

Abstract
The structural plasticity of synaptic terminals contributes to normal nervous system function but also to neural degeneration, in the form of terminal retraction, and regeneration, due to process growth. Synaptic morphological change is mediated through the actin cytoskeleton, which is enriched in axonal and dendritic terminals. Whereas the three RhoGTPases, RhoA, Cdc42 and Rac, function as upstream signaling nodes sensitive to extracellular stimuli, LIMK-cofilin activity serves as a common downstream effector to up-regulate actin turnover, which is necessary for both polymerization and depolymerization. The dual effects of LIMK activity make LIMK a potential target of therapeutic intervention for injury-induced synaptic plasticity, as LIMK inhibition can stabilize actin cytoskeleton and preserve existing structure. This therapeutic benefit of LIMK inhibition has been demonstrated in animal models of injury-induced axon retraction and neuritic sprouting by rod photoreceptors. A better understanding of the regulation of LIMK-cofilin activity and the interaction with the microtubular cytoskeleton may open new ways to promote synaptic regeneration that can benefit neuronal degenerative disease.
Keywords: LIMK, cofilin, RhoGTPases, actin turnover, structural plasticity, synaptic degeneration and regeneration
Introduction
Synapses display plasticity in response to neural activity or insult at both the pre- and postsynaptic terminals. Synaptic plasticity contributes to normal physiological functions, such as axon pathfinding by developing neurons and learning and memory storage by mature neurons, but is also invoked in response to disease and injury, and thus may lead to structural and functional abnormalities. For example, in response to the injury of retinal detachment, which is a separation of photoreceptor from retinal pigment epithelial layers, rod photoreceptors retract their synaptic terminals back towards the cell body, destroying the first synapse between photoreceptor and bipolar/horizontal cells in the visual pathway; in response to surgical reattachment, rod, and to a lesser extent cone cells grow neuritic sprouts into the inner retina where amacrine and ganglion cells reside, again potentially disrupting retinal circuitry (Lewis et al., 2002). Risk factors for retinal detachment include severe myopia, advancing age, complication from cataract surgery or trauma, and thus detachment is relatively common in the population. As actin cytoskeleton is the predominant cytoskeletal structure in all synaptic terminals, it is highly likely that some conserved mechanism is utilized by the photoreceptor in its injury response.
RhoGTPase signaling by the canonical Rho family members RhoA, Rac1, and Cdc42 is a key contributor to synaptic plasticity in part through regulation of actin turnover. Furthermore, manipulation of the actin cytoskeleton, through the use of RhoA antagonists for example, has contributed to successful therapeutic strategies for treating neuronal degeneration and promoting axon regeneration in the central nervous system (CNS) (Amano et al., 2010). The importance of Rho signaling is no less evident in the axons and terminals of the sensory neurons of the retina, the photoreceptors. Since both axon terminal retraction and sprouting are observed in isolated photoreceptors in culture and in retinal explants, these in vitro cell and tissue systems have allowed us to examine the Rho pathways during injury-induced structural plasticity of the retina. Our studies suggest that interconnections among the various pathways through the downstream effector Lim kinase (LIMK) may tie degeneration and regenerative events together and lead to new therapeutic interventions, and thus, for the visual system, preservation of vision after injury (Wang and Townes-Anderson, 2015).
RhoGTPases in Synaptic Structural Plasticity
RhoA, Rac1 and Cdc42 serve as signaling nodes by coordinating extracellular stimuli with regulation of actin polymerization, depolymerization and actomyosin contraction. Their functions are mediated through several major downstream effectors, including Rho kinase (ROCK), p21 activated kinase (Pak), LIMK and myosin light chain (MLC) (Figure 1).
Figure 1.
