

Abstract
The accumulation of unfolded proteins under endoplasmic reticulum (ER) stress leads to the activation of the multi-domain protein sensor IRE1α as part of the unfolded protein response (UPR). Clustering of IRE1α lumenal domains in the presence of unfolded proteins promotes kinase trans-autophosphorylation in the cytosol and subsequent RNase domain activation. Interestingly, there is an allosteric relationship between the kinase and RNase domains of IRE1α, which allows ATP-competitive inhibitors to modulate the activity of the RNase domain. Here, we use kinase inhibitors to study how ATP-binding site conformation affects the activity of the RNase domain of IRE1α. We find that diverse ATP-competitive inhibitors of IRE1α promote dimerization and activation of RNase activity despite blocking kinase trans-autophosphorylation. In contrast, a subset of ATP-competitive ligands, which we call KIRAs, allosterically inactivate the RNase domain through the kinase domain by stabilizing monomeric IRE1α. Further insight into how ATP-competitive inhibitors are able to divergently modulate the RNase domain through the kinase domain was gained by obtaining the first structure of apo human IRE1α in the RNase active back-to-back dimer conformation. Comparison of this structure with other existing structures of IRE1α and integration of our extensive structure activity relationship (SAR) data has led us to formulate a model to rationalize how ATP-binding site ligands are able to control IRE1α oligomeric state and subsequent RNase domain activity.
Introduction
The endoplasmic reticulum (ER) is a netlike organelle consisting of interconnected sacs and branched tubules that extend throughout the cytosol of eukaryotic cells. This organelle plays important roles in lipid biosynthesis and is the cellular origin for almost all secreted and transmembrane proteins.1 The ER maintains a distinct chemical environment–characterized by a higher calcium concentration and a more oxidizing environment–than the cytosol, which aids in the proper folding, posttranslational modification, and maturation of proteins prior to their downstream trafficking.2, 3 However, the processing capacity of the ER is not unlimited and a diversity of cellular perturbations can result in ER stress that overloads homeostatic folding capacity and results in the accumulation of misfolded proteins within the organelle. Professional secretory cells, such as plasma cells and β-cells of the endocrine pancreas, are especially sensitive to ER stress because they appear to operate at or near the limits of their secretory capacity.4, 5
Under ER stress, the accumulation of unfolded proteins exceeds an excitatory threshold within the organelle causing the activation of an integrated intracellular signal transduction pathway called the unfolded protein response (UPR).6 In mammals, the UPR contains three transmembrane sensor proteins–inositol requiring enzyme 1 alpha (IRE1α), pancreatic endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6)–that monitor the protein folding status of the ER through their lumenal domains. When unfolded proteins accumulate in the ER, these three arms of the UPR become activated and trigger specific downstream signaling cascades that affect cell fate.7–10 Upon initial activation of the UPR, IRE1α, PERK, and ATF6 work to both reduce the amount of unfolded protein arriving at the ER, through a translational block, and expand folding capacity, by increasing the size of this organelle and upregulating ER chaperones, oxidoreductases, and biosynthetic enzymes.11 However, in the event of severe and prolonged ER stress, the UPR switches from adaptive outputs to terminal signaling cascades that result in the upregulation and activation of pro-inflammatory and pro-apoptotic regimes.12, 13 Because the UPR has such profound effects on cellular fate, it is not surprising that dysregulated UPR signaling has been implicated in the etiology of numerous diseases. Accordingly, there is widespread interest in the development of pharmacological agents that modulate IRE1α, PERK, and ATF6 function.14, 15
The most ancient and well-conserved component of the UPR in mammals is the kinase/RNase IRE1α.8, 16 This multi-domain protein consists of an N-terminal lumenal domain sensor that is connected to cytosolic kinase and RNase domains through a transmembrane linker (Figure 1A). In the presence of misfolded proteins in the ER, IRE1α’s lumenal domains oligomerize, which leads to activation segment trans-autophosphorylation in the cytosolic kinase domains.17 IRE1α activation segment phosphorylation results in enhanced dimerization and the stabilization of an RNase active dimeric state18–commonly referred to as the back-to-back dimer–that positions two RNase domains close to one another, facilitating the cleavage of a 26-nucleotide intron from the XBP1 mRNA. The RNA ligase RtcB ligates the cleaved XBP1 mRNA,19, 20 yielding XBP1s, which is translated into an activated transcription factor that upregulates the expression of genes that help the ER adapt to stress.21 However, if the initial adaptive response to ER stress is unsuccessful, IRE1α’s RNase domain acts upon additional substrates, leading to extensive endonucleolytic decay of hundreds of ER-localized mRNAs.13, 22, 23 The endonucleolytic decay activity of IRE1α’s RNase domain further increases ER stress by inducing the activation of a number of pro-inflammatory and pro-apoptotic proteins and depleting mRNAs that encode for adaptive ER proteins.24, 25
Figure 1. Divergent Modulation of IRE1α RNase activity.
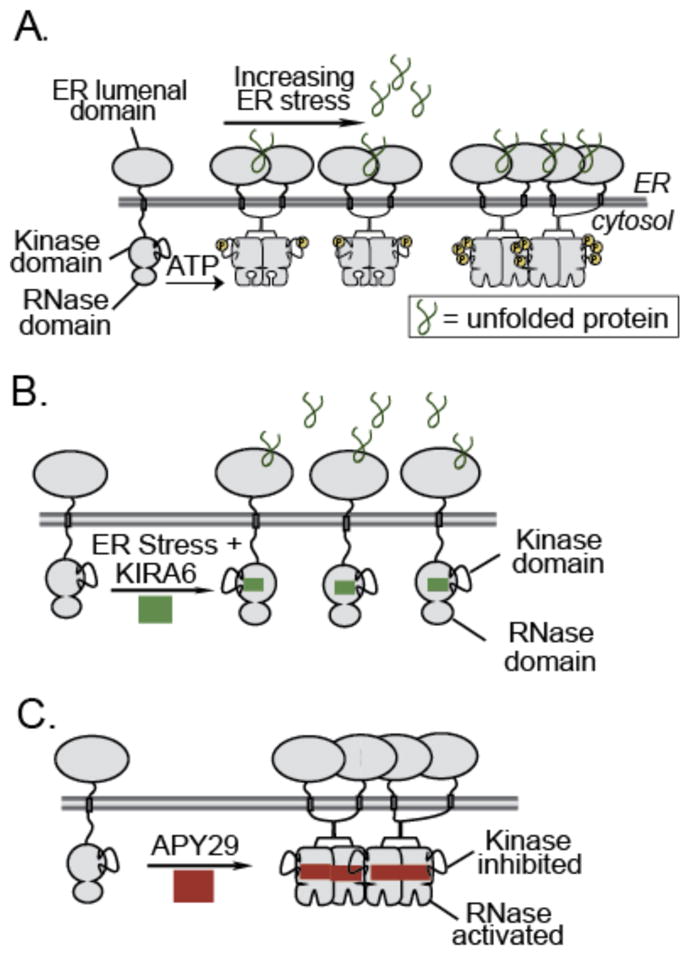
(A) Model of IRE1α activation under ER stress. IRE1α contains a stress sensing lumenal domain linked to a cytosolic kinase and RNase domain by a transmembrane linker. In the presence of unfolded proteins in the ER, IRE1α is activated to initiate an adaptive response. Prolonged ER stress leads to IRE1α oligomerization and endonucleolytic decay of ER-localized mRNAs (B) Allosteric inhibition of IRE1α’s RNase activity with an ATP-competitive inhibitor–KIRA6–under ER stress. (C) Allosteric activation of IRE1α’s RNase activity with an ATP-competitive inhibitor–APY29–in the absence of ER stress.
