


Abstract
To rationally design a productive heterologous biosynthesis system, it is essential to consider the suitability of foreign reactions for the specific endogenous metabolic infrastructure of a host. We developed a novel web server, called MRE, which, for a given pair of starting and desired compounds in a given chassis organism, ranks biosynthesis routes from the perspective of the integration of new reactions into the endogenous metabolic system. For each promising heterologous biosynthesis pathway, MRE suggests actual enzymes for foreign metabolic reactions and generates information on competing endogenous reactions for the consumption of metabolites. These unique, chassis-centered features distinguish MRE from existing pathway design tools and allow synthetic biologists to evaluate the design of their biosynthesis systems from a different angle. By using biosynthesis of a range of high-value natural products as a case study, we show that MRE is an effective tool to guide the design and optimization of heterologous biosynthesis pathways. The URL of MRE is http://www.cbrc.kaust.edu.sa/mre/.
INTRODUCTION
Recent advances in genome editing and metabolic engineering enabled a precise construction of de novo biosynthesis pathways for high-value natural products (1,2). One important design decision to make for the engineering of heterologous biosynthesis systems is concerned with which foreign metabolic genes to introduce into a given host organism (3). Although this decision must be made based on multifaceted factors, a major one is the suitability of pathways for the endogenous metabolism of a host organism, in part because the efficacy of heterologous biosynthesis is affected by competing endogenous pathways (3–5). To address this point, we developed an open-access web server called MRE (Metabolic Route Explorer) that systematically searches for promising heterologous pathways by considering competing endogenous reactions in a given host organism (Figure 1).
Figure 1.

Typical user-interface pages of MRE. (A) The query input page. (B) The summary page for the top-ranked routes. (C) The page for a graph comprising the top 10 routes. (D) The page for pathway-level information. (E) The competing reaction information page.
There are various computational tools available to guide the design of heterologous pathways with a range of scopes and functions. Table 1 summarizes features of several heterologous pathway design tools. One key difference among these design tools is the specification of host organisms. Some tools, including BNICE (6), PredPath (7) and Metabolic tinker (8), were developed to explore pathways irrespective of the consideration for host organisms. Thus, these tools cannot assess the suitability of pathways for a specific context of the endogenous metabolic system of a host organism.
Table 1. Feature summary of heterologous pathway design tools.
| Tool | Access | Chassis | Chemical transformation | Thermodynamic consideration | Ranking score | Information given for each pathway | (Ref.) |
|---|---|---|---|---|---|---|---|
| MRE | Open access web server | Many choices | Verified KEGG reactions | Boltzmann factor | Fraction of conversions via normalized Boltzmann weights | Required metabolites, EC numbers for enzymes, genes for foreign enzymes, reaction free energy, competing native reactions | – |
| FMM | Open access web server | Many choices | KEGG reactions | No | Number of reaction steps | EC numbers for enzymes, availability of each enzyme in various host organisms, suggestion for foreign enzymes | (13) |
| PHT | Open access web server | Many choices | KEGG reactions | No | Number of reaction steps | EC numbers for enzymes, local and global compound similarities for each reaction step | (14) |
| XTMS | Open access web server | E. coli | Predicted reactions | Favorability | Gene scores, reaction steps, toxicity, yield, Gibbs energy | Source compound for the retrosynthesis path, predicted reactions with EC numbers, genes for foreign enzymes, toxicity, production yield | (9) |
| Metabolic tinker | Open access web server | No host | RHEA reactions | Directionality, favorability | Net favorability | Possible reactions for each chemical transformation step and net favorability | (8) |
| PathPred | Open access web server | No host | Predicted reactions | No | Chemical similarity | Final compound of biodegradation, predicted intermediates and reactions, confidence for each predicted reaction | (7) |
| DESHARKY | Free download | E. coli | KEGG reactions | No | Growth rate | Source or target compound, EC numbers for enzymes, genes for some foreign enzymes, growth rate reduction measures | (10) |
| BNICE | Closed access | No host | Predicted reactions | No | No pathway ranking | 3-level EC number for each predicted chemical transformation | (6) |
Some other tools, on the other hand, restrict the user to use one specific host organism. For example, pathway design tools based on flux balance analysis (FBA), such as XTMS (9), DESHARKY (10), OptStrain (11) and GEM-Path (12), are specific to the Escherichia coli chassis. While FBA-based tools tend to offer rich information to evaluate de novo pathways, they demand detailed knowledge of a given metabolic system with tight reaction-flux boundaries in order to identify meaningful steady-state flux distributions among a large number of candidate solutions. Clearly, such data are only available for well-studied organisms, and this may be a major reason why FBA-based tools focus on the pathway design in E. coli.
In contrast, FMM (13) and PHT (14) allow the user to select a host organism from a large set of choices. However, these tools do not use the chassis information to rank suitable biosynthesis pathways for a given endogenous metabolic system. Instead, with this information, PHT just reports which enzymes are not natively available in the host, whereas FMM suggests the introduction of foreign enzymes for some reactions in heterologous pathways.
