

Abstract
Coxiella burnetii, the etiological agent of Q fever in humans, is an intracellular pathogen that replicates in an acidified parasitophorous vacuole derived from host lysosomes. Generation of this replicative compartment requires effectors delivered into the host cell by the Dot/Icm type IVb secretion system. Several effectors crucial for C. burnetii intracellular replication have been identified, but the host pathways coopted by these essential effectors are poorly defined, and very little is known about how spacious vacuoles are formed and maintained. Here we demonstrate that the essential type IVb effector, CirA, stimulates GTPase activity of RhoA. Overexpression of CirA in mammalian cells results in cell rounding and stress fiber disruption, a phenotype that is rescued by overexpression of wild-type or constitutively active RhoA. Unlike other effector proteins that subvert Rho GTPases to modulate uptake, CirA is the first effector identified that is dispensable for uptake and instead recruits Rho GTPase to promote biogenesis of the bacterial vacuole. Collectively our results highlight the importance of CirA in coopting host Rho GTPases for establishment of Coxiella burnetii infection and virulence in mammalian cell culture and mouse models of infection.
INTRODUCTION
The naturally obligate intracellular pathogen Coxiella burnetii is the causative agent of Q fever in humans. The agent's high infectivity, ease of spread by aerosols, and environmental stability have led to its classification as a category B select agent by the Centers for Disease Control and Prevention (1). The primary route of infection is through inhalation of contaminated aerosols, which typical results in acute Q fever, a debilitating flu-like illness that is generally self-limiting and readily resolves without antibiotic treatment. However, under some circumstances, persistent infection can lead to chronic Q fever that presents as endocarditis or hepatitis (1). While the number of reported cases of acute Q fever in the United States has been relatively low, a marked increase occurred in 1999 when Q fever became a reportable disease. A recent outbreak of Q fever in The Netherlands resulted in over 4,000 confirmed cases, with 20% of patients requiring hospitalization (2). The dramatic increase in reported cases suggests C. burnetii is an emerging pathogen and highlights our lack of understanding of C. burnetii virulence factors.
Inhalation of C. burnetii by a mammalian host results in actin-dependent endocytosis and internalization in an early endosome. While numerous intracellular pathogens actively subvert the default endocytic pathway to establish a unique host-derived vacuole, C. burnetii generally follows the default trafficking pathway to establish a Coxiella-containing vacuole (CCV) derived from the host lysosomal network. Generation of this unique replicative compartment requires active bacterial protein synthesis which drives homotypic and heterotypic vesicle fusions associated with generation of a spacious CCV that occupies the majority of the host cytoplasmic space (3–5). Manipulation of numerous host cell processes by C. burnetii is required to maintain the CCV and an intracellular environment essential for growth and replication, including inhibition of apoptosis (6), induction of autophagy (4, 7), recruitment of secretory components (3), and modulation of host kinases and phosphatases (8, 9) and involves reprograming of the host transcriptome (10).
Central to pathogenesis is a specialized type IVB secretion system (T4SS) that is homologous to the Dot/Icm secretion system of Legionella pneumophila. In Legionella, this system is composed of 26 dot/icm (defect in organelle trafficking/intracellular multiplication) genes that together form a pilus-like structure that spans the bacterial inner and outer membrane to deliver bacterial proteins into the host cell cytoplasm. Recent advances, including axenic culture (11) and genetic manipulation (12, 13), have enabled experiments that confirm that, like the Legionella secretion system, the Dot/Icm system of C. burnetii is essential for intracellular replication, CCV formation, effector translocation, and modulation of host apoptosis (14, 15).
To date, over 100 T4SS substrates have been identified in C. burnetii using a variety of techniques, including bacterial-two-hybrid (16), bioinformatics (16–19), plasmid localization (20, 21), and genomic (15) assays. Large-scale screening of these substrates demonstrated that many traffic to distinct subcellular compartments, interfere with crucial host processes, and importantly, are less functionally redundant than those of Legionella (15–18, 20–23). While phenotypes identified using these large-scale screens aid in effector characterization, the biological function of most of these substrates remains unknown. Three effector proteins—CaeA, CaeB, and AnkG—were recently reported to play a role in modulating host survival by interfering with apoptosis (6, 24), whereas a fourth effector, CvpA, was shown to engage the clathrin-mediated transport pathway (17).