IRE1α’s divergent effect on cellular fate has spurred widespread interest in the development of pharmacological modulators of this UPR sensor. As the ribonuclease activity of IRE1α appears to be the primary driver of IRE1α’s roles in the adaptive and terminal UPR, most of these efforts have focused on modulating the RNase activity of this bifunctional enzyme. Towards this end, a number of aldehyde-containing small molecules that covalently modify and inhibit the RNase domain have been developed.26–30 The allosteric relationship between the kinase and RNase domains of IRE1α–first characterized with yeast IRE1 (yIRE1)–has also made it possible to modulate RNase activity with ATP-binding site ligands.31 Ligands that interact with the ATP-binding site of IRE1α have the potential to either increase (Figure 1B) or decrease (Figure 1C) RNase activity. Indeed, all ligands that were first characterized to interact with the ATP-binding sites of yIRE1 and IRE1α, activate RNase activity despite inhibiting the kinase domain. Recently, we have reported a series of ATP-competitive inhibitors based on an imidazopyrazine scaffold, called kinase inhibiting RNase attenuators (KIRAs), that inhibit RNase activity through the kinase domain.32 Here, a systematic study of how ATP-competitive inhibitor structure affects both the kinase and RNase activities of IRE1α is presented. By performing a structure-activity relationship (SAR) analysis, we find that all imidazopyrazine-based inhibitors of IRE1α’s kinase domain also block RNase activity. However, the link between kinase and RNase domain inhibition does not appear to be a general feature of ATP-competitive ligands, as all inhibitors of IRE1α’s kinase domain that were identified in an unbiased screen of a commercially available compound collection activate RNase activity rather than inactivate it. Further insight into how ATP-competitive inhibitors are able to divergently modulate the RNase domain through the kinase domain was gained by obtaining the first structure of apo human IRE1α in the RNase active back-to-back dimer conformation. Comparison of this structure with other existing structures of IRE1α and integration of our extensive structure activity relationship (SAR) data has led us to formulate a model to rationalize how ATP-binding site ligands are able to control IRE1α oligomeric state and subsequent RNase domain activity.
Results and Discussion
Dual Enzymatic Activities of IRE1α
To better understand how specific inhibitor interactions with the ATP-binding site affect the RNase domain, a series of KIRAs based on an imidazopyrazine scaffold (Figure 2A) were tested for their effect on both enzymatic activities of IRE1α with in vitro assays. Enzymatic assays were performed using an IRE1α construct, IRE1α*, that contains only the cytosolic kinase and RNase domains of this multi-domain protein. Mass spectrometric analysis of this IRE1α* construct, which was expressed in baculovirus-infected insect cells and purified with immobilized metal ion affinity chromatography, revealed that this construct is phosphorylated on activation segment residues Ser724, Ser726, and Ser729 (Supplementary Figure S1).33, 34 Phosphorylation of these activation segment residues constitutively activates IRE1α*’s kinase and RNase domains (Supplementary Figure S2). Kinase inhibition was determined by measuring the ability of a KIRA to block IRE1α*’s phosphorylation of an exogenous protein substrate (myelin basic protein, Figure 2B). RNase inhibition was determined with an assay that uses an XBP1 mini-substrate labeled with a 5′-fluorescein and a 3′-black hole quencher (Figure 2C). KIRAs were tested for their ability to prevent mini-substrate cleavage, and the subsequent increase in fluorescence, by IRE1α.
Figure 2. Determination of IRE1α’s kinase activity and RNase activity in the presence of ATP-competitive inhibitors.

(A) Structure and proposed binding mode of parent compound 1 (KIRA3). (B) Schematic of the activity assay used to screen inhibition of IRE1α’s kinase activity. IRE1α* contains the cytosolic kinase and RNase domains of IRE1α. Kinase inhibition was determined by measuring the ability of KIRAs to block the phosphorylation of myelin basic protein (MBP). (C) RNase inhibition was determined by measuring the ability of KIRAs to block the cleavage of an XBP1 RNA mini-substrate. Cleavage of the 5′-FAM and 3′-BHQ labeled RNA substrate by IRE1α* results in release of fluorescein quenching, which can be monitored by fluorometric analysis.
SAR Analysis of KIRAs
To determine which structural elements allow KIRAs to inhibit the RNase activity of IRE1α through the ATP-binding site of the kinase domain, we performed a systematic structure-activity relationship (SAR) study using KIRA3 (1) as a benchmark (Figure 2A). The first series of analogs tested contain variable alkyl and cycloalkyl groups at the C-3 position (R1) and a fixed naphthylurea-3-trifluoromethylphenyl moiety displayed from the C-1 position (R2) of the imidazopyrazine scaffold (Figure 3). For this series, there is a ten-fold range in IC50 values for kinase and RNase activities, with the isopropyl analog (1) demonstrating the lowest potency and cyclopropylmethyl (3) demonstrating the highest. Due to cyclopropylmethyl appearing to be the most optimal R1 substituent, this group was held constant for the remainder of the analogs tested in this study. Next, we determined how replacing the naphthyl R2 group of 1 with different substituted aryl rings affects kinase and RNase inhibition. Previously, we found that inhibitors possessing a 4-naphthylamine R2 group instead of a 4-anilino moiety are significantly more potent inhibitors of IRE1α*’s RNase activity.32 Therefore, only analogs that contain substituted phenyl rings at the R2 position were generated and tested. Of this series, only the 3-fluorophenyl (7) R2 group confers sub-micromolar potency against the kinase and RNase activities of IRE1α*. Amongst mono-substituted phenyl R2 groups, 3-fluorophenyl (7) provides the most potent inhibition of IRE1α*’s kinase and RNase activities, with larger 3-chloro (9) or 3-methyl (8) substitutions proving to be less optimal.