Another main feature difference is the basis for chemical transformation of intermediate precursors that forms metabolic routes. While tools such as FMM, DESHARKY and Metabolic tinker specify chemical transformation using metabolic reaction sets from databases [e.g., KEGG (15) and RHEA (16)], other tools, including BNICE, PredPath and XTMS, predict some generalized chemical transformation rules using such curated reaction sets and apply them to expand potentially feasible metabolic routes.
Unlike these existing tools, MRE focuses on the suggestion of foreign enzymes with well-characterized activities for promising heterologous pathways by taking into account the effects of the existing, endogenous metabolic infrastructure of a host organism. To find promising biosynthesis routes from a large number of potential candidates, thermodynamic data offer useful information (17). Some existing pathway design tools, such as Metabolic tinker and XTMS, use thermodynamic data to constrain the reaction directionality or to rank pathways based on their net favorability, which does not consider competing endogenous reactions. In contrast, MRE uses thermodynamic data to rank pathways in a host-dependent manner from the perspective of the integration of new reactions into the endogenous metabolic system. In order to suggest actual foreign enzymes for the design of heterologous biosynthesis pathways, MRE only considers verified reactions as metabolic parts. For each foreign reaction in a suggested heterologous pathway, MRE generates information about endogenous reactions competing for metabolites. Since one effective approach to increase the productivity is to attenuate or eliminate competing reactions (4,5,18), MRE may also offer useful insights into how to debottleneck and optimize heterologous pathways.
MATERIALS AND METHODS
Data resources
MRE makes use of several data resources. The main ones are from the KEGG databases (15). KEGG lists around 4000 organisms, which MRE uses for the selection of a host organism. The KEGG COMPOUND database is used to identify metabolites, while the KEGG REACTION database and the ExPASy ENZYME database (19) are used to find metabolic reactions with verified activities. The eQuilibrator dataset (20) is used to obtain the reaction Gibbs energy in the standard 1M concentration setting. The KEGG RPAIR database (21) is used to restrict search space based on the relation between reactants and products. The KEGG GENES database is used for DNA sequence data for enzymatic genes, and the KEGG taxonomy mapping dataset is used to calculate taxonomic distances.
Function of MRE
To explore biosynthesis routes with MRE, the user specifies a host organism and a pair of the starting and target compounds. To increase its usability and to help the user specify organisms and compounds, MRE comes with an auto-completion feature. With advanced options, the user can override the default setting for the metabolic route search. These options include the maximum number of reaction steps (denoted by n), the number of top-ranked pathways to generate (denoted by K), and a list of compounds that are not considered as primary metabolic precursors in the search, which we call the exclusion list. By default, n and K are set to 8 and 50, respectively, while the exclusion list is based on the one from Metabolic tinker (8) and has 101 compounds that have high degrees of connectivity in its metabolic network graph, for example, water, ATP and ADP. This exclusion list can also be customized to have other compounds (e.g., CO2). In addition, MRE allows the user to constrain the chemical transformation of precursors based on RPAIR types (e.g., main, cofac and trans). These filtering schemes to constrain possible chemical transformations were reported to increase the relevance of the de novo biosynthesis route suggestion (22). By default, MRE considers chemical transformations based on main, cofac and trans RPAIR types.
Based on the input query for biosynthesis requirements, MRE generates the top-K metabolic routes, and the main result page summarizes these routes. For each metabolic route, MRE highlights whether it is endogenous or heterologous to the host organism. For each foreign reaction in a heterologous biosynthesis route, MRE predicts which metabolites may not be available in the host, and it lists exogenous genes for the corresponding enzymatic activity and suggests a list of foreign genes based on a taxonomic similarity measure whose cDNA sequences can be downloaded in the FASTA format. It also shows a list of native reactions competing for a metabolic precursor with each foreign enzymatic reaction. MRE provides a mean to visualize a specific pathway with competing endogenous reactions as well as a graph aggregating top-ranked routes.
Workflow of MRE
Figure 2 depicts the workflow of MRE. Our tool first constructs a directed graph representing a host-independent metabolic network with verified reactions. This graph comprises all metabolic reactions with verified activities found in the data source, and it is built regardless of the choice of a host organism for a biosynthesis system. It next assigns weights to the edges in the graph in a host-dependent fashion by classifying which enzymatic reactions are native and foreign in the given host organism and by using the thermodynamic data. To search for biosynthesis routes from the starting material to the product in the host, MRE explores the host-independent metabolic network with the host-dependent weighting scheme exhaustively and generates top-K biosynthesis routes.
Figure 2.

Workflow of MRE.
Host-independent metabolic network with verified reactions
To construct the host-independent metabolic network, we first identified metabolic reactions with verified activities. We categorized enzymatic reactions based on Enzyme Commission numbers (EC numbers) (23). Each EC reaction (i.e., a reaction class corresponding to each EC number) denotes a class of catalytic reactions with the same chemical transformation. To retrieve verified metabolic reactions with known enzymes, we filtered out reaction classes with partially qualified EC numbers as these partial EC reactions are unverified and can lead to misinterpretation of enzymatic activities (24). We also removed those EC reactions that do not contain any enzymes. With this filtering process, we identified 5389 complete EC reactions and 76 spontaneous reactions with verified activities.