While these observations provide some insight into how C. burnetii establishes its replicative niche, the molecular details of how the spacious CCV is formed and maintained remains largely unknown. In the present study, we identified a pathogen essential T4SS effector, CirA, which stimulates RhoA GTPase activity to promote vacuolar development. We show that in the absence of CirA, C. burnetii resides in a tight-fitting vacuole that does not expand, is attenuated in virulence, and is significantly impaired in RhoA recruitment to the CCV. Collectively our results highlight the importance of CirA in C. burnetii pathogenesis.
MATERIALS AND METHODS
Bacteria and host cell lines.
The bacteria and yeast strains used for this study are listed in Table S1 in the supplemental material. C. burnetii Nine Mile phase II (NMII) clone 4 (RSA439) was propagated in ACCM-2 under microaerophilic conditions as previously described (11). When required, 350 μg/ml kanamycin or 5 μg/ml chloramphenicol was added.
HeLa (ATCC), HEK293T (ATCC), and J774A.1 (ATCC) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). All cell lines were maintained at 37°C with 5% CO2.
Complementation of the cirA::Tn mutant.
The 1169P-Kanr cassette was PCR amplified from pMiniTn7-Kan (12) and cloned into pKM244 (18) to generate pEVS101. The open reading frame (ORF) Cbu0041 (cirA) was PCR amplified and cloned into the SalI/KpnI of pCMV-DYKDDDDK-C (Clontech) to generate a C-terminal fusion to Flag tag. The resulting fusion protein was amplified and cloned into the SalI/SphI site to generate pEVS101-CirA.
Axenically cultured RSA439 MK2 (cirA::Tn) cells (18) were washed twice and resuspended in distilled water to approximately 1010/ml. Electrocompetent bacteria were mixed with 2 μg pEVS101-CirA and electroporated as previously described (11). After 7 days, individual colonies were expanded in ACCM-2 and screened by Western blotting for expression of the Flag-tagged fusion.
To determine if plasmid complementation of RSA439 MK2 rescued the observed growth defect, HeLa or J774A.1 cells were infected with a multiplicity of infection (MOI) of 50, and genomic equivalents were isolated at 1 day and 4 days postinfection and analyzed by quantitative PCR as previously described (18). For immunofluorescence, HeLa cells were infected in triplicate with an MOI of 50, and at 5 days postinfection, cells were fixed with 4% formaldehyde for 10 min, and nuclei were visualized with 1× Hoechst. Fluorescence images were acquired with a Nikon-A1 microscope using the 60× oil immersion object, and images were processed using NIS-Elements software.
To determine if CirA is necessary for virulence, we employed a SCID mouse model of C. burnetii (NMII) infection. Five C57BL/6 SCID mice (Harlan) per group were intraperitoneally (i.p.) infected with 106 cells of wild-type RSA439, the cirA::Tn mutant, or the cirA::Tn complemented mutant. Mice were observed for clinical signs of infection daily, and at 14 days postinfection, spleens were harvested and homogenized as previously described (25). Total DNA was isolated and purified using Roche Genomic DNA kits, and genome equivalents were determined using quantitative PCR (18). All experimental procedures with animals were approved by the Texas A&M University Institutional Animal Care Use Committee and carried out in approved facilities in accordance with university and federal regulations.
Immunofluorescence.
To assess localization of CirA, HeLa cells were seeded in triplicate into 24-well glass bottom plates at 105/ml. Cells were transfected with Lipofectamine following the manufacturer's instructions. Twenty-four hours posttransfection for rounding experiments or 15 h posttransfection for colocalization studies, cells were fixed with 4% formaldehyde for 10 min and permeabilized with 0.1% Triton X-100 for 5 min, and nuclei were visualized with 1× Hoechst. To assess colocalization with Rho GTPases and to monitor suppression of toxicity, cells were cotransfected with enhanced green fluorescent protein (EGFP)-CirA and RhoA, RhoA Q63L, RhoA T19N, or Rac1 (see Table S2 in the supplemental material) and stained with anti-myc antibody (Thermo) and Alexa Fluor 555 secondary antibodies (Cell Signaling). Actin was visualized using Alexa Fluor 555- or Alexa Fluor 647-conjugated phalloidin (Life Technologies). Cells possessing at least two filaments coursing through the cytosol were considered positive for stress fibers, whereas cells possessing less than two were considered negative for stress fibers. Results were tabulated from triplicate wells with at least 50 cotransfected cells per experiment. Similar results were obtained from at least three independent experiments. For RhoA recruitment experiments, the RhoA pixel intensity by the CCV (within 2 μM) was calculated using Nikon Elements software.
