

DYT1 dystonia, an early onset generalized dystonia, also known as Oppenheim’s dystonia, is an inherited isolated dystonia characterized by progressive generalized muscle spasms and sustained postures leading to significant disability [1]. The disease is inherited in an autosomal dominant manner with incomplete penetrance (30–40 %) and typically presents in childhood [2]. Patients harbor a 3-bp (GAG) deletion in the coding region of the TOR1A gene on chromosome 9q34 that encodes the protein torsinA [3]. This deletion corresponds to loss of a single glutamate at amino acid residue 302 or 303 (torsinA ΔE) [4]. The function of wildtype torsinA has been speculative, but relatively recent studies have demonstrated its involvement in protein trafficking, quality control, secretion, and degradation (for review, see Dauer 2014 [5]). The pathogenic mechanism leading to disease as a result of this deletion is thought to likely involve disruption of sensorimotor circuit development and function [6]. Recent evidence also suggests striatal cholinergic dysfunction, or dysregulation, as a potential mechanism underlying the pathophysiology of DYT1 dystonia [7].
Various in vitro and transgenic animal models of DYT1 dystonia have demonstrated altered cell morphology and nuclear changes at the light microscopic level, including torsinA accumulation and torsinA-positive inclusions in the brainstem (see Dauer 2014 [5] and Oleas et al. 2013 [8] for review of cell culture and animal model findings, respectively). Contrary to findings in animal tissues, no consistent or specific histopathologic changes have been noted in postmortem neuropathologic studies of patients with DYT1 dystonia (Table 1). Subcellular changes (e.g., inclusions) in DYT1 brains have been described in a small human postmortem study [9], but their specificity remains controversial. That study reported the presence of perinuclear and intranuclear inclusion bodies and protein accumulation in neurons that were immunoreactive for torsinA and ubiquitin, and co-localized with choline acetyltransferase (ChAT) and a nuclear envelope marker, lamin A/C, in the midbrain and pontine reticular formation (pedunculopontine nucleus (PPN), cuneiform nucleus (CN), and periaqueductal grey (PAG)); these changes were notably absent in controls. A more recent study, however, failed to confirm the presence of immunoreactive inclusions in human DYT1 dystonia brain tissue (using antibodies to ubiquitin and p62) [10]. Additionally, two earlier studies failed to reveal inclusion bodies, aggregates, or aberrant staining for torsinA in DYT1 brains and in other dystonia (non-DYT1) cases [11, 12].
Table 1.
DYT1 dystonia neuropathology studies
| References | Subjects | Inclusions | Key antibodies | Subcellular TorsinA immunoreactivity |
|---|---|---|---|---|
| Walker et al. 2002 [11] | 1 DYT1 Dystonia subject | No | Anti-torsinA (rabbit polyclonal: AA residues 323–332) | Nucleus, cytoplasm, and dendrites in both DYT1 dystonia and control groups |
| 4 control subjects | No | |||
| Rostasy et al. 2003 [12] | 5 DYT1 Dystonia subjects | No | Anti-torsinA (rabbit polyclonal TAB1: AA residues 299–312; mouse monoclonal D-MG10: AA residues 208–249) | Cytoplasm, dendrites, and axons in both DYT1 dystonia and control groups |
| 20 control subjects | No | |||
| McNaught et al. 2004 [9] | 4 DYT1 Dystonia subjects | Yes: midbrain, pons | Anti-torsinA (rabbit polyclonal; AA residues 323–332), Anti-ubiquitin, Anti-UPC, Anti-ChAT | Perinuclear and intranuclear inclusions in DYT1 dystonia group only |
| 4 control subjects | No | |||
| Paudel et al. 2014 [10] | 7 DYT1 Dystonia subjects | No | Anti-ubiquitin, Anti-p62 | N/A |
AA amino acid, ChAT choline acetyl transferase, UPC ubiquitin-protein conjugate