We next estimated the standard reaction Gibbs energy ΔrG′° for each of these verified reactions using eQuilibrator with absolute temperature set to 298.15K. Each verified EC reaction is then split into two reactions: the forward reaction with the reaction Gibbs energy ΔrG′° and the backward reaction with the reaction Gibbs energy −ΔrG′°. Those EC reactions whose ΔrG′° could not be estimated were assigned the largest of the estimated values for both directions. We adopted this approach to take a conservative stance and avoid the suggestion of biosynthesis routes containing reactions with no thermodynamic information as much as possible. Using these reactions, we built a directed graph that models the transformation of metabolites where its vertices represent metabolites and its edges represent chemical transformations via verified metabolic reactions. Since this directed graph unifies all metabolic reactions with verified activities in the reaction databases, its structure is independent of the endogenous metabolic system of any host organism.
Host-dependent reaction weighting scheme
Using the user-specified host organism, we assigned weights to the edges of the directed graph. To assign the weight of each outgoing edge from a given compound node, we first assumed that this reaction was in the host organism and computed the probability of converting the precursor via this reaction over the competing native reactions. That is, by representing the competition for a metabolic precursor with endogenous reactions by a statistical mechanical model, we computed the probability of each reaction with ΔrG′° through the Boltzmann distribution. We next took the logarithm of this probability and assigned it as the weight of this outgoing edge. While this type of statistical mechanics modeling has been widely applied in the context of gene regulation to capture the promoter states (25–27), it is, to our knowledge, novel in the context of the biosynthesis system design. This weighting scheme depends on a host organism and models the competition for metabolic precursors with the endogenous reactions. Importantly, this competition-based weighting scheme can capture the effects of competing endogenous reactions on heterologous reactions, while a thermodynamic favorability-based weighting scheme cannot. This can make their weight assignments widely different from each other (as illustrated in Figure 3). A detailed description of this weighting scheme is given in Appendix.
Figure 3.

An illustration of differences between the thermodynamic favorability-based weighting scheme and our competition-based weighting scheme. Nodes are metabolites and edges are metabolic conversions via reactions. Red edges indicate native reactions, while blue edges indicate foreign reactions. (A) The thermodynamic favorability based approach. The value within a green oval for each edge represents the weight ΔrG′°/RT where R is the gas constant and T is the absolute temperature. (B) The competition-based approach. For each edge, the value within a pink oval represents its weight. With this scheme, edges with the same ΔrG′° value can have different weights in a host-dependent fashion. For example, the weight of C1 → C3 is ln [e1/(1 + e1 + e1)], while that of C1 → C4 is ln [e1/(1 + e1)].
Biosynthesis route search
Biosynthesis pathways of interest are often those that transform a higher fraction of a starting material to a target product. One heuristic to rank pathways based on this productivity criterion is the net favorability of pathways. At a first glance, the net thermodynamic favorability can be seen as a good measure to rank pathways based on this criterion. However, this measure can only quantify the ratio of the target concentration to the source concentration at equilibrium, which may not correspond well with the true picture of the titer of the target product, especially when a given pathway has strong competing reactions and the equilibrium concentration of the starting material is substantially lowered.
As described in the previous section, our reaction weighting scheme is based on the logarithm of normalized Boltzmann weights. Unlike thermodynamic favorability measure, this estimates a fraction of a given precursor that is converted into next intermediate metabolites. Thus, a pathway score based on the sum of all reaction weights in a given pathway can characterize the lower bound of a fraction of starting material that is converted into the product through this pathway, and this score can capture the productivity of each pathway more appropriately.
Given the metabolic network graph with host-dependent weights, MRE exhaustively searches for biosynthesis paths from the given starting material to the given product and generates top-K metabolic routes, each of which has at most n reaction steps. In this search, the compounds in the exclusion list are not considered as intermediate precursors of the product. To rank routes, MRE computes their scores by summing all reaction weights in each route and keeps K routes with the highest scores (as illustrated in Figure 4). MRE transforms the metabolic route search problem into a classical computer science problem known as K-shortest loopless path problem (28) and uses an efficient algorithm to solve it. The core part of the search was implemented in C++.
Figure 4.

An illustrative example to show differences in ranking outcomes between the thermodynamic favorability based approach and our new resource utilization competition-based approach. (A) A simplified metabolic network. Nodes are metabolites and edges are metabolic conversions. Red edges indicate native reactions, while blue edges indicate foreign reactions. The value within a green oval for each edge indicates ΔrG′°/RT where R is the gas constant and T is the absolute temperature. Here, compound C1 is the starting metabolite, and compound C6 is the target product. There are three routes for this biosynthesis. (B) Ranking of the three biosynthesis routes with the thermodynamic favorability approach (the lower the score, the better) and our competition-based approach (the higher the score, the better). For example, the score of C1 → C4 → C6 is −1 + 2 = 1 with the former, whereas it is ln [e1/(1 + e1)] + ln [e−2/(1 + e−2 + e−10)] = −2.44 with the latter.
RESULTS
Computational performance analysis in the runtime environment
To evaluate the computational performance of MRE, we measured its processing time in the runtime environment. To this end, we randomly selected 1000 reachable pairs of source and target compounds. With the setting of the largest reaction step size and the largest number of top-ranked pathways (i.e., n = 20 and K = 500), it took less than 10 s for MRE to exhaustively explore routes and process queries on average. In 95% of the samples, the processing time was less than 20 s, and even in the worst case, it was just less than 30 s. With the default setting (i.e., n = 8 and K = 50), the processing time was at most 1.36 s. Thus, we expect that the exhaustive pathway search employed in MRE will not compromise the user experience based on its processing time.