Yeast suppressor screen to identify host proteins capable of suppressing CirA toxicity.
cirA was introduced into pYesNTA2 as BamHI/SalI fragments, and toxicity was assessed by serial dilution and spotting on uracil dropout medium containing galactose as the sole carbon source (18). The pYEp13 genomic library (ATCC no. 37323) was introduced into Saccharomyces cerevisiae W303(pGal::cirA), and the resulting transformants were plated on uracil leucine dropout medium containing galactose as previously described (26). From a transformation yielding 2.0 × 106 transformants, 53 potential suppressors were isolated. To verify suppression, plasmids were isolated and retransformed into S. cerevisiae W303(pGal::cirA). Of these, 11 (pSup1 to pSup11) consistently suppressed the toxicity of CirA. Suppressor plasmids were sequenced using pYEp13 seq F (ACTACGCGATCATGGCGA) and pYEp13 seq R (TGATGCCGGCCACGATGC), and the results were analyzed using the yeast genome database (http://yeastgenome.org).
Individual ORFs from the plasmids that consistently suppressed toxicity (pSup1 to pSup7) were introduced into p415ADH (27) as BamHI/SalI or PstI/SmaI fragments. Individual plasmids were transformed into W303(pGal::cirA), and the resulting transformants were serially diluted and spotted on uracil leucine dropout medium.
GTPase assay.
GTPase assays were performed as previously described (28). His-tagged RhoA and Rac1 were purchased from Cytoskeleton, Inc., and preloaded with 10 mCi of [γ-32P]GTP. His-tagged CirA or His-tagged vector was purified from yeast as previously described (26). Preloaded Rho GTPase was mixed with His-tagged CirA, His-tagged vector, or His-tagged Rho GTPas-activating protein (GAP) as a positive control. At 5-min intervals, aliquots were removed and filtered through nitrocellulose filters. Filters were washed and dried, and the amount of radioactive nucleotide remaining was determined by scintillation counting.
Uptake assay.
To rule out differences in uptake between the strains, bacteria were stained with carboxyfluorescein succinimidyl ester (CFSE) following the manufacturer's guidelines (Life Technologies). Bacteria were enumerated as previously described (18), and an MOI of 10 was used to infect J774A.1 macrophages seeded at 105/ml in glass bottom 24-well dishes. Four hours postinfection, cells were washed 3 times with phosphate-buffered saline (PBS), and extracellular fluorescence was quenched using trypan blue. Cells were counted using a Nikon-A1 microscope using the 60× oil immersion objective lens. At least 200 cells were counted per well, with at least three wells per experiment. Similar results were obtained from at least three independent experiments. The percentage of infection was tabulated by counting the number of infected cells compared with the total number of cells.
Data analysis.
For statistical analysis, data were analyzed using one-way analysis of variance (ANOVA) or with Student's t test and generated a minimum threshold of significance of P < 0.05. When required, Dunnett's test was used as a posttest to compare all samples to the control. All statistical analyses were conducted using GraphPad Prism.
RESULTS
CirA is necessary for virulence.
We previously reported the identification of more than 80 T4SS substrates (16, 18), of which mutants with mutations in 10 of them exhibited an intracellular growth defect (18). Coinfection with wild-type bacteria rescued the growth defect for five of these transposon mutants, suggesting that the observed growth defect is due to the loss of the individual effector proteins, which we designated Coxiella effector for intracellular replication (CirA to -E).
To further substantiate that the observed growth defect is due to the loss of CirA, we complemented RSA439 MK2 (cirA::Tn) (18). As previously reported, cirA::Tn had a phenotype comparable to that of a Dot/Icm mutant and did not replicate in HeLa or J774A.1 cells (18). Plasmid complementation of cirA::Tn with an inducible N-terminally FLAG-tagged CirA resulted in visible formation of a spacious vacuole comparable to that in an intergenic control (Fig. 1A). Growth comparison, as determined by genome equivalents, demonstrated that the cirA::Tn mutant was significantly impaired in intracellular replication, a defect that was partially rescued by complementation with Flag-CirA (Fig. 1B). The partial complementation was not unexpected as overexpression of an inducible CirA would circumvent the spatial temporal regulation of this effector. Together, these results indicate that the T4SS effector CirA is needed for intracellular replication and CCV formation.
