

Abstract
Background
Anomalous levels of gonadotropin-releasing hormone (GnRH) secretion result in a variety of reproductive phenotypes associated with infertility or subfertility. The normosmic isolated hypogonadotropic hypogonadism (nIHH) is due to a failure of either GnRH pulsatile secretion in hypothalamus or its reception in pituitary. The spectrum of nIHH-associated alterations continues to expand, especially when additional ethnic populations are investigated. The aim of this study was to uncover genetic causes for nIHH in patients of Russian origin.
Methods
For two nIHH patients referred to infertility clinic, both exons and promoter sequences of 6 GnRH signaling genes were sequenced.
Results
Patient 1 was a compound heterozygote for mutations in GnRH and its receptor encoding genes, while in Patient 2 GnRHR mutations were found in homozygous state. In both patients, the coding frame of GnRHR gene harbored missense-mutation Arg139His previously described as founder mutation in Polish and Brazilan patients. IVF/ET treatments were successful, with phenotypically healthy offsprings delivered.
Conclusion
Polish founder mutation Arg139His in GnRHR was found in two nIHH patients originating from Western region of Russia. Common variant of GnRH-encoding gene, Trp16Ser, could possibly contribute to reproductive phenotypes in patients with heterozygous mutations of other GnRH signaling pathway genes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12958-016-0183-8) contains supplementary material, which is available to authorized users.
Introduction
Despite the latest increase in the volume of relevant publications, the majority of genetic variants influencing sexual development and fertility remain either unknown or poorly characterized in a majority of populations. The secretion of gonadotropin-releasing hormone (GnRH) is essential for the acquisition and maintenance of reproductive competence in mammals [1]. Importantly, anomalous levels of GnRH secretion result in a variety of reproductive phenotypes associated with infertility or subfertility.
Isolated hypogonadotropic hypogonadism, or isolated gonadotropin-releasing deficiency, is relatively rare genetic disease with an occurrence of about 1–10 cases per 100 000 births. In patients with isolated hypogonadotropic hypogonadism, serum concentrations of FSH and LH are low, while the levels of sex steroid are lacking and the puberty is delayed in absence of any other pituitary deficiencies. In about 60 % of these patients, the sense of smell is also impaired, producing either hyposmia or anosmia. The latter condition is called Kallmann syndrome (KS). Current classification of the KS includes 6 subtypes. X-linked KS (type 1) is associated with KAL1 mutation, while KS subtypes 2,3,4,5 and 6 are linked to the mutations in genes FGFR1, PROKR2, PROK2, CHD7 and FGF8, respectively [2, 3]. Patients with KAL1 mutation may also have associated disorders of a neurological or urogenital nature, thus, demonstrating more severe phenotype [1–3].
In the remaining 40 % patients, the sense of smell is unchanged. The normosmic isolated hypogonadotropic hypogonadism (nIHH) is due to a failure of either gonadotropin-releasing hormone (GnRH) pulsatile secretion in hypothalamus or its reception in pituitary. In nIHH patients, causal mutations were identified in genes GnRH1, GnRHR, KISS1, KISS1R, TAC3 and TAC3R [2]. Importantly, the spectrum of nIHH-associated alterations continues to expand, especially when additional ethnic populations are investigated.
Here we present our observations and successful treatment of two non-consanguineous Russian female patients diagnosed with nIHH. Prior to the referral to fertility clinic, each patient underwent about 10 years of hormone replacement therapy. In both patients, IVF/ET treatment was successful, with phenotypically healthy offspring delivered. In each patient, six candidate genes (GnRH1, GnRHR, KISS1, KISS1R, TAC3, TAC3R) were analyzed by sequencing. Patient 1 was a compound heterozygote for mutations in gonadotropin-releasing hormone encoding gene (GnRH1) and in gene encoding for its receptor (GnRHR), while in Patient 2 GnRHR mutations were found in homozygous state. In both patients, the coding frame of GnRHR gene harbored missense-mutation Arg139His previously described as founder mutation in Polish and Brazilan patients (Beneduzzi D et al., 2014) and in some other unrelated patients across the globe (Laitinen EM et al, 2012).