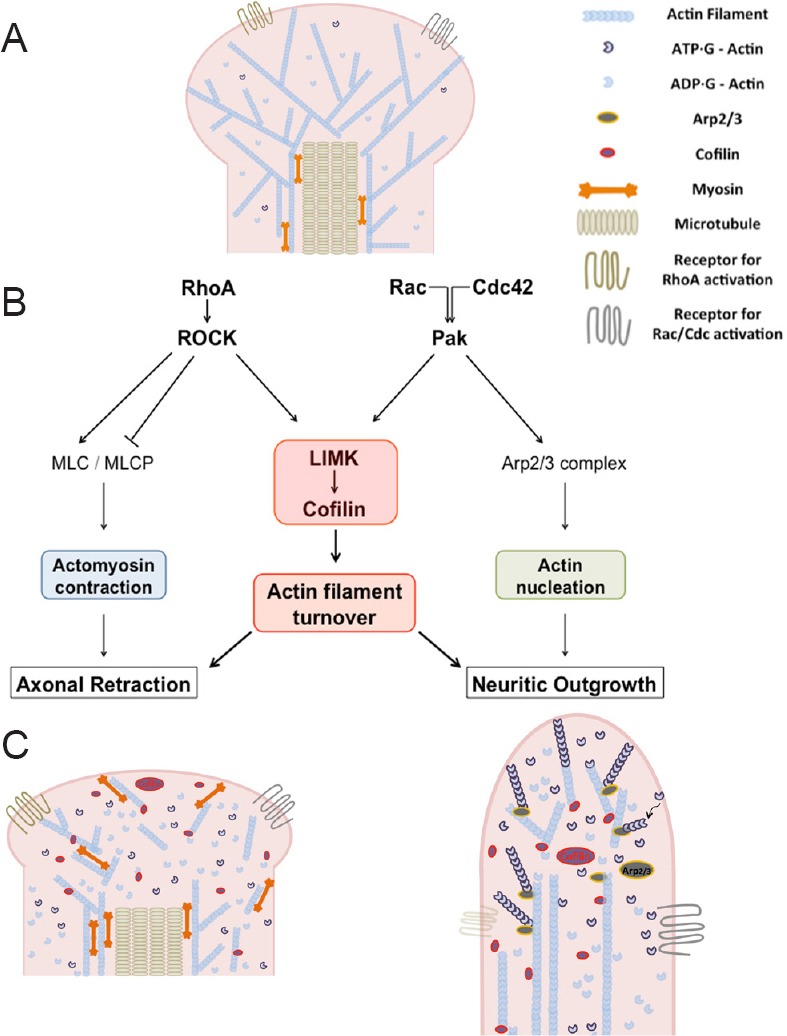
LIMK and the actin cytoskeleton.
(A) Diagram of rod photoreceptor axon terminal with intracellular cytoskeletal structure. (B) Diagram of proposed pathways involved in injured photoreceptor synaptic plasticity. Actomyosin contraction and actin filament turnover and branching, are suggested to contribute to plasticity after injury. RhoA, Rac and Cdc42, Rho GTPases; ROCK: Rho kinase: Pak: p21-activated kinase; LIMK, LIM kinase; MLC: myosin light chain; MLCP: myosin light chain phosphatase; Arp2/3: actin-related proteins 2 and 3. (C) Left: diagram of retracting axon terminal with dissembled actin cytoskeleton; right: diagram of process growth with newly assembled actin filaments.
The first functional study of Rho GTPases was described in a temperature-sensitive Saccharomyces cerevisiae mutant. Defective budding indicated that a Cdc42 mutant might induce abnormal actin dynamics; this was confirmed by a microinjection study in fibroblasts. In neurons, RhoA promotes actin depolymerization through LIMK activity and generates contractile force through phosphorylation of MLC, both of which lead to growth cone collapse and axon retraction. Rac1 instead promotes actin polymerization and controls the formation of lamellipodia and membrane ruffling whereas Cdc42 regulates the formation of filopodia (Nobes and Hall, 1995).
During nerve cell development, all three RhoGTPases play critical roles in actin turnover and rearrangement which promote the growth and retraction of filopodia and lamellipodia necessary for pathfinding by axonal growth cones and for the formation of precise synaptic connections. In mature neurons, RhoGTPases continue to regulate plasticity. Perhaps most well known is the role of RhoGTPases in the shape changes of dendritic spines in response to neural activity. The role of RhoGTPases in neuronal injury is less well understood. In pig retina, active RhoA, RhoA-GTP, increases immediately after detachment and remains elevated for at least 24 hours (Wang and Townes-Anderson, unpublished data). At the same time rod presynaptic terminals begin to round up and separate from their postsynaptic partners. We demonstrated the contribution of RhoA signaling to the injury-induced axonal retraction of rod photoreceptors using inhibition of ROCK, a serine/threonine kinase and direct effector of RhoA, in isolated photoreceptors, as well as in retinal explants. Inhibition of ROCK significantly reduced axon retraction of photoreceptors in detached retina (Fontainhas and Townes-Anderson, 2011). This work indicated that there was a link in rod cells between RhoA and injury-induced presynaptic structural change.