Case study
As a case study, we applied MRE to search for pathways for various biosynthesis specifications using either E. coli K-12 MG1655 or Saccharomyces cerevisiae as the host organism. Table 2 summarizes the top-ranked heterologous pathways that MRE discovered. This shows that, in biosynthesis of a range of high-value natural products, MRE was able to identify pathways that are known to be productive. We also analyzed the results by comparing them with those from four open-access web servers that can design heterologous biosynthesis pathways, namely, FMM (13), Metabolic tinker (8), PHT (14) and XTMS (9). To explore biosynthesis pathways with these tools, we used their default configurations.
Table 2. Top-ranked pathways identified by MRE for various biosynthesis specifications.
| Biosynthesis specification | Results of top-ranked pathway identified by MRE | Comparison with existing tools | |||||
|---|---|---|---|---|---|---|---|
| Source | Target | Host | Steps | Necessary foreign enzymes | Remark | Found a patha | Match with MREb |
| L-tyrosine | naringenin | E. coli | 4 | 4.3.1.23, 6.2.1.12, 2.3.1.74, | Recovered a known route (29) | FMM, XTMS | FMM |
| (C00082) | (C00509) | (ECO) | 5.5.1.6 | as the top route | |||
| glycerol | 1,3-PDO | E. coli | 2 | 4.2.1.30, 1.1.1.202 | Recovered a known route (30) | FMM, PHT | FMM, PHT |
| (C00116) | (C02457) | (ECO) | as the top route | ||||
| glycerol | R-1,2-PDO | yeast | 5 | 4.2.3.3, 1.1.1.79, 1.1.1.77 | Recovered a known route (31) | MT | MT |
| (C00116) | (C02912) | (SCE) | as the top route | ||||
| acetyl-CoA | artemisinic acid | yeast | 10 | 2.5.1.92, 4.2.3.50, 4.2.3.82, | Recovered a known route (32), | none | none |
| (C00024) | (C20309) | (SCE) | 4.2.3.24, 1.14.13.158 | and predicted better ones | |||
| L-tyrosine | resveratrol | E. coli | 3 | 4.3.1.23, 6.2.1.12, 2.3.1.95 | Recovered a known route (33) | FMM | FMM |
| (C00082) | (C03582) | (ECO) | as the top route | ||||
| D-xylose | xylitol | E. coli | 2 | 1.1.1.21, 1.1.1.307 | Recovered two known routes (34) | FMM, PHT | FMM, PHT |
| (C00181) | (C00379) | (ECO) | as the top routes | ||||
| PRPP | histidine | E. coli | 8 | 2.6.1.27 | Predicted | ||
| better and shorter routes | FMM, MT | none | |||||
| (C00119) | (C00135) | (ECO) | than a known native route (35) | ||||
| chorismate | tryptophan | yeast | 5 | none | Predicted the native route (36) as | FMM, MT, | FMM |
| (C00251) | (C00078) | (SCE) | the best, and found shorter routes | PHT | |||
For each biosynthesis specification, the source and target compounds are specified in KEGG ID, and the host organism is in KEGG organism code. For each pathway, the number of reaction steps and the necessary foreign enzymes(in EC number) are specified. Comparison with FMM (13), Metabolic tinker (MT) (8), PHT (14) and XTMS (9) is also shown. For each tool, its default setting was used, except for the configuration of a pathway length, which was set to accommodate known pathways.
aTools that have identified at least one path for a given biosynthesis specification.
bTools whose top-ranked pathway is the same as the top-ranked one from MRE.
Biosynthesis of naringenin
Naringenin is a plant secondary metabolite, which is reported to have various health benefits (37), including high antioxidant capacities (38) and significant antiviral effects on the hepatitis C virus (39). Owing to inefficiencies in the production of naringenin from natural plant sources, metabolic engineering to have an efficient microbial synthesis of this high-value natural product is thought to be a commercially viable alternative (29,40).
In this analysis, we selected L-tyrosine (KEGG compound ID: C00082), an aromatic non-essential amino acid, as the starting material since a state-of-the-art heterologous naringenin production from L-tyrosine in an E. coli strain is known (see Figure 5A). This heterologous biosynthesis route comprises four foreign enzymatic reactions. To analyze the performance of MRE in comparison with other tools, we applied two open-access biosynthesis pathway web servers, Metabolic tinker (8) and XTMS (9). Since these two recently developed tools also rely on reaction thermodynamic data for their pathway ranking, we can also analyze the effects on our competition-based ranking scheme.
Figure 5.

A heterologous biosynthesis pathway to produce naringenin from L-tyrosine in an E. coli host. (A) The structure of an experimentally derived biosynthesis pathway (29). The EC number for each reaction is indicated below the arrow. The KEGG compound ID of each metabolite is framed in red. The abbreviations are: tyrosine ammonia lyase (TAL); 4-coumarate:CoA ligase (4CL); chalcone synthase (CHS) and chalcone isomerase (CHI). (B) The information of the top-ranked biosynthesis pathway in MRE.