Subjects and methods
Patients
Both patients were referred to infertility clinic due to primary amenorrhea in absence of eating disorders or strenuous exercises, and infertility. Karyotypes were 46, XX, with normal sense of smell. Both patients received hormone replacement therapy with 2 mg oestradiol hemihydrate and a combination of 2 mg oestradiol hemihydrate/10 mg dydrogesterone (Femoston 2/10 mg, Abbott, Netherlands). Other characteristics of the patients and the results of their MRI and ultrasound examinations are shown in Table 1.
Table 1.
Anthropometric and clinical characteristics of nIHH patients and their partners for IVF with ICSI
| Characteristics | Patient 1 | Patient 2 |
|---|---|---|
| Age at first visit to fertility clinic | 28 years | 31 years |
| Pubarche (spontaneous) | at 14 years | at 15 years |
| Heights | 170 cm | 156 cm |
| Weight | 65 kg | 53 kg |
| Breast development by Tanner scale | Stage V | Stage IV |
| Sense of odors | Normal | Normal |
| Karyotype | 46, XX | 46, XX |
| MRI of the pituitary | Size: 10 mm x 6 mm x16 mm. Signs of signal reduction in the right half of the posterior pituitary (4 mm x 3 mm x 2 mm), depicting a cyst. | Size: 3.5 mm x 5 mm x 9 mm showing signs of moderate pituitary hypoplasia |
| Vaginal ultrasound |
Uterus: 27 mm x 43 mm x 30 mm Endometrium: 2.3 mm Right ovary: 18 mm x 16 mm with 4 follicles (6 to 8 mm). Left ovary: 17 mm x 18 mm with 5 follicles (6 to 7 mm). Moderate hypoplasia of the uterus and ovaries. |
Uterus: 35 mm x 22 mm x30 mm Endometrium: 1.8 mm Right ovary: 21 mm x 18 mm with 3 follicles (3 to 5 mm) Left ovary: 17 mm x 18 mm with 5 follicles (6 to 7 mm). Moderate hypoplasia of the ovaries. |
| Partner characteristics for IVF with ICSI |
Ejaculate volume: 2 ml Sperm concentration: 39x106/ml, Motile sperms: 3 % Normal forms by Kruger strict criteria: 2 %. |
Ejaculate volume: 5 ml Sperm concentration: 31x106/ml Motile sperms: None Normal forms by Kruger strict criteria: 1 %. |
In vitro fertilization (IVF) with intracytoplasmic sperm injection (ICSI)
In partners of both patients, the spermograms showed insufficient sperm quality (Table 1), thus justifying in vitro fertilization (IVF) with intracytoplasmic sperm injection (ICSI).
In both patients, oocytes were obtained by standard techniques within 36 h after administration of ovulatory dose of hCG (10,000 IU, Pregnyl, MSD, Netherlands), by follicle aspiration with 19G needle under ultrasound control. Obtained cumulus-oocyte complexes were placed in culture media (Universal IVF Medium, Origio, Denmark) for 2 h, then moved to medium containing recombinant human hyaluronidase (ICSI Cumulase, Origio, Denmark) for one minute, following by relocation into culture medium. To estimate the quality of oocytes, their shape and the presence of first polar body (metaphase II) were assessed. Living sperm cells selected for ICSI were immobilized in a drop of 10 % polyvinylpyrrolidone then injected into oocyte located at position 6 or 12 o’clock.
DNA sequencing
Genomic DNA was isolated from the blood of two nIHH patients using phenol-chloroform technique. Exonic and promoter sequences for genes GNRH1 (NG_016457.1), GNRHR (NG_009293.1), KISS1 (NG_032151.1), KISS1R (NG_008277.1), TAC3 (NG_021398.1) and TAC3R (NG_023344.1) were amplified by PCR using primers located outside of intron-exon borders (Additional file 1: Table S1), the resultant PCR products were purified with DyEx columns (QIAGEN) and bidirectionally sequenced on the ABI PRISM®310 Genetic Analyzer (Applied Biosystems). Sequence data were analyzed with Chromas (Technelysium Pty.) software version 1.51 and compared with the RefSeq sequences indicated above.