Here, we sought to analyze genetically confirmed DYT1 brain specimens with antibodies to torsinA, ubiquitin protein conjugate (UPC), and ChAT in an attempt to identify the previously reported intracellular immunoreactive protein inclusions/aggregates in the midbrain as well as in other cholinergic nuclei, including the striatum. Our study included six brain samples from DYT1 patients (mean age 83.0 ± 9.1 years; all female), procured from the University of Maryland Brain and Tissue Bank (BTB); demographic and autopsy data from four of our DYT1 subjects matched those reported previously [10] and most likely represent tissue from the same subjects. In our study, three patients were clinically symptomatic and the remaining were non-manifesting carriers of the DYT1 mutation. Control brain tissue samples from seven subjects without clinical evidence of dystonia or other movement disorders were matched for age and sex (mean age 83.4 ± 8.0 years; all female). Formalin-fixed paraffin-embedded (FFPE) tissue sections from the striatum, motor and sensory cortices in all patients and controls, and from the brainstem (midbrain/pons, including PPN, CN, and PAG) in five of six DYT1 cases and all controls were evaluated. Routine hematoxylin-eosin staining was unrevealing. Immunohistochemistry (IHC) with antibodies directed to the following antigens were used: torsinA (Chemicon, MAB5550), ChAT (Millipore, AB144P), and ubiquitin protein conjugate (Millipore, 09–408). Additionally, IHC with anti-beta amyloid (Dako, M0872), anti-phospho-tau (AT8, Pierce, MN1020), and anti-alpha synuclein (Cell Signaling, 2647) antibodies along with Bielschowsky silver staining excluded significant neurodegenerative disease. Double staining IHC was performed for torsinA and ChAT using an automated immunostainer (Leica Bond-Max, Buffalo Grove, IL). Western blot analysis was performed with anti-torsinA on fresh-frozen control human brain tissue lysates and appropriate molecular weight labeling for the torsinA protein (37 kDa) was confirmed (Fig. 1). To further evaluate intracellular chromogen (3,3-diaminobenzidine and Fast Red) localization, multispectral imaging (CRi Nuance V.2.8, Woburn, MA) was employed on double-labeled fixed tissue stained with torsinA and ChAT.
Fig. 1.
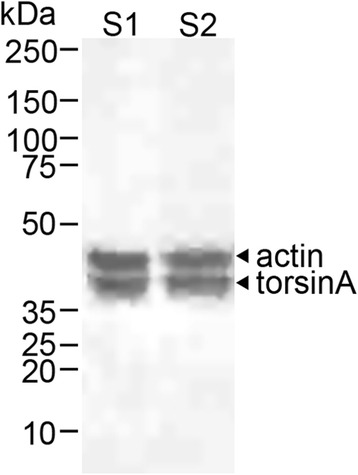
Specificity of the torsinA antibody MAB5550 by Western blot analysis. This Western blot of two control human brain homogenates (labeled as S1 and S2) was performed with anti-torsinA and anti-actin primary antibodies. The location and corresponding molecular weight for torsinA (37 kDa) is shown. Actin (42 kDa) was used as a loading control
No specific cytoplasmic, perinuclear, or intranuclear torsinA-immunoreactive inclusions were identified in our DYT1 tissues. Immunoreactivity for torsinA, ChAT, and UPC in both DYT1 and control groups yielded similar results and predominantly showed normal staining patterns for these endogenous cellular proteins. In both groups, torsinA demonstrated granular cytoplasmic staining in all regions evaluated, as well as punctate immunoreactivity in the neuropil (Fig. 2a). In addition to its widespread neuroanatomic distribution, cellular localization studies have shown that wildtype torsinA is limited to neuronal cytoplasm (endoplasmic reticulum), nuclear envelope, and processes. It should be noted that occasional variations in the staining pattern of torsinA were seen in both DYT1 and control tissues. Apparent cytoplasmic aggregation of the torsinA protein was seen in both groups, with these changes most prominent in large striatal cholinergic interneurons (Fig. 2b, c, d). Inclusion body-like immunoreactivity seen in a control subject (Fig. 2e) exemplifies the non-specific nature of these changes. Occasional neurons in the striatum in two DYT1 patients demonstrated perinuclear accumulation or accentuation of torsinA (Fig. 2f, g); this change was rare and not seen in control tissue. The staining patterns and distribution for ChAT and UPC were similar between both groups. ChAT showed diffuse expression in neuronal cell bodies and processes, while UPC demonstrated predominantly cytoplasmic immunoreactivity without evidence of inclusion bodies in the aforementioned brain regions.