Although RhoA-ROCK and Rac/Cdc42-Pak pathways each has unique substrates, evidence indicates they both modulate LIMK-cofilin activity. Our recent study of photoreceptor axonal plasticity in injured retina revealed that while ROCK inhibition reduced axon retraction, LIMK inhibition reduced both axonal retraction and neuritic growth. This finding suggested that the two seemingly opposite events, degeneration and regeneration, may actually utilize the same machinery. We speculated that understanding the mechanism of LIMK-cofilin regulated actin turnover, including the crosstalk with other signaling pathways, may result in new therapeutic strategies for nerve cell regeneration.
LIMK, Cofilin and Actin Turnover Regulation
The LIM kinase family includes LIMK1 and LIMK2. LIMK1 is expressed mainly in the CNS, but also in heart and skeletal muscle, whereas LIMK2 is expressed in all tissues. LIMK has two N-terminal LIM domains and a C-terminal kinase domain, with a PDZ domain in-between. While LIM and PDZ domains serve as a constitutive inhibitor, the C-terminal contains an activation loop, in which threonines (Thr508/505) reside. Phosphorylation of Thr508/505 by either ROCK or Pak is essential for LIMK activation (Manetti, 2012).
Upon activation, LIMK regulates actin filament turnover through phosphorylation of its substrate, cofilin, at serine 3 (Arber et al., 1998). Cofilin, including cofilin1 and cofilin2, binds to actin filaments. Severing of actin filaments occurs at the junction between undecorated and cofilin-decorated regions, probably due to unequal filament stability between these two regions. Cofilin has a higher binding affinity to ADP-actin, the subunit which makes up mature actin filaments. Thus, cofilin promotes change in the actin cytoskeleton by severing of mature actin filaments into fragments and drives monomer actin release.
When LIMK phosphorylates cofilin, it is released from actin. Slingshot (SSH), a phosphatase, dephosphorylates cofilin and restores its ability to bind actin. However, this does not mean that LIMK plays the role of inhibitor or SSH of activator for cofilin-induced severing of actin filaments. Dephosphorylation is necessary for cofilin to bind and hence begin the process of severing actin filaments, but phosphorylation is equally necessary in order for cofilin to be released from filaments, ready to bind and sever other filaments. Mathematical simulations indicate that LIMK-dependent cofilin phosphorylation can actually amplify cofilin activity locally, in cooperation with other regulatory processes such as cofilin dephosphorylation. This model explains what many studies have reported: LIMK activity, which phosphorylates cofilin, is necessary for cell motility and morphological change (Bravo-Cordero et al., 2013).
Although LIMK-cofilin mediated actin turnover is important for disassembly of existing actin networks, LIMK also participates in assembly of new actin filaments. When RhoA-ROCK activity is dominant, the LIMK-cofilin pathway serves to promote retraction, presumably through severing of actin-filaments in collaboration with actomyosin contraction due to ROCK-MLC/MLCP activity (see Figure 1) (Wang and Townes-Anderson, 2015). When Rac/Cdc42-Pak activity is high, actin polymerization and growth predominate (Manetti, 2012). Cdc42, together with PIP2, binds and activates WASP (Wiskott-Aldrich syndrome protein); Rac activates WAVE (WASP-family verprolin-homologous protein). WASP/WAVE then recruits Arp2/3, which facilitates addition of new branches to existing filaments. LIMK contributes to polymerization and growth because elongation and formation of new branches in the actin meshwork require actin fragments with barbed ends, which are generated when LIMK-cofilin severs actin filaments. The force of elongation of actin filaments and assembly of new branches drive membrane forward at the leading edge.