Figure 3. SAR analysis of KIRA analogs (1-32).

Half maximum inhibitory concentration (IC50) values for kinase and RNase activity (mean ± S.E.M, n=3). *Compound 30 utilizes a tert-butyl group at the R1 position rather than a cyclopropyl methyl moiety.
Our original strategy to identify ATP-competitive inhibitors that allosterically inhibit the RNase activity of IRE1α was to screen compounds previously shown to stabilize an inactive conformation (DFG-Asp-out conformation) of the conserved Asp-Phe-Gly motif (DFG-motif) found at the base of the activation segment (type II inhibitors). Indeed, the original hit from this screen, and all subsequent potent KIRAs, contains an aryl-urea moiety, which is a known pharmacophore for a number of type II inhibitors that stabilize the DFG-out inactive conformation.35–39 When type II inhibitors of this class are bound to the DFG-out form of protein kinases, the aryl group of the aryl-urea moiety occupies a hydrophobic pocket that is formed upon movement of the Phe side chain of the DFG-motif, and the urea moiety serves as a linker that forms hydrogen bonds with a conserved glutamic acid on helix-αC, and with the backbone of the DFG-motif. In order to test whether these canonical DFG-out-stabilizing interactions are necessary for allosteric inhibition of IRE1α’s RNase domain, the aryl (R3) and urea (R4) moieties of 1 were systematically varied. As shown in Figure 3, diverse 3- and 4-substituted phenyl groups are tolerated at the R3 position (12-21), with all but one phenylurea demonstrating IC50 values for RNase and kinase inhibition that are within 10-fold of the most potent KIRA, 3. Notably, inhibitors 20 and 21, which contain the 4-methylpiperazin-1-yl group from the DFG-out-stabilizing, clinically-approved drug imatinib, are potent inhibitors of the kinase and RNase activities of IRE1α*. Inhibitors that possess a benzyl (23) or phenethyl (24) R3 group are poorer inhibitors of IRE1α*. Furthermore, inhibitors with heteroaryl (22) or cycloakyl (25-28) groups at the R3 position also poorly inhibit IRE1α*’s enzymatic activities. Finally, we explored whether a urea linker at the R4 position is absolutely necessary for allosteric inhibition of the RNase domain through the ATP-binding site of IRE1α. In this series, analogs that contain either an R4 urea linkage that is not able to form a hydrogen bond donor and acceptor (29 and 30) or an ether linkage (31 and 32) are very poor inhibitors of IRE1α*. Thus, a urea linkage appears to be optimal for bridging the C-3 naphthyl group and the hydrophobic R3 substituent. However, despite the low potencies of non-urea containing analogs, all compounds that inhibit the kinase activity of IRE1α* show at least some degree of RNase inactivation. Therefore, a urea is not strictly necessary for linking kinase inhibition to inactivation of the RNase domain.
Correlation Between Kinase and RNase Inhibiton
Throughout this SAR study, we observed that almost all inhibitors that block the kinase activity of IRE1α also inhibit RNase activity with a similar potency. Despite being based on an ATP-competitive inhibitor scaffold, we further confirmed that KIRAs do not directly inhibit the RNase domain by demonstrating that ATP is able to dose dependently restore RNase activity in the presence of an inhibitory concentration of 2 (Supplementary Figure S3). Furthermore, plotting the kinase and RNase IC50 values for the inhibitors shown in Figure 3 shows strong correlation (R2 = 0.88) that spans almost three orders of magnitude in potency (Figure 4A). Despite the correlation in inhibitory potencies, the fact that KIRAs allosterically modulate IRE1α‘s RNase activity through the kinase domain leaves open the possibility that only partial RNase inhibition may result from quantitative ATP-binding site occupancy by an inhibitor. Therefore, representative KIRAs were tested against IRE1α at a single concentration and the residual amount of kinase and RNase activity was measured (Figure 4B). For a majority of the inhibitors tested, the observed residual RNase activity is within +/− 5% of the residual kinase activity (Figure 4C). Therefore, the magnitude of kinase and RNase inhibition is tightly coupled for these compounds. In contrast, for compounds 26-28, the amount of residual RNase activity is 30–50% greater than the remaining kinase activity. For example, in the presence of 60 μM of inhibitor 28 IRE1α retains <2% of its kinase activity but ~35% of its RNase activity. A similar disparity in inhibition levels was observed for inhibitors 26 and 27. This disparity does not appear to be solely due to the lower potencies of 26-28, as inhibitors 24, 29, and 32 are also poor inhibitors of IRE1α’s kinase activity but show similar levels of RNase inhibition. The common distinguishing feature of compounds 26-28 that differentiates them from the other KIRAs tested is that they possess smaller substituents at the R3 position. Smaller substituents at this position may only lead to partial displacement of the structural element that is necessary for RNase inhibition.
Figure 4. Inter-relationship between the kinase and RNase domains of IRE1α.

(A) Correlation plot of the kinase and RNase IC50 values for KIRAs. The R2 value is 0.88. Inhibitors 26, 27, and 30-32 were excluded from this analysis because kinase and RNase IC50 vales could not be accurately determined. (B) Residual kinase and RNase activity of various inhibitors at a concentration in which the ATP-binding site is either >90% occupied (inhibitors 3, 6-8, 12, 20, and 25) or at a concentration of 60 μM* for less potent inhibitors (24, 26-28, 29, and 32) (mean ± S.E.M, n=3). *A maximum inhibitor concentration of 60 μM was selected due to limits on inhibitor solubility. (C) Difference in percent residual RNase activity and kinase activity [value = (% residual RNase activity) – (% residual kinase activity)].