Fig. 2.
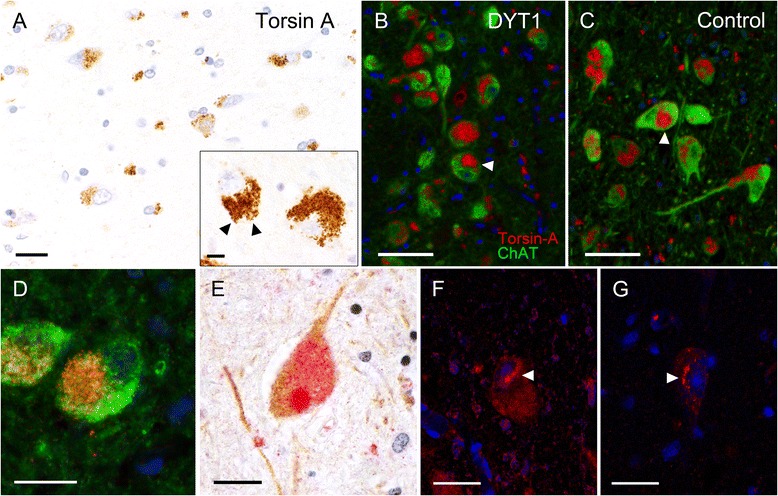
Cellular distribution of torsinA and ChAT in DYT1 and control striatum. a Normal staining pattern and cellular distribution of torsinA in the human striatum (scale bar, 20 μm). TorsinA shows granular cytoplasmic staining (inset, arrowheads; scale bar, 10 μm). b, c, d Fluorescence-like multispectral image (gated immunohistochemical chromogen signal) of torsinA protein (red) reveals circumscribed collections in the cytoplasm of both DYT1 and control neurons (arrowheads; scale bars, 20 μm). ChAT (green) shows diffuse immunoreactivity within the cytoplasm and processes in both DYT1 and controls. e This section of an unaffected control shows an inclusion-like aggregate immunoreactive to torsinA (red) (scale bar, 20 μm). f, g Also identified was perinuclear accumulation of torsinA (red) seen in DYT1 tissue, thought to be a response to cellular stress (scale bars, 20 μm)
Our results add to the growing body of evidence that there are no consistent torsinA immunoreactive protein inclusions associated with, or specific to, DYT1 dystonia in humans. In cell culture studies of neural cells transfected with mutant torsinA, cells demonstrated large perikaryal inclusions ultrastructurally composed of spheroid whorled membranes [13]. The location of the collections appeared to be mutation status-related, with overexpression of mutant torsinA forming inclusions adjacent to the nuclear membrane and overexpressed wildtype protein aggregating within the cytoplasm. This latter feature is in keeping with our findings but was nonspecific and may possibly represent a response of wildtype torsinA to cellular stress (e.g., agonal state). The perinuclear accentuation of torsinA immunoreactivity we observed has previously been described in cultured cells exposed to oxidative stress [14]. Lastly, it has been suggested that endogenous torsinA levels may be lower in vivo as compared to those in transfected cells due to overexpression in cell culture models, which may affect subcellular localization of torsinA [15].