Ethical approval
Ethical approval was obtained from the local Institutional Review Board at Department of Obstetrics and Gynecology, State Pediatric Medical University, St.-Petersburg, Russia.
Results
Clinical outcomes
Upon initial nIHH diagnosis, both patients received combined hormone replacement therapy. Table 2 lists endogenous hormone values before the onset of reproduction assistance treatments.
Table 2.
Hormone values before the onset of reproduction assistance treatments
| Hormone | Patient 1 | Patient 2 | |
|---|---|---|---|
| 1 attempt | 2 attempt | ||
| FSH | 0.1 IU/L | 0.75 IU/L | 0.3 IU/L |
| LH | 0.1 IU/L | 0.26 IU/L | 0.1 IU/L |
| TSH | 5.4 μIU/ml | 1.34 μIU/ml | 2.24 μIU/ml |
| Estradiol | 12 pg/ml | 12 pg/ml | NA |
| Prolactin | 202 mIU/ml | 350 mIU/ml | 153.8 mIU/ml |
| Inhibin B | 8.8 pg/ml | NA | NA |
| AMH | 0.1 ng/ml | 1.96 ng/ml | 0.93 ng/ml |
FSH follicle stimulating hormone, LH luteinizing hormone, TSH thyroid-stimulating hormone, AMH anti-mullerian hormone, NA not assayed
Patient 1
To correct the levels of TSH, patient received L-thyroxin supplementation till her levels of TSH were brought to 2.8 nIU/ml. In IVF cycle, she received injections of human menopausal gonadotropin (300 IU, Menopur, Ferring) for 11 days. After administration of human chorionic gonadotropin (Pregnyl, MSD) at a dose of 10000 IU, twelve oocytes of good quality were retrieved. After fertilization by standard ICSI and 3 days of culture, two eight-cells embryos were transferred to uterine cavity, while three remaining good quality embryos were frozen. One gestational sac was detected in uterine cavity three weeks later.
After typical course of pregnancy, patient had uneventful spontaneous delivery of a girl, at week 39, with birth weight of 3150 g and length of 51 cm, Apgar scores were 9/9.
Patient 2
Ovarian stimulation was initiated with 225 IU of human menopausal gonadotropin in daily injections for 13 days, followed by induction of ovulation by human chorionic gonadotropin (10000 IU). Five good quality oocytes were retrieved. After conventional ICSI, two blastocysts of good quality were transferred into uterine cavity, while two remaining embryos were frozen. After four weeks, a viable pregnancy was detected. At 9 weeks of gestation, this pregnancy was lost. The cytogenetic analysis of miscarried embryo showed normal karyotype 46, XX. Subsequent transfer of frozen embryos was not successful.
One year after first attempt, another ovarian stimulation was performed, with 300 IU of human menopausal gonadotropin accompanied by FSH and LH for 15 days. After total FSH dose of 4275 IU, three oocytes were retrieved. After ICSI fertilization, two good quality embryos (A8 and A6) were transferred in the uterus of the patient.
Early and mid-pregnancy course was typical. Patient went into preterm birth due to placental abruption at week 37. The delivery of a boy was by C-section. Weight of a newborn was 3000 g, length was 50 cm, and Apgar scores were 8/9.
Genetic analysis of the probands
Bidirectional sequencing was performed for 6 loci (GnRH, GnRHR, KISS1, KISS1R, TAC3 and TAC3R) to cover all exons and their borders. In both patients, the mutations in GnRH and GnRHR genes were found (Table 3). In Patient 1 genome, a compound heterozygosity state was detected, with one copy of the GnRH1 gene exon 1 (g. G6757C; p. W16S) and one copy of GnRHR gene exon 3 (g. C20426T; p. T269M) mutated. Additionally, another mutation (g. G7167A; p. Arg139His) was identified in exon 1 of the GnRHR gene in the same patient.