FIG 1.

CirA is necessary for virulence. (A) Confocal images of HeLa cells infected at an MOI of 50 for 5 days with an intergenic Tn mutant (Ig0179-0180), the cirA::Tn mutant, and the cirA::Tn complemented (comp) mutant. Images were captured with a Nikon A1 confocal microscope with at least 500 infected cells observed per experiment. Data are representative of three independent experiments. (B) Replication of wild-type RSA439, the cirA::Tn mutant, and the complemented mutant in HeLa cells infected with an MOI of 50. Genome equivalents (GE) were determined at 4 days using quantitative PCR. Data are representative of three independent experiments. (C) Five C57BL/6 mice per group were i.p. infected, and at 14 days postinfection, spleens were harvested, and bacterial burden was determined by quantitative PCR. (B) Statistical analyses were tabulated using Student's t tests and generated a statistical difference of P < 0.01 (*) for the cirA::Tn mutant compared to the complemented mutant. (C) Statistical analyses were tabulated using one-way ANOVA and generated a statistically significant difference of P < 0.0001 (***) compared to RSA439 or P < 0.05 (*) compared to the CirA::Tn complemented mutant.
To determine if CirA is necessary for virulence, we utilized a SCID mouse model of C. burnetii infection. In line with previous studies (25), mice infected with wild-type RSA439 exhibited a high bacterial burden in the spleen. In contrast, mice infected with the cirA::Tn mutant exhibited a significantly reduced splenic bacterial burden (Fig. 1C), which was not statistically different from infection with the icmX::Tn mutant, a Dot/Icm secretion system mutant previously established as necessary for replication in cell culture (18). Complementation of the cirA::Tn mutant resulted in a significantly higher bacterial burden in the spleen compared to both the cirA::Tn and icmX::Tn mutants. Of note, CirA is the first C. burnetii T4SS substrate to be shown to be essential for virulence in an animal model. Furthermore, our results show for the first time that the Dot/Icm secretion system is required for virulence. Collectively these results establish CirA as a key C. burnetii virulence factor and validate the SCID mouse model as a crucial tool for screening C. burnetii transposon mutants.
Overexpression of CirA leads to cell rounding and disruption of stress fibers.
To develop a mechanistic model for CirA's role in disease, HeLa cells were transfected with EGFP-CirA. At 15 h posttransfection, CirA predominately localized to the plasma membrane with small puncta distributed throughout the cytoplasm and in close proximity to the membrane. In contrast, cells expressing EGFP alone exhibited a uniform localization throughout the cell. Further characterization of CirA overexpression in either Hek293 or HeLa cells at later time points (15 to 24 h posttransfection) (Fig. 2A) resulted in a dramatic phenotype characterized by rapid cell rounding, membrane blebbing, decreased puncta, and detachment from the culture dish (see Movie S1 in the supplemental material). By 24 h posttransfection, 77% of CirA-expressing cells had rounded and detached, whereas only 13% of cells transfected with the negative control (EGFP alone) had rounded, and these cells did not show signs of membrane blebbing or detachment (Fig. 2B; see Movie S2 in the supplemental material). Phalloidin staining revealed that cells transfected with CirA did not exhibit the robust stress fiber formation seen in cells transfected with EGFP alone (Fig. 2C). Only 24% of cells transfected with EGFP-CirA possessed stress fibers, whereas 85% of EGFP-transfected cells exhibited robust stress fiber formation. Collectively these results suggest that overexpression of CirA perturbs the actin cytoskeleton of the host cell.
FIG 2.

Overexpression of CirA in mammalian cells leads to disruption of the cytoskeleton. (A) HeLa cells were transiently transfected with EGFP-CirA or EGFP, and stress fibers were visualized by staining with Alexa Fluor 555-phalloidin. Scale bars in merged images are 10 μm. (B and C) Rounding and stress fiber disruption were assessed 24 h posttransfection by counting at least 150 transfected cells. Data are representative of three independent experiments. Statistical analyses were tabulated using Student's t test and generated a statistically significant difference of P < 0.0001 (***) for EGFP-CirA compared to EGFP alone.
CirA targets the Rho GTPase RhoA.