Potential reasons for recurrent failure to demonstrate inclusions in human DYT1 tissue have been noted [5, 10, 12] and deserve mention here. Foremost, DYT1 dystonia is rare and tissue availability for sufficient sample size analysis is very limited; this is a limitation in the context of an incompletely penetrant disease, as disease-specific changes may be related to clinically apparent disease. Notably, while our evaluation was already limited to six DYT1 patients, only five included brainstem tissue (including the PPN, CN, and PAG) for evaluation. Second, studies involving human and animal tissues have failed to recapitulate the immunoreactive protein aggregates observed in culture cells overexpressing torsinA (see Dauer [5] for review), supporting the suspected lack of neurodegeneration in the disease. The human tissue studies that failed to identify torsinA inclusions used various anti-torsinA antibodies directed against different epitopes, which further supports that lack of inclusion body formation in DYT1 dystonia. Furthermore, there is strong evidence linking altered cholinergic striatal function, and its inputs from other structures such as the brainstem, cerebellum and thalamus, to the underlying pathophysiology in DYT1 dystonia (for review see Eskow Jaunarajs et al. [16]). Indeed, postmortem analyses of the putamen of DYT1 subjects have shown decreased levels of cholinergic markers [7].
In conclusion, there is little evidence supporting the presence of specific cellular morphologic changes in DYT1 dystonia at the light microscopic level in human tissue. The significance and specificity of the changes we observed are best addressed with larger postmortem studies combining histopathology and disease-specific cell models, such as patient-specific induced pluripotent stem cells (iPSCs). Use of iPSC-derived neurons would allow the subcellular localization of torsinA, and any inclusions, to be explored in living cells without altering the level of torsinA expression.
Acknowledgments
We are grateful for the contribution of the patients (and their families) who donated their tissues for research studies. The authors would like to thank Michael Feldman for performing the Western blot.
Authors’ contributions
DP wrote the manuscript, contributed to the study design, performed the imaging analysis, and assisted in the histologic examination. KM co-wrote the manuscript, contributed to the study design, and assisted in performing the histologic examination. NE and SR performed the immunohistochemical and histochemical staining. ST, BDB, and MH conceptualized the study, gathered the materials, and edited the manuscript. ARC performed the histologic examination and conceptualized the study. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Financial support
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and the National Institute of Neurological Disorders and Stroke.
The NIH Office of Human Subject Research Protection has determined that this study is exempt from Institutional Review Board review.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Contributor Information
Drew Pratt, Email: Drew.pratt@nih.gov.
Karin Mente, Email: Karin.mente@nih.gov.
Shervin Rahimpour, Email: shervin.rahimpour@duke.edu.
Nancy A. Edwards, Email: EdwardsN@ninds.nih.gov
Sule Tinaz, Email: sule.tinaz@yale.edu.
Brian D. Berman, Email: Brian.berman@ucdenver.edu
Mark Hallett, Email: Hallettm@ninds.nih.gov.
Abhik Ray-Chaudhury, Email: Abhik.ray-chaudhury@nih.gov.