Given this biosynthesis requirement, Metabolic tinker and PHT were not able to find any pathways, while XTMS generated a predicted pathway with hypothetical reactions as its top-ranked candidate. In contrast, the top-ranked route from MRE and FMM was identical to the state of the art. The pathway information given by MRE indicates that the third reaction in the pathway, which transforms p-coumaroyl-CoA into naringenin chalcone, is a bottleneck and competes for the availability of cofactor malonyl-CoA with a more favorable native reaction involved in the fatty acid biosynthesis in the E. coli host (Figure 5B). This suggests that an increase in the concentration of malonyl-CoA or the inhibition of the fatty acid biosynthesis could enhance the productivity of this naringenin biosynthesis pathway. Indeed, previous studies demonstrated that both an increase in the availability of malonyl-CoA in the host and a decrease in the activities in the fatty acid pathway can increase the naringenin titers (29,41). While FMM was also able to identify the heterologous naringenin biosynthesis pathway that MRE found, the pathway information given by FMM was not helpful to find an optimization target as FMM does not have a feature to quantify the effects of competing reactions in the host.
Production of value-added chemicals from glycerol
Glycerol is a readily available and relatively inexpensive chemical compound that can be generated in large amounts as a byproduct of biodiesel and bioethanol production processes (42,43). Because of its economic viability and long-term sustainability, fermentative production of high-value materials from glycerol has gained much attention recently (44). By using glycerol as the starting material, we searched for pathways for the production of two value-added chemicals, 1,3-propanediol (1,3-PDO), a commodity chemical mainly used to make polyester fiber, and 1,2-propanediol (1,2-PDO), another high-demand commodity chemical used to make a wide range of products including antifreeze, thermoset plastics and cosmetics.
We first applied MRE to search for pathways for the production of 1,3-PDO in E. coli chassis. The top-ranked pathway that MRE identified is a known two-step heterologous pathway (42), which requires the introduction of a glycerol dehydratase gene and a 1,3-propanediol dehydrogenase gene in the host (Figure 6A). Since the first glycerol dehydratase reaction competes for the utilization of glycerol against several native reactions including glycerol kinase, MRE predicts that this can be a target for productivity optimization. Metabolic tinker and XTMS were not able to find any pathways for the 1,3-PDO production, whereas FMM and PHT found the same pathway that MRE identified.
Figure 6.

Pathway-level graphs generated in MRE. (A) The top-ranked pathway for the production of 1,3-PDO from glycerol in E. coli. (B) The top-ranked pathway for the production of R-1,2-PDO from glycerol in yeast. (C) A known pathway and (D) top-ranked pathway for the production of artemisinic acid from acetyl-CoA in yeast. In these graphs, oval nodes represent compounds, while box nodes represent reactions. For compound nodes, red nodes are the starting material, green nodes are the target products and yellow nodes are other compounds. For reaction nodes, purple nodes are native reactions, cyan nodes are foreign reactions and gray nodes are competing native reactions.
We next applied MRE to search for pathways for the synthesis of R-1,2-PDO in the yeast chassis. We found that the top-ranked pathway (Figure 6B) was a known synthesis pathway for 1,2-PDO (31). In this pathway, glycerol is first converted to dihydroxyacetone phosphate (DHAP) via two native enzymatic reactions. Methylglyoxal synthase then transforms DHAP into methylglyoxal, which is, in turn, converted into (R)-lactaldehyde. Finally, lactaldehyde reductase is used to produce R-1,2-PDO from (R)-lactaldehyde. FMM and PHT were not able to find any pathways that convert glycerol into R-1,2-PDO, whereas Metabolic tinker identified the same pathway that MRE found as the top-ranked one. Since XTMS focuses on the E. coli chassis, we applied this tool to search for heterologous R-1,2-PDO production pathways in E. coli; however, no pathways were found.
Production of artemisinic acid
Artemisinic acid is an intermediate precursor for antimalaria drug artemisinin (32,45), and its production is often celebrated as one of the early success stories of the combination of metabolic engineering and synthetic biology (46). This engineered biosynthesis pathway utilizes the endogenous mevalonate pathway in budding yeast to transform acetyl-CoA into farnesyl pyrophosphate (FPP), which is then converted into artemisinic acid with heterologous amorphadiene synthase and three-step oxidation reactions (32,45).
To see if MRE could recover this engineered pathway, we applied it to explore pathways for the production of artemisinic acid from acetyl-CoA in yeast. We found that one of the top-ranked pathways that MRE generated was this known heterologous pathway (Figure 6C). Interestingly, the pathway that MRE identified as the top candidate (Figure 6D) was slightly different from the previously engineered pathway. The difference lies in how isopentenyl pyrophosphate (IPP) is converted into farnesyl pyrophosphate (FPP). In the top-ranked path, IPP is first converted into (2Z,6Z)-farnesyl diphosphate (Z,Z-FPP). This route is chosen because IPP is a precursor of a thermodynamically highly favorable native reaction, and the conversion reaction from IPP to Z,Z-FPP is much more favorable than that from IPP to FPP, enabling a higher fraction of IPP to be utilized in the route. By using Z,Z-FPP as the precursor, this route introduces three foreign carbon-oxygen lyases to form FPP. FMM, Metabolic tinker and PHT were not able to find any pathways. XTMS found a partial pathway that converts FPP into artemisinic acid, albeit it is for the E. coli chassis.