We previously reported that CirA was toxic when heterologously expressed in yeast (18). To identify the host proteins targeted by CirA, we conducted a yeast suppressor screen to identify proteins that, when overexpressed, were capable of suppressing CirA toxicity. An S. cerevisiae strain expressing CirA from a galactose-inducible promoter was transformed with the pYEp13 yeast genomic library (26). From a transformation yielding 2.0 × 106 transformants, 53 colonies were isolated, 11 (pSup1 to pSup11) of which consistently suppressed the toxicity of CirA (Fig. 3A). Sequencing revealed over half of these clones contained identical plasmids (pSup1 to pSup7), carrying five full-length yeast orf genes and a truncated mms1 gene (Fig. 3A and B). Individual expression of each full-length orf gene revealed that only the small GTPase Rho1 was capable of suppressing CirA toxicity, suggesting that Rho1 is a cellular target of CirA (Fig. 3C).
FIG 3.
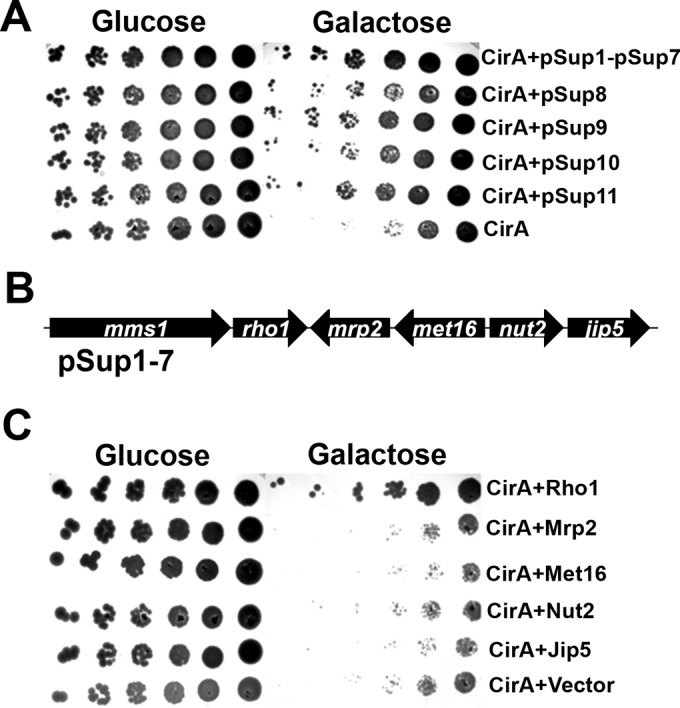
Overexpression of Rho1 suppresses the toxicity of CirA in yeast. (A) S. cerevisiae W303, overexpressing CirA from a galactose-inducible promoter, was transformed with the yeast genomic library pYEp13. Eleven clones (pSup1 to pSup11) were isolated that consistently suppressed the toxicity of CirA. (B) Yeast orf genes present in pSup1 to pSup7. (C) Individual expression of the full-length orf genes in pSup1 revealed that Rho1 is capable of suppressing CirA toxicity. Data are representative of at least three independent experiments.
To assert that our observations in yeast are predictive of mammalian targets, we cotransfected HeLa cells with EGFP-CirA and the mammalian Rho GTPase RhoA (mammalian homology of Rho1) or Rac1. As shown in Fig. 4A to C, coexpression of CirA with RhoA, but not Rac1, rescued stress fiber formation and inhibited rounding to levels comparable to those in EGFP-transfected cells. Furthermore, the Rho GTPase RhoA, but not Rac1, colocalized with CirA (Fig. 5). No colocalization between EGFP and any of the GTPases was noted. These results further support that CirA interacts with the Rho GTPase RhoA.
FIG 4.

Overexpression of RhoA in mammalian cells rescues cell rounding and stress fiber formation. (A to C) HeLa cells were cotransfected with EGFP-CirA or GFP alone or myc-tagged RhoA or Rac1, and stress fibers were visualized by staining with Alexa Fluor 647-conjugated phalloidin. Scale bars in merged images are 10 μm. (B and C) To determine if the GTPase state of Rho is necessary for suppression of CirA toxicity, cells were cotransfected with constitutively active (Q63L) or dominant-negative (T19N) RhoA. Rounding and stress fiber disruption were determined 24 h posttransfection by counting at least 50 transfected cells from triplicate wells. Data are representative of at least three independent experiments. Statistical analyses were tabulated using one-way ANOVA and generated a statistically significant difference of P < 0.0001 (***) or P < 0.05 (**) compared to EGFP alone.