General Effects of Kinase Inhibitors on the RNase Domain
Above, we demonstrated that all KIRAs that interact with the ATP-binding site of IRE1α also provide at least some level of RNase inhibition. However, almost all other ligands that have been reported to interact with the ATP-binding site of IRE1α increase RNase activity rather than decrease it.14, 28, 31, 32, 40, 41 Therefore, we were curious whether the KIRAs are unique in their dual inhibitory properties or if there are other general classes of kinase inhibitors that are also capable of blocking IRE1α‘s kinase and RNase activities. To address this question, a diverse panel of 378 commercially-available kinase inhibitors (Selleckchem Kinase Inhibitor Library)–containing both type I and II inhibitors–was screened for their ability to block the kinase activity of IRE1α (Figure 5). Of the 378 compounds tested, 15 inhibited the kinase activity of IRE1α with an IC50 of less than 10 μM (Figure 5). The 13 most potent kinase inhibitors were then tested for their ability to inhibit the RNase activity of IRE1α*. In contrast to the KIRAs, none of the 13 IRE1α kinase inhibitors reduced the RNase activity of IRE1α* (Figure 6A). In the absence of any observed RNase inhibition of phosphorylated IRE1α*, the same 13 compounds were tested for their ability to activate the RNase domain of a minimally active IRE1α construct, dP-IRE1α*, which is quantitatively dephosphorylated and has low basal RNase activity (Supplementary Figure S2). Incubation of dP-IRE1α* with 10 μM of each inhibitor increased the cleavage of the XBP1 mini-substrate by 5–15-fold (Figure 6B). Thus, all of the IRE1α kinase inhibitors identified in this screen are activators, rather than inactivators, of dP-IRE1α*’s RNase domain. These results are consistent with the fact that all the inhibitors identified in this screen contain a type I pharmacophore, which binds to the active conformation of protein kinase ATP-binding sites. Indeed, co-crystal structures of eight of the inhibitors shown in Figure 5 have been reported (Supplementary Figure S4), and in all cases the bound kinase target’s ATP-binding site is in an active conformation.
Figure 5. Structures and IC50 values for compounds from the Selleckchem Kinase Inhibitor Library that inhibit the kinase activity of IRE1α >50% at a concentration of 10 μM.

Values shown are mean ± S.E.M (n=3).
Figure 6. Diverse ATP-competitive inhibitors activate the RNase domain of dP-IRE1α*.
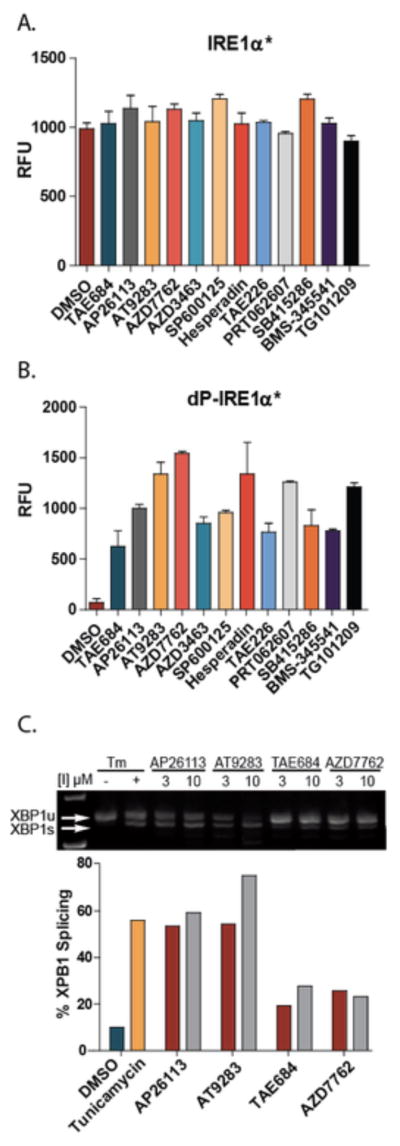
(A) Endpoint fluorescence of IRE1α*-catalyzed cleavage of the XBP1 mini-substrate in the presence of DMSO or 10 μM of the inhibitor listed. Values shown are mean ± S.E.M (n=3). (B) Endpoint fluorescence of dP-IRE1α*-catalyzed cleavage of the XBP1 mini-substrate in the presence of DMSO or 10 μM of the inhibitor listed. Values shown are mean ± S.E.M (n=3). dP-IRE1α* was generated by treating IRE1α* with lambda phosphatase. (C) Ethidium bromide-stained agarose gel of XBP1 cDNA amplicons from INS-1 cells treated with inhibitors at the indicated concentrations for 2 hours. For the positive control, INS-1 cells were treated with 500 ng/ml Tm for 4 hr. Ratiometric quantitation of XBP1s/(XBP1u + XBP1s) is shown at the bottom.
As a truncated form of IRE1α was used for the in vitro studies described above, we next used a cell-based assay to determine whether activating kinase inhibitors have similar effects on the full-length IRE1α transmembrane protein (Figure 6C). To do this, INS-1 rat insulinoma cells, which are derived from insulin-producing pancreatic β-cell tumors, were treated with two concentrations (3 and 10 μM) of four of the most potent IRE1α-activating inhibitors (AT9283, AP26113, TAE684, and AZD7762). Consistent with the ability of type I kinase inhibitors to activate full-length IRE1α, treatment with each inhibitor led to an increase in spliced cellular XBP1 mRNA levels in the absence of ER stress. Strikingly, incubating INS-1 cells with AP26113 and AT9283 led to an equal or greater level of spliced XBP1 mRNA than treatment with 0.5 μg/mL of the ER stress agent tunicamycin (Tm), which is a protein glycosylation inhibitor that leads to acute ER stress.