Table 3.
The genotypes of the patients and their family members as assessed by bidirectional sequencing
| Gene | Family 1 | Family 2 | ||||
|---|---|---|---|---|---|---|
| P1 | P1’s mother | P1’s offspring (girl) | P2 | P2’s mother | P2’s offspring (boy) | |
| GNRH1 exon 1 | g.6757 G > C rs6185 Heterozygous |
wt | wt | wt | wt | wt |
| g.6891 T > G rs2709608 Homozygous intronic variant | g.6891 T > G rs2709608 Homozygous intronic variant | na | g.6891 T > G rs2709608 Homozygous intronic variant | g.6891 T > G rs2709608 Homozygous intronic variant | na | |
| GNRHR exon 1 | g. 7167 G > A rs104893842 Heterozygous |
g. 7167 G > A rs104893842 Heterozygous |
wt | g. 7167 G > A rs104893842 Homozygous |
g. 7167 G > A rs104893842 Heterozygous |
g. 7167 G > A rs104893842 Heterozygous |
| GNRHR exon 3 | g. 20426 C > T Heterozygous | wt | wt | wt | wt | wt |
P1 Patient 1, P2 Patient 2, wt wild-type, na not assessed
In patient 2, homozygous mutation in exon1 of the GnRHR gene (g. G7167A; p. Arg139His) was detected along with another homozygous intronic substitution (g. T6891G).
No mutations were detected in exons or promoter areas of KISS1, KISS1R, TAC3 and TAC3R.
The mutations contributing to nIHH condition of the patients are summarized in Table 3 and Fig. 1 (a-c).
Fig. 1.
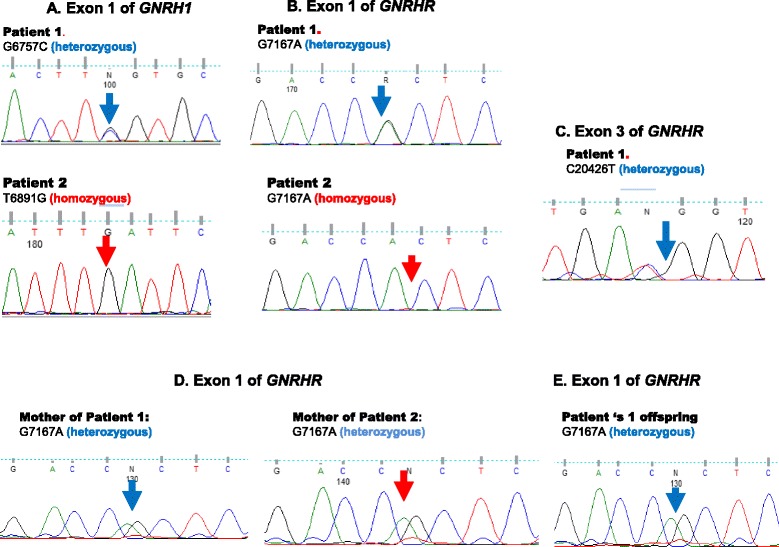
Sanger sequencing chromatograms of mutated loci in patients and their family members. a Sequencing chromatogram of the exon 1 of GNRH1 gene in Patient 1 and Patient 2 genomes. b Sequencing chromatogram of the exon 1 of GNRHR gene in Patient 1 and Patient 2 genomes. c Sequencing chromatogram of the exon 3 of GNRHR gene in Patient 1 genomes. d Sequencing chromatogram of the exon 1 of GNRHR gene in the genomes of mothers of Patient 1 and Patient 2. e Sequencing chromatogram of the exon 1 of GNRHR gene in the genome of Patient 1 offspring
Genetic analysis of the relatives of probands
Affected exons of GnRH and GnRHR genes were bidirectionaly sequenced in DNA samples in mothers and live offsprings of both probands.
