76].Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro OM, Deberardinis RJ, Boothman DA. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 2015; 34:3908-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Guan F, Handa K, Hakomori SI. Specific glycosphingolipids mediate epithelial-to-mesenchymal transition of human and mouse epithelial cell lines. Proc Natl Acad Sci USA 2009; 106:7461-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Todeschini AR, Dos Santos JN, Handa K, Hakomori SI. Ganglioside GM2-tetraspanin CD82 complex inhibits met and its cross-talk with integrins, providing a basis for control of cell motility through glycosynapse. J Biol Chem 2007; 282:8123-33; PMID:17215249; http://dx.doi.org/ 10.1074/jbc.M611407200 [DOI] [PubMed] [Google Scholar]
- [79].Chang J, Erler J. Hypoxia-mediated metastasis. Adv Exp Med Biol 2014; 772:55-81; PMID:24272354; http://dx.doi.org/ 10.1007/978-1-4614-5915-6_3 [DOI] [PubMed] [Google Scholar]
- [80].Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci 2012; 125:5591-6; PMID:23420197; http://dx.doi.org/ 10.1242/jcs.116392 [DOI] [PubMed] [Google Scholar]
- [81].Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996; 86:353-64; PMID:8756718; http://dx.doi.org/ 10.1016/S0092-8674(00)80108-7 [DOI] [PubMed] [Google Scholar]
- [82].Ruan K, Song G, Ouyang G. Role of hypoxia in the hallmarks of human cancer. J Cell Biochem 2009; 107:1053-62; PMID:19479945; http://dx.doi.org/ 10.1002/jcb.22214 [DOI] [PubMed] [Google Scholar]
- [83].Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 2001; 93:266-76; PMID:11181773; http://dx.doi.org/ 10.1093/jnci/93.4.266 [DOI] [PubMed] [Google Scholar]
- [84].Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006; 441:437-43; PMID:16724055; http://dx.doi.org/ 10.1038/nature04871 [DOI] [PubMed] [Google Scholar]
- [85].Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res 1996; 56:4509-15; PMID:8813149 [PubMed] [Google Scholar]
- [86].Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 2007; 26:225-39; PMID:17440684; http://dx.doi.org/ 10.1007/s10555-007-9055-1 [DOI] [PubMed] [Google Scholar]
- [87].Weinmann M, Belka C, Plasswilm L. Tumour hypoxia: impact on biology, prognosis and treatment of solid malignant tumours. Onkologie 2004; 27:83-90; PMID:15007254; http://dx.doi.org/ 10.1159/000075611 [DOI] [PubMed] [Google Scholar]
- [88].Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003; 3:721-32; PMID:13130303; http://dx.doi.org/ 10.1038/nrc1187 [DOI] [PubMed] [Google Scholar]
- [89].Kannan A, Krishnan A, Ali M, Subramaniam S, Halagowder D, Sivasithamparam ND. Caveolin-1 promotes gastric cancer progression by up-regulating epithelial to mesenchymal transition by crosstalk of signalling mechanisms under hypoxic condition. Eur J Cancer 2014; 50:204-15; PMID:24070739; http://dx.doi.org/ 10.1016/j.ejca.2013.08.016 [DOI] [PubMed] [Google Scholar]
- [90].Ren T, Zhang W, Liu X, Zhao H, Zhang J, Zhang J, Li X, Zhang Y, Bu X, Shi M, et al.. Discoidin domain receptor 2 (DDR2) promotes breast cancer cell metastasis and the mechanism implicates epithelial-mesenchymal transition programme under hypoxia. J Pathol 2014; 234:526-37; PMID:25130389; http://dx.doi.org/ 10.1002/path.4415 [DOI] [PubMed] [Google Scholar]
- [91].Cho KH, Choi MJ, Jeong KJ, Kim JJ, Hwang MH, Shin SC, Park CG, Lee HY. A ROS/STAT3/HIF-1alpha signaling cascade mediates EGF-induced TWIST1 expression and prostate cancer cell invasion. Prostate 2014; 74:528-36; PMID:24435707; http://dx.doi.org/ 10.1002/pros.22776 [DOI] [PubMed] [Google Scholar]
- [92].Ide T, Kitajima Y, Miyoshi A, Ohtsuka T, Mitsuno M, Ohtaka K, Koga Y, Miyazaki K. Tumor-stromal cell interaction under hypoxia increases the invasiveness of pancreatic cancer cells through the hepatocyte growth factor/c-Met pathway. Int J Cancer J Int Du Cancer 2006; 119:2750-9; http://dx.doi.org/ 10.1002/ijc.22178 [DOI] [PubMed] [Google Scholar]
- [93].Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al.. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704-15; PMID:18485877; http://dx.doi.org/ 10.1016/j.cell.2008.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PloS one 2008; 3:e2888; PMID:18682804; http://dx.doi.org/ 10.1371/journal.pone.0002888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Ann Rev Med 2007; 58:267-84; PMID:17002552; http://dx.doi.org/ 10.1146/annurev.med.58.062105.204854 [DOI] [PubMed] [Google Scholar]
- [96].Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nature Rev Cancer 2009; 9:265-73; http://dx.doi.org/ 10.1038/nrc2620 [DOI] [PubMed] [Google Scholar]
- [97].Chou YF, Chen HH, Eijpe M, Yabuuchi A, Chenoweth JG, Tesar P, Lu J, McKay RD, Geijsen N. The growth factor environment defines distinct pluripotent ground states in novel blastocyst-derived stem cells. Cell 2008; 135:449-61; PMID:18984157; http://dx.doi.org/ 10.1016/j.cell.2008.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al.. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010; 12:468-76; PMID:20418870; http://dx.doi.org/ 10.1038/ncb2048 [DOI] [PubMed] [Google Scholar]