DISCUSSION
In this paper, we introduced MRE, an open-access biosynthesis design tool, that searches for promising metabolic routes for a given biosynthesis specification and suggests exogenous enzymes for heterologous biosynthesis pathways based on the infrastructure of an endogenous metabolic system. A main limitation of MRE is its reliance on the data sources (mainly KEGG) to mine verified metabolic reactions and to search for biosynthesis routes based on them. Indeed, while painstaking effort has resulted in a large collection of annotated metabolic reaction data, among the 9910 reactions in the KEGG REACTION database (Release 76.0), we found 1272 with no EC numbers, 1079 with partial EC numbers and 2170 with no annotations for associated genes. While this deficiency can prevent MRE from finding promising biosynthesis pathways, we expect the number of verified reactions in KEGG to increase over time and this issue to be alleviated eventually. At the same time, we are considering an option to also integrate other metabolic reaction databases such as Rhea (16) in a future release.
Several existing tools took an approach to expand a list of metabolic parts in hand by defining specific transformation rules (6,7,9), albeit such rules can be subjective (47). To design biosynthesis systems, this approach relies on the prediction of metabolic parts with specific metabolic activities, which may or may not exist. Thus, the design of biosynthesis systems via this top-down approach may require the de novo design of unnatural proteins to achieve specific metabolic activities. MRE was, on the other hand, developed to suggest actual enzymes for heterologous pathways. Thus, it takes a complementary, bottom-up approach in which a biosynthesis system is designed by using well-characterized metabolic parts. To this end, we made a conscious decision to use only verified reactions.
Here, by using the biosynthesis of a range of high-value natural products as a case study, we have shown that MRE can suggest promising heterologous biosynthesis pathways and provide useful information to pinpoint bottlenecks of pathways. In summary, with the host-dependent competition-based pathway ranking scheme along with the suggestion of foreign enzymes with competing endogenous reactions, MRE is expected to offer novel insights into the design and optimization of heterologous biosynthesis systems.
Acknowledgments
We thank Elad Noor for his help with eQuilibrator. This research made use of the resources of the computer clusters at KAUST.
APPENDIX
Mathematical description of the host-dependent reaction weighting scheme
To derive a mathematical description of the weighting scheme, we consider a scenario in which to generate weights for edges in the reactions transforming precursor C. Here, let 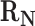 be a set of native reactions that can transform C in a given host organism. For each reaction r that can transform C, we set
be a set of native reactions that can transform C in a given host organism. For each reaction r that can transform C, we set 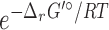 as its Boltzmann factor. Then, we define f(r), the normalized Boltzmann factor for r, as follows:
as its Boltzmann factor. Then, we define f(r), the normalized Boltzmann factor for r, as follows:
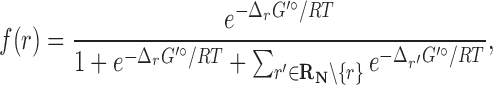 |
(1) |
where R is the gas constant and T is the absolute temperature. That is, those reactions that are not in the host organism do not affect the calculation of the Boltzmann distribution. If 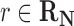 , then f(r) is simply based on the Boltzmann distribution of the native reaction system transforming compound C. On the other hand, if
, then f(r) is simply based on the Boltzmann distribution of the native reaction system transforming compound C. On the other hand, if 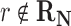 , then f(r) is based on the Boltzmann distribution of the reaction system that contains all native reactions transforming C and foreign reaction r. With this scheme, in our graph, every edge that transforms C in reaction r has the weight log f(r).
, then f(r) is based on the Boltzmann distribution of the reaction system that contains all native reactions transforming C and foreign reaction r. With this scheme, in our graph, every edge that transforms C in reaction r has the weight log f(r).
FUNDING
King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) [URF/1/1976-04]. Funding for open access charge: King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) [URF/1/1976-04].
Conflict of interest statement. None declared.