Structure of apo dP-IRE1α*
Crystal structures of IRE1 have been highly informative in determining which interactions are necessary for stabilizing RNase active IRE1 dimers and for providing insight into the conformational changes that occur in the kinase domain in transitioning from a monomeric to an RNase active dimeric state.18, 28, 41–44 During our attempts to obtain a structure of a KIRA bound to IRE1α we obtained the structure of human apo dP-IRE1α* to a resolution of 2.6 Å (Supplementary Table S1). Even though apo dP-IRE1α* is not phosphorylated, it forms an RNase active back-to-back dimer under the crystallization conditions used. The asymmetric unit of the crystal contains a dP-IRE1α* monomer, featuring a canonical bilobal kinase domain, and a globular α-helix rich RNase domain (Figure 7A, 7B). In the back-to-back dimer, the dP-IRE1α* kinase domain adopts an active kinase conformation: the aspartate (Asp711) of the DFG motif (Asp711-Phe712-Gly713) faces into the active site (DFG-in), and glutamate (Glu612) of helix-αC forms a salt bridge with the catalytic lysine (Lys599), typical of the helix-αC-in conformation (Figure 7C). Despite being dephosphorylated, the activation segment in dP-IRE1α* is completely resolved and adopts an extended active conformation. Interestingly, the beginning of the activation segment (Gly713, Cys715, Lys717) engages with helix-αC (Arg611, Glu618) and appears to anchor it in the helix-αC-in conformation (Figure 7C). Because helix-αC is a major component of the back-to-back dimer interface, its stabilization in the helix-αC-in orientation promotes dimerization and RNase activation. Phosphorylation of the activation segment would further stabilize the extended active conformation of this flexible structural element, resulting in enhanced dimerization and subsequent RNase activation. A novel feature of our dP-IRE1α* kinase domain structure is the presence of two cesium ion (Cs+) binding sites, CsB1 and CsB2 (Figure 7 and Supplementary Figure S5). These cesium ions stabilize the extended activation segment, and dimer interface residues near helix-αC, into conformations conducive to back-to-back dimerization. Our structure of dP-IRE1α* shows that activation segment phosphorylation is not absolutely necessary for adopting the active kinase conformation and the RNase active back-to-back dimer.
Figure 7. Apo human dP-IRE1α* adopts an RNase active back-to-back dimer promoted by cesium mediated stabilization of the active kinase conformation.

(A) Two apo human dP-IRE1α* protomers (PDB: 5HGI) adopt an active kinase conformation that is stabilized by Cs+ ions (green spheres), allowing the protomers to align parallel in a back-to-back dimer configuration comparable to the RNase active yIRE1 structure (PDB: 3FBV). Each protomer consists of a protein kinase domain (grey) and an RNase domain (purple). Protein kinase regulatory elements such as the P-loop (red), helix-αC (orange), and activation segment (blue) are highlighted. The helix-loop-element dubbed the HLE (cyan), a catalytic motif in the RNase domain is also shown. The arrow denotes that the position of helix-αE has been hidden in order to reveal one of the Cs+ ions that stabilize the activation segment. A two-fold symmetry axis is indicated by the black line. (B) Cartoon schematic illustrating that cesium and coupling between helix-αC and the activation segment helps mediate stabilization of the active kinase hallmarks (helix-αC-in, extended activation segment), which are associated with the RNase active back-to-back dimer. Cartoon features with reduced opacity denotes those which are behind the dimer interface plane that is parallel to the two-fold symmetry axis (black line). (C) A close-up view of the dP-IRE1α* kinase active site, presenting the canonical kinase active conformation hallmarks and key salt bridge interactions that couple the activation segment with helix-αC.
Protomers of neighboring asymmetric units of apo dP-IRE1α* form a back-to-back dimer that is similar in overall orientation as the RNase active yeast IRE1 structure (PDB: 3FBV) (Figure 7A, Supplementary Figures S6A, B and Tables S2, S3).18, 40 Analysis of the alignment of the protomers in dimeric IRE1α indicates that the dP-IRE1α* protomers align in a parallel and symmetrical fashion for both the kinase and RNase domains, like the active yIRE1 dimer (Supplementary Figures S6C), confirming that our dP-IRE1α* structure resembles a fully RNase active state. This contrasts with the pre-active dimer configuration of a recently reported human dP-IRE1α structure (PDB: 4Z7H).42 in which the protomers are not aligned fully parallel, and the RNase domains do not complement to form the active conformation. While the overall quaternary structure is highly conserved between dP-IRE1α* and active yIRE1, sequence divergence and insertions cause secondary structure differences that change the details of stabilizing interactions for the activation segment and helix-αC. Similarly, only three dimer interactions in the back-to-back dimer interface are conserved (Supplementary Figures S7, S8 and Tables S2, S3).
ATP-Competitive Inhibitors Modulate RNase Activity by Affecting Helix-αC Conformation
To mechanistically rationalize how KIRAs are able to inhibit the RNase activity of IRE1α, we compared our active apo dP-IRE1α* structure to two different inactive mammalian IRE1α structures: (1) dephosphorylated IRE1α bound to ADP (PDB: 3P23)17 and (2) dephosphorylated IRE1α bound to a KIRA recently developed by Amgen (compound 33) (PDB: 4U6R)43 (Figure 8 and Supplementary Figure S9). Analysis of these structures shows that the most direct mechanism to inhibit IRE1α’s RNase activity is through the displacement of IRE1α’s helix-αC from the active conformation (Figure 8 and Supplementary Figure S9). This movement of helix-αC would disrupt important contacts that contribute to the interface of the RNase active back-to-back dimer, likely resulting in monomeric IRE1α and subsequent loss of RNase activity. The ATP-competitive KIRA developed by Amgen (compound 33) appears to exploit this mechanism of RNase inhibition (PDB: 4U6R).43 Compound 33 projects an arylsulfonamide moiety towards IRE1α*’s helix-αC, which causes a ~2 Å displacement from the active conformation (Figure 8B and Supplementary Figure S9). This shift of IRE1α*’s helix-αC from the active conformation disrupts part of the back-to-back dimer interface of the kinase domain and results in potent RNase inhibition. Furthermore, incubation of a saturating concentration of 33 with phosphorylated IRE1α* resulted in suppression of dimer formation in an in vitro crosslinking assay (Supplementary Figure S10). Although the KIRAs characterized in this study possess aryl-urea moieties that are common in ligands that stabilize a flipped activation segment DFG-motif (DFG-out), it is likely they also stabilize IRE1α*’s helix-αC-out conformation based on their inhibition of dimer formation of phosphorylated IRE1α* in the same crosslinking assay (Figure 8C and Supplementary Figure S11). For many of the KIRAs shown in Figure 3, this could be accomplished by directly displacing helix-αC from an active conformation like compound 33–although a larger movement than what is observed in structure 4U6R would likely be necessary to accommodate bulkier aryl-urea moieties (Supplementary Figure S12). Consistent with this notion, a co-crystal structure of compound 1 bound to an off target kinase–Src tyrosine kinase–that we have determined shows this kinase in the helix-αC-out conformation, demonstrating that aryl-ureas are able to stabilize this inactive form (Supplementary Figure S12). Overlay of Src·1 with our active apo dP-IRE1α* structure reveals that the aryl-urea moiety of 1 is sterically incompatible with the helix-αC-in conformation (Supplementary Figure S13). Thus, it is likely that our KIRAs and compound 33 lead to very similar conformational changes in the ATP-binding site of IRE1α and have similar mechanism of RNase inhibition.