References
- 1.Greene P, Kang UJ, Fahn S. Spread of symptoms in idiopathic torsion dystonia. Mov Disord. 1995;10:143–52. doi: 10.1002/mds.870100204. [DOI] [PubMed] [Google Scholar]
- 2.Risch NJ, Bressman SB, deLeon D, Brin MF, Burke RE, Greene PE, Shale H, Claus EB, Cupples LA, Fahn S. Segregation analysis of idiopathic torsion dystonia in Ashkenazi Jews suggests autosomal dominant inheritance. Am J Hum Genet. 1990;46:533–8. [PMC free article] [PubMed] [Google Scholar]
- 3.Ozelius L, Kramer PL, Moskowitz CB, Kwiatkowski DJ, Brin MF, Bressman SB, Schuback DE, Falk CT, Risch N, de Leon D, et al. Human gene for torsion dystonia located on chromosome 9q32-q34. Neuron. 1989;2:1427–34. doi: 10.1016/0896-6273(89)90188-8. [DOI] [PubMed] [Google Scholar]
- 4.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–8. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 5.Dauer W. Inherited isolated dystonia: clinical genetics and gene function. Neurotherapeutics. 2014;11:807–16. doi: 10.1007/s13311-014-0297-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang CC, Tanabe LM, Jou S, Chi F, Dauer WT. TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J Clin Invest. 2014;124:3080–92. doi: 10.1172/JCI72830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pappas SS, Darr K, Holley SM, Cepeda C, Mabrouk OS, Wong JM, LeWitt TM, Paudel R, Houlden H, Kennedy RT, Levine MS, Dauer WT. Forebrain deletion of the dystonia protein torsinA causes dystonic-like movements and loss of striatal cholinergic neurons. Elife. 2015;4:e08352. doi: 10.7554/eLife.08352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oleas J, Yokoi F, DeAndrade MP, Pisani A, Li Y. Engineering animal models of dystonia. Mov Disord. 2013;28:990–1000. doi: 10.1002/mds.25583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McNaught KS, Kapustin A, Jackson T, Jengelley TA, Jnobaptiste R, Shashidharan P, Perl DP, Pasik P, Olanow CW. Brainstem pathology in DYT1 primary torsion dystonia. Ann Neurol. 2004;56(4):540–7. doi: 10.1002/ana.20225. [DOI] [PubMed] [Google Scholar]
- 10.Paudel R, Kiely A, Li A, Lashley T, Bandopadhyay R, Hardy J, Jinnah HA, Bhatia K, Houlden H, Holton JL. Neuropathological features of genetically confirmed DYT1 dystonia: investigating disease-specific inclusions. Acta Neuropathol Commun. 2014;2:159. doi: 10.1186/s40478-014-0159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker RH, Brin MF, Sandu D, Good PF, Shashidharan P. TorsinA immunoreactivity in brains of patients with DYT1 and non-DYT1 dystonia. Neurology. 2002;58:120–4. doi: 10.1212/WNL.58.1.120. [DOI] [PubMed] [Google Scholar]
- 12.Rostasy K, Augood SJ, Hewett JW, Leung JC, Sasaki H, Ozelius LJ, Ramesh V, Standaert DG, Breakefield XO, Hedreen JC. TorsinA protein and neuropathology in early onset generalized dystonia with GAG deletion. Neurobiol Dis. 2003;12:11–24. doi: 10.1016/S0969-9961(02)00010-4. [DOI] [PubMed] [Google Scholar]
- 13.Hewett J, Gonzalez-Agosti C, Slater D, Ziefer P, Li S, Bergeron D, Jacoby DJ, Ozelius LJ, Ramesh V, Breakefield XO. Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum Mol Genet. 2000;9:1403–13. doi: 10.1093/hmg/9.9.1403. [DOI] [PubMed] [Google Scholar]
- 14.Hewett J, Ziefer P, Bergeron D, Naismith T, Boston H, Slater D, Wilbur J, Schuback D, Kamm C, Smith N, Camp S, Ozelius LJ, Ramesh V, Hanson PI, Breakefield XO. TorsinA in PC12 cells: localization in the endoplasmic reticulum and response to stress. J Neurosci Res. 2003;72:158–68. doi: 10.1002/jnr.10567. [DOI] [PubMed] [Google Scholar]
- 15.Harata NC. Current gaps in the understanding of the subcellular distribution of exogenous and endogenous protein TorsinA. Tremor Other Hyperkinet Mov (N Y) 2014;4:260. doi: 10.7916/D8JS9NR2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eskow Jaunarajs KL, Bonsi P, Chesselet MF, Standaert DG, Pisani A. Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia. Prog Neurobiol. 2015;127–128:91–107. doi: 10.1016/j.pneurobio.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]