In the genome of mother of Patient 1, heterozygous mutation of GNRHR exon 1 (g. 7167 G > A, rs104893842) was found, while GNRHR exon 3 was mutation free. No mutations were found in GNRH1 exon 1. Mother of Patient 2 was also heterozygous by GNRHR exon 1 mutation (g. 7167 G > A, rs104893842). Mothers of both patients were also homozygous for g.6891 T > G variant in gene GNRH1 (rs2709608).
The genotype of the offspring of patient 1 was wild-type for all informative loci, while the genome of the offspring of patient 2 carried heterozygous mutation g. 7167 G > A (rs104893842). In offspring, the state of the g.6891 T > G polymorphism in gene GNRH1 (rs2709608) was not assessed. Observed genotypes are described in Table 3 and Fig. 1 (d, e).
Discussion
In this study, the GnRH1, GnRHR, KISS1, KISS1R, TAC3 and TAC3R genes were analyzed in two unrelated nIHH patients successfully treated for infertility. In both patients, Arg139His substitutions were identified within the receptor for hypothalamic gonadotropin-releasing hormone. In patient 1, this mutation was found in heterozygous state, while patient 2 was homozygous. Mutation p. Arg139His is located in a conserved DRS motif between the third transmembrane loop and the second intracellular loop. It completely inactivates the GnRH-binding activity of the receptor and prevents GnRH-induced stimulation of inositol phosphate accumulation in vitro [4].
In a recent study of Choi et al., R139H mutation was found to share a common founder core haplotype 220 kB in size. Interestingly, eight out of 15 carriers of this founder haplotype were of Polish origin [5]. In our study, both patients originated from Western region of Russia. In particular, Patient 1 and her mother were born in Pskov area that was under the winter-long siege by the Polish Army during the final stage of the Livonian War of 1558–1583. Hence, it is logical to conclude that p. Arg139His most likely was inherited from Polish founders. Also, it is tempting to speculate that R139 codon of GNRHR gene is a hotspot region for receptor-inactivating mutations, as the same codon was earlier found to contain another p.R139C mutation identified in two unrelated cases of Turkish origin [6, 7] and in Brazilian nIHH patients, where it was confirmed to associate with different founder haplotype [8].
Interestingly, GNRHR gene of Patient 1 also possessed a heterozygous T269M substitution that previously described in ClinVar as pathogenic in a nIHH patient of Asian/Indian origin. As the mother of patient 1 had no mutation in this locus, this substitution should be of either paternal origin or de novo. An absence of the mutation in Patient’s 1 husband and offspring genotypes suggests that Patient 1 genome is a cis-compound heterozygote for R139H and T269M mutations. In turn, that means that second allele of GNRHR in Patient 1 genome remains mutation free, and that one copy of wild type allele able to compensate for the deficiency of the receptor for GnRH binding only partially, at least in presence of pituitary deficiency and/or GnRH1 variant.
Indeed, the genome of Patient 1 had yet another heterozygous substitution, Trp16Ser [rs6185], in GnRH1. This variant was previously reported as polymorphism examined for its minor Ser16 allele association with decreased bone mineral density [9] and shorter disease-free survival in breast cancer patients [10]. Another recent study investigated this variant in polycystic ovarian syndrome (PCOS) and showed that homozygous or heterozygous carrier of Ser16 allele demonstrate somewhat more benign phenotype as compared with non-carriers, with lower follicle counts, as well as lower levels of testosterone, free androgen index and fasting insulin [11]. On the other hand, the presence of Ser16 variant had no influence on gonadotrophic hormone levels [11]. It would be tempting to speculate that Ser16 variant of hypothalamic gonadotropin-releasing hormone has lower potency than its wild-type variant, and could be a contributing factor to the reproductive phenotype of patient 1.