Figure 8. The conformation of helix-αC in the kinase domain affects IRE1α oligomerization state and RNase domain activity.

(A) The kinase domain active site of apo human dP-IRE1α* adopts an active conformation. Because helix-αC-in is a conformation conducive to dimerization, this allows the formation of RNase active back-to-back dimers as shown by the cartoon below. (B) In contrast, human IRE1α bound to the ATP-competitive Amgen KIRA (compound 33) (PDB: 4U6R), presents an inactive kinase conformation because the helix-αC-out conformation is adopted. The cartoon below indicates that back-to-back dimers are incompatible with this inactive kinase conformation due helix-αC-out steric clashes at the dimer interface (Supplementary Figure S9). (C) Quantification of the ratios of dimeric/oligomeric to monomeric IRE1α* (mean ± S.E.M, n=3). IRE1α* (15 μM) was incubated with DMSO or the KIRA shown (100 μM) and then treated with the chemical crosslinker disuccinimidyl suberate ([DSS] = 250 μM). (Supplementary Figure S11) (D) Quantification of the ratios of dimeric/oligomeric to monomeric IRE1α* (mean ± S.E.M, n=3). dP-IRE1α* (15 μM) was incubated with DMSO or the KIRA/Activator shown (100 μM) and then treated with 250 μM DSS. (Supplementary Figure S11).
Based on the observed interactions between IRE1α*’s helix-αC and the base of the activation segment in our dP-IRE1α* structure, we propose that inhibitors that stabilize the DFG-out conformation of the DFG-motif could also lead to helix-αC displacement and RNase inhibition by disrupting the interactions between the activation segment and helix-αC (Figure 7C). These interactions appear to anchor helix-αC in the active conformation and the crankshaft rotation of the DFG motif that yields the DFG-out conformation would collapse the activation segment into an inactive disordered conformation and destabilize the helix-αC-in conformation as exemplified in existing inactive IRE1α structures (PDB: 3P23, 4PL3).17, 28 Indeed, in the structure of dephosphorylated IRE1α* bound to ADP (PDB: 3P23), which contains a helix-αC displaced 7.4 Å from the active conformation, the Glu618-Lys717, and Arg611-Gly713, and Cys715-backbone salt bridges are absent and the activation segment is completely disordered (Supplementary Figure S12D). Therefore, KIRAs may be able to prevent the RNase active back-to-back dimer and inhibit the RNase activity of IRE1α by displacing the activation segment, resulting in the disruption of contacts that anchor helix-αC in the active conformation. This model is especially appealing for KIRAs 20 and 21, which contain extended 4-methylpiperazin-1-yl aryl-urea substituents–like the DFG-out-stabilizing inhibitor imatinib–that are unlikely to be accommodated in the relatively confined pockets created by the movement of helix-αC in structures 4U6R and 3P23 (Figure 8B and Supplementary Figure S12). One caveat to this model is that any KIRAs that stabilize the DFG-out inactive conformation of IRE1α must lack the canonical hydrogen bond with Glu612 in helix-αC because this interaction would likely lead to increased dimer formation and subsequent RNase activation.
Our data in Figure 6 suggest that ATP-competitive kinase inhibitors activate the RNase domain unless they displace either helix-αC or the DFG-motif from the active conformation. For all IRE1α RNase activators (Figure 5) that co-crystal structures are available for, the bound kinase target is in the active, helix-αC-in conformation (Supplementary Figure S4). Similarly, these activators could be computationally docked to the active structure of dP-IRE1α*, while aryl-ureas KIRAs cannot (Supplementary Figure S14). Consistent with the RNase activators in Figure 5 leading to stabilization of the helix-αC-in conformation all compounds tested led to increased dimerization/oligomerization of dP-IRE1α* (Figure 8D). However, this stabilization does not appear to be through a direct interaction with either the DFG-motif or helix-αC, as none of the crystal structures or docked inhibitor structures show direct interactions with these structural motifs. Instead, binding of inhibitors to the ATP-binding site complements a cluster of hydrophobic amino acids (“catalytic spine”) that promotes the closure of the active site between the kinase N- and C-lobes, which stabilizes the active helix αC-in conformation (Supplementary Figure S15).45
Conclusions
The ATP-binding sites of protein kinases are allosterically coupled to distal binding and regulatory sites in the kinase domain.46 These allosteric networks allow communication between the ATP-binding and protein substrate-binding sites and are important for the regulation of kinase catalytic activity.47, 48 Biochemical and structural studies have defined how binding at allosteric regulatory sites and post-translational modifications are able to affect kinase catalytic activity through the modulation of ATP-binding site conformation.49 These kinase allosteric networks are bidirectional in that conformation-selective ATP-competitive inhibitors are able to influence distal sites in the kinase domain.49–52 In this study, we have explored the allosteric relationship between the kinase and RNase domains of the ER sensor protein IRE1α. By screening a panel of commercially available kinase inhibitors, we find that most ATP-competitive inhibitors that interact with the kinase domain of IRE1α activate RNase activity rather than inactivate it. Diverse ATP-competitive ligands show variable levels of RNase activation of dephosphorylated IRE1α, which has very low basal RNase activity. Furthermore, these ATP-competitive IRE1α activators increase cellular XBP1 mRNA splicing in the absence of ER stress. All tested kinase inhibitors that increase the activity of the RNase domain also promote dimerization of the dephosphorylated form of IRE1α. Thus, most ATP-competitive kinase inhibitors strengthen the dimer interface that is necessary for RNase activity. The general characteristic of demonstrating enhanced dimerization in the presence of most ATP-competitive ligands is shared by RAF kinase.54 For RAF, the propensity to dimerize in the presence of ATP-competitive inhibitors is believed to be responsible for the paradoxical activation of the RAF->MEK->Erk signaling pathway that is observed in certain cell lines. Interestingly, inhibitors that overcome paradoxical RAF pathway activation may prevent dimerization by enforcing a more pronounced adoption of the helix-αC-out conformation.55
In contrast to most inhibitors of IRE1α’s kinase activity, a series of imidazopyrazine-based inhibitors that we have developed–called KIRAs–lead to allosteric inhibition of the RNase domain. For inhibitors based on the imidazopyrazine KIRA scaffold, IC50s for kinase and RNase inhibition are highly correlated, highlighting the strong degree of coupling between both enzymatic activities of IRE1α. Furthermore, for all but a few of the KIRAs characterized, the level of ATP-binding site occupancy directly correlates with the degree of RNase inhibition, demonstrating that most KIRAs show complete rather than partial allosteric inhibition. The observed ability of KIRAs to block IRE1α dimerization/oligomerization, which is necessary for RNase activity, provides insight into the mechanism of allosteric inhibition. By stabilizing an ATP-binding site conformation that is incompatible with dimer formation, KIRAs are able to directly translate kinase domain interactions into RNase domain inhibition. This mechanism of allosteric RNase inhibition appears to be shared by the only other KIRA series that have been developed to date.44 These KIRAs–exemplified by inhibitor 33–are based on a completely different scaffold and contain an aryl sulfonamide instead of an aryl-urea, yet still inhibit RNase activity through the kinase domain and suppress formation of IRE1α dimers/oligomers. ATP-competitive RNase activators are able to competitively restore the RNase activity of IRE1α* in the presence of an inhibitory concentration of our KIRAs (2 and 20) or 33 (Supplementary Figure S16), further highlighting the similarities between these inhibitor classes.