FIG 5.

Ectopically expressed EGFP-CirA colocalizes with Rho GTPases RhoA but not Rac1. Representative micrographs of HeLa cells cotransfected with EGFP-CirA or EGFP (green) and myc-tagged RhoA or Rac1 (red). White boxes denote the areas shown in the insets. Scale bars in merged images are 10 μm. Data are representative of three independent experiments with at least 100 cotransfected cells per experiment.
To establish whether suppression of cell rounding and disruption of stress fibers requires a specific activation state of the Rho GTPase, we cotransfected cells with a GTP-locked (Q63L) or GDP-locked (T19N) RhoA. Cotransfected cells overexpressing a GTP-locked RhoA exhibited reduced cell rounding and robust stress fiber formation compared with CirA alone or cells cotransfected with a GDP-locked RhoA (Fig. 4B and C). The finding that suppression of CirA induced cell rounding and stress fiber disruption requires the GTP-bound form of RhoA suggests that CirA inhibits Rho signaling by inactivating Rho GTPases.
CirA stimulates RhoA GTPase activity.
To establish if CirA is able to stimulate RhoA, we employed a GTPase assay. His-tagged CirA, purified from yeast, was incubated with recombinant His-tagged RhoA or Rac1 preloaded with [γ-32P]GTP. Aliquots were removed at 5-min intervals over 15 min, and the amount of protein bound radioactive nucleotide captured by the filter was measured by scintillation counting. As shown in Fig. 6, His-tagged CirA robustly stimulated the GTPase activity of RhoA similar to the p50 subunit of the Rho GAP-positive control. No activity was detected with Rac1 or when any of the Rho GTPases were incubated with purified His-tagged protein expressed by vector alone from yeast. These results indicate that CirA can stimulate RhoA GTPase activity.
FIG 6.

CirA stimulates RhoA GTPase activity on RhoA. Activity of His-tagged CirA or His-tagged vector toward RhoA or Rac1 was determined using a GTPase assay. Data are representative of three independent experiments.
CirA is dispensable for uptake.
Several pathogenic bacteria secrete effector proteins or toxins that target Rho GTPases to modulate uptake (29, 30). However, the T4SS of C. burnetii may not be active until at least 8 h postinfection and requires CCV acidification and endocytic maturation (31) for appropriate function. This observation theoretically precludes T4SS substrates from playing a role in bacterial uptake or initial stages of endocytic trafficking. We speculate that it is therefore highly unlikely that CirA interacts with Rho GTPases to modulate bacterial uptake. To test this hypothesis, we compared uptake of CFSE-labeled bacteria by J774Al.1 macrophages. As predicted, uptake was not impacted by CirA mutation and was only impaired for the predicted enhC::Tn positive control, a protein homolog previously established to be involved in Legionella uptake (Fig. 7) (32). This observation strongly indicates that CirA does not engage RhoA to modulate bacterial uptake.
FIG 7.
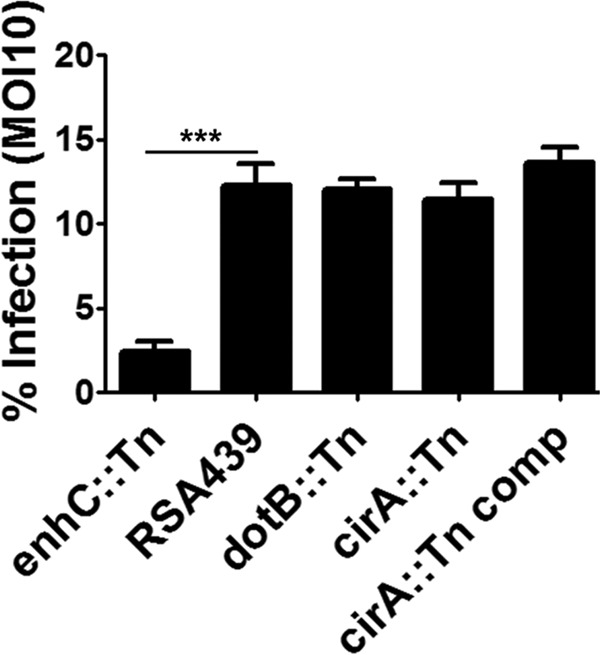
CirA is dispensable for bacterial uptake. J774A.1 macrophages were infected with CFSE-labeled bacteria for 4 h at an MOI of 10. Only the enhC::Tn positive control was deficient in uptake. Data are representative of three independent experiments with at least 600 cells compared from triplicate samples. Statistical analysis were tabulated using one-way ANOVA and generated a statistically significant difference of P < 0.0001 (***).