In isolated photoreceptors, inhibition of either ROCK, Pak or LIMK reduced axon retraction (Wang and Townes-Anderson, 2015). The combined treatment of ROCK and Pak inhibition had an additive effect due perhaps to increased inhibition of LIMK or to inhibition of both LIMK and MLC activity. Phosphorylated LIMK (p-LIMK), the active form of LIMK, was present in rod axon terminals, precisely the area that undergoes dramatic morphological change after injury. Additionally, we observed a correlation between the injury of retinal detachment and increased cofilin phosphorylation, mediated by ROCK-LIMK signaling, in detached porcine retina (Wang and Townes-Anderson, 2014). These data together confirm the contribution of LIMK activity to photoreceptor axon retraction.
Injury-induced neuritic growth by photoreceptors also responded to LIMK activity. Active (phosphorylated) LIMK was found at the leading edge of growing lamellipodia, filopodia, and in new varicosities of cultured photoreceptors. Inhibition of LIMK significantly reduced process growth; Pak inhibition resulted in a similar effect (Wang and Townes-Anderson, 2014, 2015). These results demonstrate that Pak-LIMK activity promotes neuritic growth in isolated photoreceptors.
The involvement of LIMK in neuronal growth and degeneration has also been reported in other studies. LIMK expression and cofilin phosphorylation increase during neuritogenesis whereas semaphorin 3A-induced growth cone collapse is mediated through cofilin phosphorylation by LIMK1. Examination of the brains of patients with Alzheimer’s disease (AD) showed increased p-LIMK positive neurons in the areas affected with AD pathology (Heredia et al., 2006). Treating primary hippocampal neurons in culture with fibrillar amyloid β induced dramatic reorganization of actin filaments, neuritic dystrophy and cell death. The cells demonstrated increased levels of p-LIMK1 and p-cofilin, whereas inhibition of LIMK-regulated cofilin phosphorylation prevented actin filament reorganization and neuronal degeneration induced by fibrillar amyloid β treatment.
Our studies of the ROCK/Pak-LIMK pathway in synaptic plasticity of injured photoreceptors, therefore, highlight what may be a conserved mechanism for neuronal structural change: LIMK activity is required for both retraction and outgrowth, characteristics of neuronal degeneration and regeneration respectively.
LIMK as a Therapeutic Target in the Retina and Future Research Directions
As we describe above, LIMK-regulated actin turnover is crucial to both axon retraction and neuritic growth of photoreceptors in response to injury. Thus LIMK inhibition has the therapeutic potential for reducing pathological axonal plasticity of rod photoreceptors after retinal injury, stabilizing the photoreceptor-to-bipolar synapse, and preserving retinal circuitry. Unlike antagonists to RhoA or ROCK, which have been used in the central nervous system to increase regenerative axon growth and which in isolated rod cells promote presynaptic varicosity development (Fontainhas and Townes-Anderson, 2008), antagonists to LIMK preserve existing structure: BMS-5, a highly potent and noncytotoxic LIMK inhibitor, with high selectivity for both LIMK 1 & 2, significantly reduced axon retraction and process growth of rod photoreceptor after injury (Wang and Townes-Anderson, 2015). LIMK inhibition therefore may be a useful addition to the Rho drug arsenal for nervous system repair.
LIMK’s effect is expressed through the regulation of cofilin activity. However, phosphorylation of cofilin by LIMK must be accompanied by dephosphorylation for actin turnover to proceed. SSH, the major phosphatase for cofilin, is under dual regulation by calcium signaling, activated by calcineurin and deactivated by Ca2+/calmodulin-dependent protein kinase II (CaMKII). It has been proposed that the calcium binding proteins act together as a switch, to control dephosphorylation of cofilin. Activation and intracellular targeting of SSH is regulated by coronin 1B. Other regulators include PKD (protein kinase D), and the 14-3-3 family of regulatory proteins. In addition, SSH can dephosphorylate LIMK. Improved understanding of the mechanisms of cofilin dephosphorylation may contribute to more effective therapeutics with LIMK antagonists (Bravo-Cordero et al., 2013).