Importantly, reproductive interventions were successful in both nIHH patients despite the presence of GnRH resistance which is typical for women who have mutations in GNRHR [3, 12]. In nIHH patients, decreased circulating Anti-Mullerian Hormone (AMH) concentrations are not indicative of a decreased follicular reserve. An administration of human menopausal gonadotropin aids in overcoming ovarian insufficiency by inducing maturation of ovarian follicles. Women with nIHH should be informed that their infertility is treatable. In cases of homozygous founder mutations, mothers should be advised about the possibility that the chances for their offspring to inherit nIHH may increase if their partner comes from the same geographical region. However, the autosome recessive mode of inheritance along with non-life threatening, treatable nature of the nIHH condition make pre-implantation diagnostics unnecessary in the majority of cases.
Conclusions
Polish founder mutation Arg139His in GnRHR was found in two nIHH patients originating from Western region of Russia. Common variant of GnRH-encoding gene, Trp16Ser, could possibly contribute to reproductive phenotypes in patients with heterozygous mutations of other GnRH signaling pathway genes. Women with nIHH should be informed that their infertility is treatable.
Acknowledgements
The authors wish to gratefully acknowledge proofread by Jerome Glasser.
Funding
This study was not funded.
Availability of data and material
Not applicable.
Authors’ contributions
NZ, MS, AB and KB conceived the design of the study. NZ carried the sequencing reactions. AB, MS and NZ analyzed and interpreted the data. NZ, MS, AB and KB drafted the manuscript and revised it critically for important intellectual content. All authors critically revised the manuscript and approved the final version.
Authors’ information
NZ is a physician specializing in medical genetics. MS and AB perform basic and applied research in the area of medical genetics. KB is a physician specializing in human reproduction.
Competing interests
NZ, MS and AB declare no competing interests. KB is an employee of reproductive clinic “Genesis”.
Consent for publication
Both patients consented for publishing their individual data in de-identified form.
Ethics approval and consent to participate
The study protocol was approved by Genesis Internal Review Board. Written informed consent was provided by all patients at inception.
Abbreviations
- AMH
Anti-Mullerian hormone
- FSH
Follicle stimulating hormone
- GnRH
Gonadotropin-releasing hormone
- GnRHR
Gonadotropin-releasing hormone receptor
- hCG
Human chorionic gonadotropin
- ICSI
Intracytoplasmic sperm injection
- IVF/ET
In Vitro Fertilization/Embryo Transfer
- KS
Kallmann syndrome
- LH
Luteinizing hormone
- nIHH
Normosmic isolated hypogonadotropic hypogonadism
- PCR
Polymerase chain reaction
- TSH
Thyroid-stimulating hormone.
Additional file
Primers for bidirectional Sanger sequencing in nIHH patients. (DOCX 15 kb)
Contributor Information
Nikolay Zernov, Email: nzernov01@gmail.com.
Mikhail Skoblov, Email: mskoblov@gmail.com.
Ancha Baranova, Phone: 1-571-334-1145, Email: aancha@gmail.com, Email: abaranov@gmu.edu, http://cscmd.cos.gmu.edu.
Konstantin Boyarsky, Email: boyarsky@pochta.ru.