Rho GTPase recruitment to the CCV requires CirA.
In addition to regulating cytoskeletal rearrangements, Rho GTPases play an integral role in regulating vesicle trafficking through endoplasmic reticulum to the Golgi compartment and endocytic and exocytic transport pathways (33). Importantly, RhoA but not Rac1 is recruited to the CCV and plays an integral role in CCV formation (34). Immunofluorescence analysis of infected HeLa cells revealed that CCVs harboring mutant cirA::Tn bacteria were significantly impaired in their ability to recruit RhoA relative to cells infected with intergenic or cirA::Tn complemented strains (Fig. 8), suggesting that CirA modulates Rho GTPases to promote CCV maturation. We repeated these localization studies with antibody recognizing native RhoA to eliminate potential artifacts inherent in overexpressed, transiently transfected cells. An example of RhoA localization during infection by the wild type is shown in Fig. S3 in the supplemental material.
FIG 8.
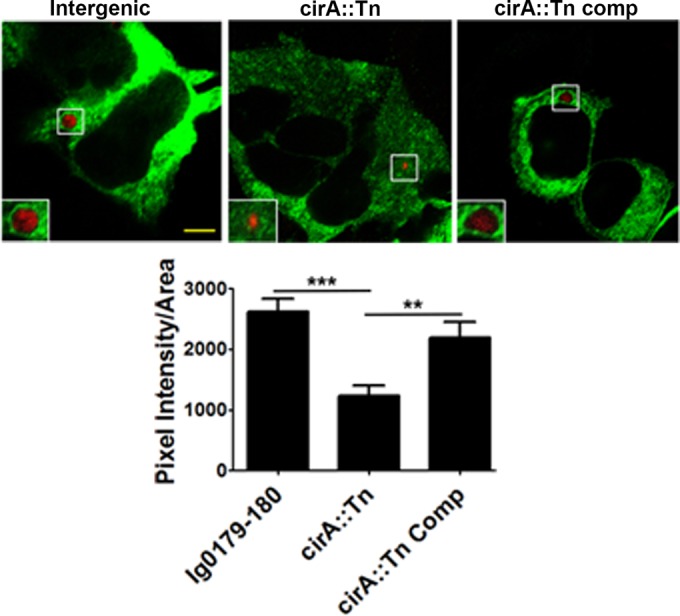
RhoA recruitment to the CCV requires CirA. HeLa cells, infected with an MOI of 100 for 48 h, were transfected with myc-tagged RhoA. Data are representative of two independent experiments with at least 100 infected transfected cells per well and three wells per experiment. RhoA recruitment was quantitated as described in Materials and Methods. Scale bars are 5 μm. White boxes denote the areas shown in the insets. Statistical analyses were tabulated using one-way ANOVA and generated a statistically significant difference of P < 0.0001 (***) compared to Ig0179-180 or P < 0.01 (**) compared to the cirA::Tn complemented mutant (Comp).
DISCUSSION
We previously demonstrated that the T4SS effector CirA is essential for intracellular replication and CCV formation (18). CirA is highly conserved among C. burnetii pathotypes, suggesting that it may be essential for establishment of this obligate intracellular pathogen niche. In the present study, we sought to characterize the role of CirA in pathogenesis. To do so, we employed a yeast suppressor screen to identify host proteins targeted by CirA. We found that CirA is highly toxic in yeast and causes cytopathic effects (stress fiber disruption and rounding) in mammalian cells, and these effects can be suppressed by overexpression of the Rho GTPase RhoA. This suppression requires the GTP-bound state of the GTPase, suggesting that the function of CirA is to interfere with cellular RhoA signaling. Biochemical assays indicate CirA stimulates RhoA GTPase activity. Importantly, we identified CirA as the first C. burnetii T4SS effector protein essential for growth and pathogenesis in an animal model.