- [99].Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer 2009; 9:665-74; PMID:19693095; http://dx.doi.org/ 10.1038/nrc2714 [DOI] [PubMed] [Google Scholar]
- [100].Hazlehurst LA, Enkemann SA, Beam CA, Argilagos RF, Painter J, Shain KH, Saporta S, Boulware D, Moscinski L, Alsina M, et al.. Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res 2003; 63:7900-6; PMID:14633719 [PubMed] [Google Scholar]
- [101].Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999; 93:1658-67; PMID:10029595 [PMC free article] [PubMed] [Google Scholar]
- [102].Hazlehurst LA, Argilagos RF, Dalton WS. Beta1 integrin mediated adhesion increases Bim protein degradation and contributes to drug resistance in leukaemia cells. Br J Haematol 2007; 136:269-75; PMID:17233818; http://dx.doi.org/ 10.1111/j.1365-2141.2006.06435.x [DOI] [PubMed] [Google Scholar]
- [103].Klein B, Zhang XG, Lu ZY, Bataille R. Interleukin-6 in human multiple myeloma. Blood 1995; 85:863-72; PMID:7849308 [PubMed] [Google Scholar]
- [104].Zhong Z, Wen Z, Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994; 264:95-8; PMID:8140422; http://dx.doi.org/ 10.1126/science.8140422 [DOI] [PubMed] [Google Scholar]
- [105].Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernández-Luna JL, Nuñez G, et al.. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999; 10:105-15; PMID:10023775; http://dx.doi.org/ 10.1016/S1074-7613(00)80011-4 [DOI] [PubMed] [Google Scholar]
- [106].Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al.. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012; 44:852-60; PMID:22751098; http://dx.doi.org/ 10.1038/ng.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000; 2:76-83; PMID:10655586; http://dx.doi.org/ 10.1038/35000025 [DOI] [PubMed] [Google Scholar]
- [108].Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 2005; 8:197-209; PMID:16169465; http://dx.doi.org/ 10.1016/j.ccr.2005.07.009 [DOI] [PubMed] [Google Scholar]
- [109].Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2:442-54; PMID:12189386; http://dx.doi.org/ 10.1038/nrc822 [DOI] [PubMed] [Google Scholar]
- [110].Zhu LF, Hu Y, Yang CC, Xu XH, Ning TY, Wang ZL, Ye JH, Liu LK. Snail overexpression induces an epithelial to mesenchymal transition and cancer stem cell-like properties in SCC9 cells. Lab Invest J Tech Methods Pathol 2012; 92:744-52; http://dx.doi.org/ 10.1038/labinvest.2012.8 [DOI] [PubMed] [Google Scholar]
- [111].Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, Sorokin A, Ovchinnikov LP, Davicioni E, Triche TJ, et al.. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell 2009; 15:402-15; PMID:19411069; http://dx.doi.org/ 10.1016/j.ccr.2009.03.017 [DOI] [PubMed] [Google Scholar]
- [112].Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 2004; 18:1131-43; PMID:15155580; http://dx.doi.org/ 10.1101/gad.294104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, Sadik H, Argani P, Wagner P, Vahdat LT, et al.. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res 2012; 72:1384-94; PMID:22282653; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer 2010; 10:147-56; PMID:20075923; http://dx.doi.org/ 10.1038/nrc2789 [DOI] [PubMed] [Google Scholar]
- [115].Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W, Frank MH. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 2005; 65:4320-33; PMID:15899824; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-3327 [DOI] [PubMed] [Google Scholar]
- [116].Walsh N, Kennedy S, Larkin AM, Tryfonopoulos D, Eustace AJ, Mahgoub T, Conway C, Oglesby I, Collins D, Ballot J, et al.. Membrane transport proteins in human melanoma: associations with tumour aggressiveness and metastasis. Br J Cancer 2010; 102:1157-62; PMID:20234362; http://dx.doi.org/ 10.1038/sj.bjc.6605590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Saxena M, Stephens MA, Pathak H, Rangarajan A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis 2011; 2:e179; PMID:21734725; http://dx.doi.org/ 10.1038/cddis.2011.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000; 406:532-5; PMID:10952316; http://dx.doi.org/ 10.1038/35020106 [DOI] [PubMed] [Google Scholar]
- [119].Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet 2007; 8:341-52; PMID:17440531; http://dx.doi.org/ 10.1038/nrg2101 [DOI] [PubMed] [Google Scholar]
- [120].Rafii S, Lyden D. S100 chemokines mediate bookmarking of premetastatic niches. Nat Cell Biol 2006; 8:1321-3; PMID:17139281; http://dx.doi.org/ 10.1038/ncb1206-1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Wels J, Kaplan RN, Rafii S, Lyden D. Migratory neighbors and distant invaders: tumor-associated niche cells. Genes Dev 2008; 22:559-74; PMID:18316475; http://dx.doi.org/ 10.1101/gad.1636908 [DOI] [PMC free article] [PubMed] [Google Scholar]