REFERENCES
- 1.Luo Y., Li B.-Z., Liu D., Zhang L., Chen Y., Jia B., Zeng B.-X., Zhao H., Yuan Y.-J. Engineered biosynthesis of natural products in heterologous hosts. Chem. Soc. Rev. 2015;44:5265–5290. doi: 10.1039/c5cs00025d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jakočiūnas T., Jensen M.K., Keasling J.D. CRISPR/Cas9 advances engineering of microbial cell factories. Metab. Eng. 2016;34:44–59. doi: 10.1016/j.ymben.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Prather K.L.J., Martin C.H. De novo biosynthetic pathways: rational design of microbial chemical factories. Curr. Opin. Biotechnol. 2008;19:468–474. doi: 10.1016/j.copbio.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Solomon K.V., Moon T.S., Ma B., Sanders T.M., Prather K. L.J. Tuning primary metabolism for heterologous pathway productivity. ACS Synth. Biol. 2013;2:126–135. doi: 10.1021/sb300055e. [DOI] [PubMed] [Google Scholar]
- 5.Ida K., Ishii J., Matsuda F., Kondo T., Kondo A. Eliminating the isoleucine biosynthetic pathway to reduce competitive carbon outflow during isobutanol production by Saccharomyces cerevisiae. Microb. Cell Fact. 2015;14:62. doi: 10.1186/s12934-015-0240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hatzimanikatis V., Li C., Ionita J.A., Henry C.S., Jankowski M.D., Broadbelt L.J. Exploring the diversity of complex metabolic networks. Bioinformatics. 2005;21:1603–1609. doi: 10.1093/bioinformatics/bti213. [DOI] [PubMed] [Google Scholar]
- 7.Moriya Y., Shigemizu D., Hattori M., Tokimatsu T., Kotera M., Goto S., Kanehisa M. PathPred: an enzyme-catalyzed metabolic pathway prediction server. Nucleic Acids Res. 2010;38:W138–W143. doi: 10.1093/nar/gkq318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McClymont K., Soyer O.S. Metabolic tinker: an online tool for guiding the design of synthetic metabolic pathways. Nucleic Acids Res. 2013;41:e113. doi: 10.1093/nar/gkt234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carbonell P., Parutto P., Herisson J., Pandit S.B., Faulon J.-L. XTMS: pathway design in an eXTended metabolic space. Nucleic Acids Res. 2014;42:W389–W394. doi: 10.1093/nar/gku362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodrigo G., Carrera J., Prather K.J., Jaramillo A. DESHARKY: automatic design of metabolic pathways for optimal cell growth. Bioinformatics. 2008;24:2554–2556. doi: 10.1093/bioinformatics/btn471. [DOI] [PubMed] [Google Scholar]
- 11.Pharkya P., Burgard A.P., Maranas C.D. OptStrain: a computational framework for redesign of microbial production systems. Genome Res. 2004;14:2367–2376. doi: 10.1101/gr.2872004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campodonico M.A., Andrews B.A., Asenjo J.A., Palsson B.O., Feist A.M. Generation of an atlas for commodity chemical production in Escherichia coli and a novel pathway prediction algorithm, GEM-Path. Metab. Eng. 2014;25:140–158. doi: 10.1016/j.ymben.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 13.Chou C.-H., Chang W.-C., Chiu C.-M., Huang C.-C., Huang H.-D. FMM: a web server for metabolic pathway reconstruction and comparative analysis. Nucleic Acids Res. 2009;37(suppl 2):W129–W134. doi: 10.1093/nar/gkp264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rahman S.A., Advani P., Schunk R., Schrader R., Schomburg D. Metabolic pathway analysis web service (Pathway Hunter Tool at CUBIC) Bioinformatics. 2005;21:1189–1193. doi: 10.1093/bioinformatics/bti116. [DOI] [PubMed] [Google Scholar]
- 15.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgat A., Axelsen K.B., Lombardot T., Alcántara R., Aimo L., Zerara M., Niknejad A., Belda E., Hyka-Nouspikel N., Coudert E. Updates in Rhea—a manually curated resource of biochemical reactions. Nucleic Acids Res. 2014;43:D459–D464. doi: 10.1093/nar/gku961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ataman M., Hatzimanikatis V. Heading in the right direction: thermodynamics-based network analysis and pathway engineering. Curr. Opin. Biotechnol. 2015;36:176–182. doi: 10.1016/j.copbio.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 18.Avalos J.L., Fink G.R., Stephanopoulos G. Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat. Biotechnol. 2013;31:335–341. doi: 10.1038/nbt.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bairoch A. The ENZYME database in 2000. Nucleic Acids Res. 2000;28:304–305. doi: 10.1093/nar/28.1.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flamholz A., Noor E., Bar-Even A., Milo R. eQuilibrator–the biochemical thermodynamics calculator. Nucleic Acids Res. 2012;40:D770–D775. doi: 10.1093/nar/gkr874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimizu Y., Hattori M., Goto S., Kanehisa M. Generalized reaction patterns for prediction of unknown enzymatic reactions. Genome Inform. 2008;20:299. [PubMed] [Google Scholar]
- 22.Faust K., Croes D., van Helden J. Metabolic pathfinding using RPAIR annotation. J. Mol. Biol. 2009;388:390–414. doi: 10.1016/j.jmb.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Webb E.C. Enzyme nomenclature 1992: Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes. San Diego: Academic Press; 1992. [Google Scholar]