We have resolved an apo dP-IRE1α* structure that adopts a back-to-back dimer resembling the active yIRE1 dimer. Furthermore, the dP-IRE1α* kinase domain contains the signatures of an active conformation, a prerequisite for IRE1α back-to-back dimerization and subsequent RNase activity. The active helix-αC-in conformation is critical for RNase domain activity because it allows IRE1α back-to-back dimerization to occur. Salt bridge interactions also exist between helix-αC and the activation segment, indicating coupling between these regulatory elements. Because these regulatory elements are coupled, the destabilization/stabilization of either one can likely lead to destabilization/stabilization of the other, allowing for divergent modulation of RNase activity (Supplementary Figure S17). As helix-αC is a dynamic element in the ATP-binding site that is a major component of the dimer interface it is tempting to speculate that type I ATP-competitive RNase activators of IRE1α exert their influence by stabilizing the active conformation of helix-αC. None of the activators described in this study, for which kinase-bound structures are available, directly interact with helix-αC, so this influence would have to be of an indirect nature. In contrast, KIRAs can inhibit the RNase activity of phosphorylated IRE1α by displacing helix-αC from an active conformation to an inactive conformation that is incompatible with back-to-back dimer formation. This displacement can either occur directly by inhibitors displaying substituents that would clash with the active helix-αC-in conformation, or indirectly, by stabilizing an inactive activation segment that disrupts the anchoring of helix-αC.
Supplementary Material
Acknowledgments
This work was supported by the NIH (R01GM086858 (D.J.M.), R01DK100623 (D.J.M. and F.R.P.), R01DK080955 (Papa), R01DK095306 (Papa), P01HL108794 (Papa and Backes)), Camille and Henry Dreyfus Foundation (D.J.M.) and the Burroughs-Wellcome Career Award in the Biomedical Sciences (CABS) (Papa). Data presented in this publication was collected on beamlines X25 and X29 at the National Synchrotron Light Source at the Brookhaven National Laboratory, and we wish to thank all staff involved. The use of this facility is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-98CH10886.
Footnotes
Conflict of interest statement: Bradley J. Backes, Feroz R. Papa, and Dustin J. Maly are scientific co-founders, equity holders, and paid consultants for OptiKira L. L. C.
References
- 1.Anelli T, Sitia R. Protein quality control in the early secretory pathway. EMBO J. 2008;27:315–327. doi: 10.1038/sj.emboj.7601974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Anken E, Braakman I. Versatility of the endoplasmic reticulum protein folding factory. Crit Rev Biochem Mol Biol. 2005;40:191–228. doi: 10.1080/10409230591008161. [DOI] [PubMed] [Google Scholar]
- 3.Merksamer PI, Trusina A, Papa FR. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell. 2008;135:933–947. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Anken E, Pena F, Hafkemeijer N, Christis C, Romijn EP, Grauschopf U, Oorschot VM, Pertel T, Engels S, Ora A, Lastun V, Glockshuber R, Klumperman J, Heck AJ, Luban J, Braakman I. Efficient IgM assembly and secretion require the plasma cell induced endoplasmic reticulum protein pERp1. Proc Natl Acad Sci U S A. 2009;106:17019–17024. doi: 10.1073/pnas.0903036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 7.Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. 2015;17:829–838. doi: 10.1038/ncb3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 10.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 12.Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol. 2011;23:143–149. doi: 10.1016/j.ceb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maly DJ, Papa FR. Druggable sensors of the unfolded protein response. Nat Chem Biol. 2014;10:892–901. doi: 10.1038/nchembio.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- 16.Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, Collins I, Davies FE, Pearl LH. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011;30:894–905. doi: 10.1038/emboj.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee KP, Dey M, Neculai D, Cao C, Dever TE, Sicheri F. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell. 2008;132:89–100. doi: 10.1016/j.cell.2007.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Y, Liang FX, Wang X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol Cell. 2014;55:758–770. doi: 10.1016/j.molcel.2014.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosmaczewski SG, Edwards TJ, Han SM, Eckwahl MJ, Meyer BI, Peach S, Hesselberth JR, Wolin SL, Hammarlund M. The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep. 2014;15:1278–1285. doi: 10.15252/embr.201439531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 22.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 24.Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, Papa FR, Oakes SA. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338:818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, Koong AC. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, Hu Y, Fabre C, Minami J, Ohguchi H, Kiziltepe T, Ikeda H, Kawano Y, French M, Blumenthal M, Tam V, Kertesz NL, Malyankar UM, Hokenson M, Pham T, Zeng Q, Patterson JB, Richardson PG, Munshi NC, Anderson KC. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood. 2012;119:5772–5781. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanches M, Duffy NM, Talukdar M, Thevakumaran N, Chiovitti D, Canny MD, Lee K, Kurinov I, Uehling D, Al-awar R, Poda G, Prakesch M, Wilson B, Tam V, Schweitzer C, Toro A, Lucas JL, Vuga D, Lehmann L, Durocher D, Zeng Q, Patterson JB, Sicheri F. Structure and mechanism of action of the hydroxy-aryl-aldehyde class of IRE1 endoribonuclease inhibitors. Nat Commun. 2014;5:4202. doi: 10.1038/ncomms5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Volkmann K, Lucas JL, Vuga D, Wang X, Brumm D, Stiles C, Kriebel D, Der-Sarkissian A, Krishnan K, Schweitzer C, Liu Z, Malyankar UM, Chiovitti D, Canny M, Durocher D, Sicheri F, Patterson JB. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. The Journal of biological chemistry. 2011;286:12743–12755. doi: 10.1074/jbc.M110.199737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, Harding HP. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109:E869–878. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papa FR, Zhang C, Shokat K, Walter P. Bypassing a kinase activity with an ATP-competitive drug. Science. 2003;302:1533–1537. doi: 10.1126/science.1090031. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Perera BG, Hari SB, Bhhatarai B, Backes BJ, Seeliger MA, Schurer SC, Oakes SA, Papa FR, Maly DJ. Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat Chem Biol. 2012;8:982–989. doi: 10.1038/nchembio.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prischi F, Nowak PR, Carrara M, Ali MM. Phosphoregulation of Ire1 RNase splicing activity. Nat Commun. 2014;5:3554. doi: 10.1038/ncomms4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itzhak D, Bright M, McAndrew P, Mirza A, Newbatt Y, Strover J, Widya M, Thompson A, Morgan G, Collins I, Davies F. Multiple autophosphorylations significantly enhance the endoribonuclease activity of human inositol requiring enzyme 1alpha. BMC Biochem. 2014;15:3. doi: 10.1186/1471-2091-15-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, Regan J. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9:268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 36.Hari SB, Perera BG, Ranjitkar P, Seeliger MA, Maly DJ. Conformation-selective inhibitors reveal differences in the activation and phosphate-binding loops of the tyrosine kinases Abl and Src. Acs Chem Biol. 2013;8:2734–2743. doi: 10.1021/cb400663k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seeliger MA, Ranjitkar P, Kasap C, Shan Y, Shaw DE, Shah NP, Kuriyan J, Maly DJ. Equally potent inhibition of c-Src and Abl by compounds that recognize inactive kinase conformations. Cancer Res. 2009;69:2384–2392. doi: 10.1158/0008-5472.CAN-08-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ranjitkar P, Brock AM, Maly DJ. Affinity reagents that target a specific inactive form of protein kinases. Chem Biol. 2010;17:195–206. doi: 10.1016/j.chembiol.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okram B, Nagle A, Adrián FJ, Lee C, Ren P, Wang X, Sim T, Xie Y, Wang X, Xia G, Spraggon G, Warmuth M, Liu Y, Gray NS. A general strategy for creating “inactive-conformation” abl inhibitors. Chem Biol. 2006;13:779–786. doi: 10.1016/j.chembiol.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 40.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Stroud RM, Zhang C, Shokat KM, Walter P. Cofactor-mediated conformational control in the bifunctional kinase/RNase Ire1. BMC Biol. 2011;9:48. doi: 10.1186/1741-7007-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joshi A, Newbatt Y, McAndrew PC, Stubbs M, Burke R, Richards MW, Bhatia C, Caldwell JJ, McHardy T, Collins I, Bayliss R. Molecular mechanisms of human IRE1 activation through dimerization and ligand binding. Oncotarget. 2015;6:13019–13035. doi: 10.18632/oncotarget.3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harrington PE, Biswas K, Malwitz D, Tasker AS, Mohr C, Andrews KL, Dellamaggiore K, Kendall R, Beckmann H, Jaeckel P, Materna-Reichelt S, Allen JR, Lipford JR. Unfolded Protein Response in Cancer: IRE1alpha Inhibition by Selective Kinase Ligands Does Not Impair Tumor Cell Viability. ACS Med Chem Lett. 2015;6:68–72. doi: 10.1021/ml500315b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korennykh AV, Korostelev AA, Egea PF, Finer-Moore J, Stroud RM, Zhang C, Shokat KM, Walter P. Structural and functional basis for RNA cleavage by Ire1. BMC Biol. 2011;9:47. doi: 10.1186/1741-7007-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tong M, Seeliger MA. Targeting conformational plasticity of protein kinases. ACS Chem Biol. 2015;10:190–200. doi: 10.1021/cb500870a. [DOI] [PubMed] [Google Scholar]
- 47.Foda ZH, Shan Y, Kim ET, Shaw DE, Seeliger MA. A dynamically coupled allosteric network underlies binding cooperativity in Src kinase. Nat Commun. 2015;6:5939. doi: 10.1038/ncomms6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 2011;42:9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agius MP, Soellner MB. Modulating noncatalytic function with kinase inhibitors. Chem Biol. 2014;21:569–571. doi: 10.1016/j.chembiol.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 49.Krishnamurty R, Brigham JL, Leonard SE, Ranjitkar P, Larson ET, Dale EJ, Merritt EA, Maly DJ. Active site profiling reveals coupling between domains in SRC-family kinases. Nat Chem Biol. 2013;9:43–50. doi: 10.1038/nchembio.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Register AC, Leonard SE, Maly DJ. SH2-catalytic domain linker heterogeneity influences allosteric coupling across the SFK family. Biochemistry. 2014;53:6910–6923. doi: 10.1021/bi5008194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leonard SE, Register AC, Krishnamurty R, Brighty GJ, Maly DJ. Divergent modulation of Src-family kinase regulatory interactions with ATP-competitive inhibitors. ACS Chem Biol. 2014;9:1894–1905. doi: 10.1021/cb500371g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hari SB, Merritt EA, Maly DJ. Conformation-selective ATP-competitive inhibitors control regulatory interactions and noncatalytic functions of mitogen-activated protein kinases. Chem Biol. 2014;21:628–635. doi: 10.1016/j.chembiol.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kung JE, Jura N. Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure. 2016;24:7–24. doi: 10.1016/j.str.2015.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holderfield M, Nagel TE, Stuart DD. Mechanism consequences of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640–645. doi: 10.1038/bjc.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, Tsang G, Marimuthu A, Cho H, Wu G, Wang W, Fong D, Nguyen H, Shi S, Womack P, Nespi M, Shellooe R, Carias H, Powell B, Light E, Sanftner L, Walters J, Tsai J, West BL, Visor G, Rezaei H, Lin PS, Nolop K, Ibrahim PN, Hirth P, Bollag G. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–586. doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.