In addition to actin regulation, LIMK also contributes to regulation of microtubule dynamics though a clear mechanism and the immediate substrates are yet to be investigated. Compared to actin filaments, microtubules are relatively stiff. Within neurons, they bundle together to provide transport tracks and to support the lengthy projections of axons. Some actin filaments also exist in axons, but it is the axon terminals where actin filaments are enriched and regulate structural change. At the region where axon shaft meets axon terminal, microtubule and actin filaments interact with each other through motor proteins, such as myosin and dynein. Although many studies on synaptic plasticity focus on actin dynamics because of the flexibility and quick response of the actin cytoskeleton to environmental change, axon retraction cannot occur without microtubule disassembly. Thus, it is important to consider how to modify microtubular structure as well.
It has been suggested that LIMK activity results in microtubule destabilization. In human endothelial cells, for instance, LIMK was found to co-localize with microtubules. Down-regulation of LIMK activity interrupted thrombin-induced microtubule destabilization as well as actin polymerization, whereas activation by ROCK2 caused a decrease in LIMK-tubulin and an increase in LIMK-actin interaction (Gorovoy et al., 2005). Thus, an additional therapeutic benefit of LIMK lies in the fact that it may be a common regulator of both actin and microtubule dynamics. Specifically, inhibition of LIMK may result in an overall stabilization of cytoskeletal structure, including both actin at the synaptic terminal and microtubules in the axon.
More studies are required to define the temporal and spatial relationships of LIMK with other cytoskeletal regulators, such as Pak, ROCK, and SSH, and to increase our understanding of the emerging role for LIMK as a bi-functional structural modifier and therapeutic tool.
Footnotes
Funding: This work was supported by NIH grant EY021542 and the F.M. Kirby Foundation.
Conflicts of interest: None declared.
References
- 1.Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010;67:545–554. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 3.Bravo-Cordero JJ, Magalhaes MA, Eddy RJ, Hodgson L, Condeelis J. Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol. 2013;14:405–415. doi: 10.1038/nrm3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fontainhas AM, Townes-Anderson E. RhoA and its role in synaptic structural plasticity of isolated salamander photoreceptors. Invest Ophthalmol Vis Sci. 2008;49:4177–4187. doi: 10.1167/iovs.07-1580. [DOI] [PubMed] [Google Scholar]
- 5.Fontainhas AM, Townes-Anderson E. RhoA inactivation prevents photoreceptor axon retraction in an in vitro model of acute retinal detachment. Invest Ophthalmol Vis Sci. 2011;52:579–587. doi: 10.1167/iovs.10-5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorovoy M, Niu J, Bernard O, Profirovic J, Minshall R, Neamu R, Voyno-Yasenetskaya T. LIM kinase 1 coordinates microtubule stability and actin polymerization in human endothelial cells. J Biol Chem. 2005;280:26533–26542. doi: 10.1074/jbc.M502921200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heredia L, Helguera P, de Olmos S, Kedikian G, Sola Vigo F, LaFerla F, Staufenbiel M, de Olmos J, Busciglio J, Caceres A, Lorenzo A. Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer’s disease. J Neurosci. 2006;26:6533–6542. doi: 10.1523/JNEUROSCI.5567-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis GP, Charteris DG, Sethi CS, Fisher SK. Animal models of retinal detachment and reattachment: identifying cellular events that may affect visual recovery. Eye (Lond) 2002;16:375–387. doi: 10.1038/sj.eye.6700202. [DOI] [PubMed] [Google Scholar]
- 9.Manetti F. LIM kinases are attractive targets with many macromolecular partners and only a few small molecule regulators. Med Res Rev. 2012;32:968–998. doi: 10.1002/med.20230. [DOI] [PubMed] [Google Scholar]
- 10.Nobes CD, Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem Soc Trans. 1995;23:456–459. doi: 10.1042/bst0230456. [DOI] [PubMed] [Google Scholar]
- 11.Wang W, Townes-Anderson E. Regulation of actin dynamics by RhoA-LIMK-cofilin during axon retraction by rod photoreceptors after retinal injury. (2014 ASCB/IFCB Meeting abstracts #1279) Molecular Biology of the Cell. 2014;25:3987. [Google Scholar]
- 12.Wang W, Townes-Anderson E. LIM Kinase, a newly identified regulator of presynaptic remodeling by rod photoreceptors after injury. Invest Ophthalmol Vis Sci. 2015;56:7847–7858. doi: 10.1167/iovs.15-17278. [DOI] [PMC free article] [PubMed] [Google Scholar]