References
- 1.Bry-Gauillard H, Trabado S, Bouligand J, Sarfati J, Francou B, Salenave S, Chanson P, Brailly-Tabard S, Guiochon-Mantel A, Young J. Congenital hypogonadotropic hypogonadism in females: clinical spectrum, evaluation and genetics. Ann Endocrinol (Paris) 2010;71:158–62. doi: 10.1016/j.ando.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 2.Valdes-Socin H, Rubio Almanza M, Tomé Fernández-Ladreda M, Debray FG, Bours V, Beckers A. Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes. Front Endocrinol (Lausanne) 2014;5:109. doi: 10.3389/fendo.2014.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R, Young J. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11:547–64. doi: 10.1038/nrendo.2015.112. [DOI] [PubMed] [Google Scholar]
- 4.Ulloa-Aguirre A, Janovick JA, Leaños-Miranda A, Conn PM. Misrouted cell surface GnRH receptors as a disease aetiology for congenital isolated hypogonadotrophic hypogonadism. Hum Reprod Update. 2004;10(2):177–92. doi: 10.1093/humupd/dmh015. [DOI] [PubMed] [Google Scholar]
- 5.Choi JH, Balasubramanian R, Lee PH, Shaw ND, Hall JE, Plummer L, Buck CL, Kottler ML, Jarzabek K, Wołczynski S, Quinton R, Latronico AC, Dode C, Ogata T, Kim HG, Layman LC, Gusella JF, Crowley WF., Jr Expanding the Spectrum of Founder Mutations Causing Isolated Gonadotropin-Releasing Hormone Deficiency. J Clin Endocrinol Metab. 2015;100:E1378–85. doi: 10.1210/jc.2015-2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gurbuz F, Kotan LD, Mengen E, Siklar Z, Berberoglu M, Dokmetas S, Kılıçlı MF, Güven A, Kirel B, Saka N, Poyrazoğlu Ş, Cesur Y, Doğan M, Özen S, Özbek MN, Demirbilek H, Kekil MB, Temiz F, Önenli Mungan N, Yüksel B, Topaloğlu AK. Distribution of gene mutations associated with familial normosmic idiopathic hypogonadotropic hypogonadism. J Clin Res Pediatr Endocrinol. 2012;4:121–6. doi: 10.4274/Jcrpe.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Topaloglu AK, Lu ZL, Farooqi IS, Mungan NO, Yuksel B, O'Rahilly S, Millar RP. Molecular genetic analysis of normosmic hypogonadotropic hypogonadism in a Turkish population: identification and detailed functional characterization of a novel mutation in the gonadotropin-releasing hormone receptor ene. Neuroendocrinology. 2006;84:301–8. doi: 10.1159/000098147. [DOI] [PubMed] [Google Scholar]
- 8.Beneduzzi D, Trarbach EB, Min L, Jorge AA, Garmes HM, Renk AC, Fichna M, Fichna P, Arantes KA, Costa EM, Zhang A, Adeola O, Wen J, Carroll RS, Mendonça BB, Kaiser UB, Latronico AC, Silveira LF. Role of gonadotropin-releasing hormone receptor mutations in patients with a wide spectrum of pubertal delay. Fertil Steril. 2014;102:838–846. doi: 10.1016/j.fertnstert.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwasaki H, Emi M, Ezura Y, Ishida R, Kajita M, Kodaira M, Yoshida H, Suzuki T, Hosoi T, Inoue S, Shiraki M, Swensen J, Orimo H. Association of a Trp16Ser variation in the gonadotropin releasing hormone signal peptide with bone mineral density, revealed by SNP-dependent PCR typing. Bone. 2003;32:185–90. doi: 10.1016/S8756-3282(02)00949-3. [DOI] [PubMed] [Google Scholar]
- 10.Piersma D, Themmen AP, Look MP, Klijn JG, Foekens JA, Uitterlinden AG, Pols HA, Berns EM. GnRH and LHR gene variants predict adverse outcome in premenopausal breast cancer patients. Breast Cancer Res. 2007;9:R51. doi: 10.1186/bcr1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valkenburg O, Uitterlinden AG, Piersma D, Hofman A, Themmen AP, de Jong FH, Fauser BC, Laven JS. Genetic polymorphisms of GnRH and gonadotrophic hormone receptors affect the phenotype of polycystic ovary syndrome. Hum Reprod. 2009;24:2014–22. doi: 10.1093/humrep/dep113. [DOI] [PubMed] [Google Scholar]
- 12.Seminara SB, Beranova M, Oliveira LM, Martin KA, Crowley WF, Jr, Hall JE. Successful use of pulsatile gonadotropin-releasing hormone (GnRH) for ovulation induction and pregnancy in a patient with GnRH receptor mutations. J Clin Endocrinol Metab. 2000;85(2):556–62. doi: 10.1210/jcem.85.2.6357. [DOI] [PubMed] [Google Scholar]