- 24.Green M., Karp P. Genome annotation errors in pathway databases due to semantic ambiguity in partial EC numbers. Nucleic Acids Res. 2005;33:4035–4039. doi: 10.1093/nar/gki711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ackers G.K., Johnson A.D., Shea M.A. Quantitative model for gene regulation by λ phage repressor. Proc. Natl. Acad. Sci. U.S.A. 1982;79:1129–1133. doi: 10.1073/pnas.79.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arkin A., Ross J., McAdams H. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells. Genetics. 1998;149:1633–1648. doi: 10.1093/genetics/149.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuwahara H., Myers C.J., Samoilov M.S. Temperature control of fimbriation circuit switch in uropathogenic Escherichia coli: quantitative analysis via automated model abstraction. PLoS Comput. Biol. 2010;6:e1000723. doi: 10.1371/journal.pcbi.1000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yen J.Y. Finding the K shortest loopless paths in a network. Manag. Sci. 1971;17:712–716. [Google Scholar]
- 29.Santos C.N.S., Koffas M., Stephanopoulos G. Optimization of a heterologous pathway for the production of flavonoids from glucose. Metab. Eng. 2011;13:392–400. doi: 10.1016/j.ymben.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Tang X., Tan Y., Zhu H., Zhao K., Shen W. Microbial conversion of glycerol to 1,3-propanediol by an engineered strain of Escherichia coli. Appl. Environ. Microbiol. 2009;75:1628–1634. doi: 10.1128/AEM.02376-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeon E., Lee S., Kim D., Yoon H., Oh M., Park C., Lee J. Development of a Saccharomyces cerevisiae strain for the production of 1,2-propanediol by gene manipulation. Enzyme Microb. Technol. 2009;45:42–47. [Google Scholar]
- 32.Ro D.-K., Paradise E.M., Ouellet M., Fisher K.J., Newman K.L., Ndungu J.M., Ho K.A., Eachus R.A., Ham T.S., Kirby J., et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 33.Mei Y.-Z., Liu R.-X., Wang D.-P., Wang X., Dai C.-C. Biocatalysis and biotransformation of resveratrol in microorganisms. Biotechnol. Lett. 2015;37:9–18. doi: 10.1007/s10529-014-1651-x. [DOI] [PubMed] [Google Scholar]
- 34.Cirino P.C., Chin J.W., Ingram L.O. Engineering Escherichia coli for xylitol production from glucose-xylose mixtures. Biotechnol. Bioeng. 2006;95:1167–1176. doi: 10.1002/bit.21082. [DOI] [PubMed] [Google Scholar]
- 35.Alifano P., Fani R., Liò P., Lazcano A., Bazzicalupo M., Carlomagno M.S., Bruni C.B. Histidine biosynthetic pathway and genes: structure, regulation, and evolution. Microbiol. Rev. 1996;60:44–69. doi: 10.1128/mr.60.1.44-69.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toyn J.H., Gunyuzlu P.L., White W.H., Thompson L.A., Hollis G.F. A counterselection for the tryptophan pathway in yeast: 5-fluoroanthranilic acid resistance. Yeast. 2000;16:553–560. doi: 10.1002/(SICI)1097-0061(200004)16:6<553::AID-YEA554>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 37.Hollman P.C., Katan M.B. Bioavailability and health effects of dietary flavonols in man. Arch. Toxicol. Suppl. 1998;20:237–248. doi: 10.1007/978-3-642-46856-8_21. [DOI] [PubMed] [Google Scholar]
- 38.Cavia-Saiz M., Busto M.D., Pilar-Izquierdo M.C., Ortega N., Perez-Mateos M., Muñiz P. Antioxidant properties, radical scavenging activity and biomolecule protection capacity of flavonoid naringenin and its glycoside naringin: a comparative study. J. Sci. Food Agr. 2010;90:1238–1244. doi: 10.1002/jsfa.3959. [DOI] [PubMed] [Google Scholar]
- 39.Nahmias Y., Goldwasser J., Casali M., van Poll D., Wakita T., Chung R.T., Yarmush M.L. Apolipoprotein B–dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology. 2008;47:1437–1445. doi: 10.1002/hep.22197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fowler Z.L., Koffas M.A. Biosynthesis and biotechnological production of flavanones: current state and perspectives. Appl. Microbiol. Biotechnol. 2009;83:799–808. doi: 10.1007/s00253-009-2039-z. [DOI] [PubMed] [Google Scholar]
- 41.Leonard E., Yan Y., Fowler Z.L., Li Z., Lim C.-G., Lim K.-H., Koffas M.A. Strain improvement of recombinant Escherichia coli for efficient production of plant flavonoids. Mol. Pharm. 2008;5:257–265. doi: 10.1021/mp7001472. [DOI] [PubMed] [Google Scholar]
- 42.Yazdani S.S., Gonzalez R. Anaerobic fermentation of glycerol: a path to economic viability for the biofuels industry. Curr. Opin. Biotechnol. 2007;18:213–219. doi: 10.1016/j.copbio.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 43.Quispe C.A., Coronado C.J., Carvalho J.A., Jr Glycerol: Production, consumption, prices, characterization and new trends in combustion. Renew Sustain. Energy Rev. 2013;27:475–493. [Google Scholar]
- 44.Clomburg J.M., Gonzalez R. Anaerobic fermentation of glycerol: a platform for renewable fuels and chemicals. Trends Biotechnol. 2013;31:20–28. doi: 10.1016/j.tibtech.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 45.Paddon C.J., Westfall P.J., Pitera D., Benjamin K., Fisher K., McPhee D., Leavell M., Tai A., Main A., Eng D., et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature. 2013;496:528–532. doi: 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- 46.Paddon C.J., Keasling J.D. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 2014;12:355–367. doi: 10.1038/nrmicro3240. [DOI] [PubMed] [Google Scholar]
- 47.Martin C.H., Nielsen D.R., Solomon K.V., Prather K. L.J. Synthetic metabolism: engineering biology at the protein and pathway scales. Chem. Biol. 2009;16:277–286. doi: 10.1016/j.chembiol.2009.01.010. [DOI] [PubMed] [Google Scholar]