Saccharomyces cerevisiae has emerged as a valuable model to characterize bacterial virulence factors due to the conservation of many pathways between yeast and mammals, genetic tractability, and ease of use (35). Heterologous expression of bacterial effector proteins in yeast has been exploited in numerous studies to identify effectors that impair crucial host processes (15, 18, 26, 36, 37). In an attempt to understand the function of these toxic effectors, yeast genetic screens have been employed to identify host proteins that, when overexpressed, suppress the toxicity of the effector protein (26, 38, 39). One key study showed that the lethality associated with L. pneumophila AnkX was suppressed by overexpression of components involved in membrane trafficking, which allowed the investigators to determine that AnkX modifies Rab1 by phosphorylcholination (26). This screening tool has since been extended to characterize two other effectors: LecE, an effector that manipulates phospholipid biosynthesis (39), and Ceg14, an effector that inhibits actin polymerization (38).
Based on the successful application of the yeast suppressor screens for the characterization of Legionella effectors, we chose to employ this tool as a means to identify the host pathways targeted by CirA. Using this approach, we demonstrated that the toxicity of CirA could be suppressed by overexpression of the Rho GTPase Rho1, suggesting this is the pathway targeted by CirA. Rho GTPases are targeted by many bacterial effector proteins, including Yersinia pseudotuberculosis YopE (30), Salmonella enterica serovar Typhimurium SopE (40), Vibrio cholerae RTX toxin (41), and Escherichia coli EspH (29), indicating this is a conserved target of bacterial pathogens (42). Bacterial virulence factors and toxins regulate Rho activity using at least one of three methods (42): (i) through indirect regulation of localization and activation by mimicking Rho GAPs, guanine nucleotide exchange factors (GEFs), or guanine nucleotide dissociation inhibitors (GDIs) (30, 43); (ii) direct regulation through posttranslational modifications, including ADP-ribosylation (44), adenylation (45), deamidation (46), and glucosylation (47); (iii) or through targeting upstream regulators (29, 48). Consequently these modifications affect Rho GTPase activity and ultimately impact host signaling pathways, resulting in inhibition of immune function or endocytosis, generating a more permissive host for the pathogen.
Many pathogenic bacteria target cytoskeletal components, including actin, intermediate filaments, and microtubules, and to modulate invasion, establish a replicative niche, and facilitate cell-to-cell spread through actin-based motility, as well as for dissemination (49–51). Specifically, several pathogens secrete T3S effectors that target Rho GTPases to modulate uptake. Salmonella releases at least four effector proteins to modulate actin rearrangements through Rho GTPases: SopE and SopE2 directly activate GTPases by mimicking Rho GEFs (52, 53), whereas SopB indirectly activates Rho GTPases through its interaction with inositol phosphatase (54). Following actin polymerization, SptP downregulates cytoskeletal rearrangements induced by other type III secretion (T3S) effectors by acting as a Rac1 and Cdc42 GAP. This tightly balanced cycle acts to rebuild the cytoskeleton following invasion (40). In contrast to the model exemplified by Salmonella, Yersinia secretes an array of antiphagocytic T3S effectors that disrupts the cytoskeleton to block uptake: most notable is YopE, which targets RhoA, Rac1, and Cdc42 by mimicking Rho GAPs (30). While targeting of Rho GTPases to modulate uptake is a common theme among secreted effectors, C. burnetii CirA uniquely targets Rho GTPases post-uptake. Our results suggest that CirA may be involved in formation of the spacious CCV as both F-actin and Rho GTPases are recruited to the CCV and are crucial for normal development (34). F-actin assembles on phagosomes, which serves as a delivery network on which lysosomes or late endosomes can progress to ultimately fuse with phagosomes (34, 55). This suggests that recruitment of Rho GTPases may facilitate heterotypic fusions with endosomes and lysosomes as well as homotypic fusions with other small CCVs and may serve as a method for delivering membrane to the growing vacuole (34).
Regulation of numerous cell processes, including vesicle trafficking, phagocytosis, cell motility, and cell adhesion, requires rapid remodeling of the actin cytoskeleton (33, 56). These processes are tightly controlled and regulated by numerous signaling molecules, including Rho GTPases, which provide directionality for trafficking of cargo on actin tracks. In yeast, Golgi compartment-derived vesicles are directed toward the emerging bud through dramatic reorganization of the actin cytoskeleton (57). This redirection of vesicle trafficking requires trafficking of both the GTPase and its cognate regulators in the vesicle to the designated target site. For instance, the Rho GAP bud emergence protein 3 (bem3) from yeast resides in recycling endosomes and actively facilitates delivery of secretory vesicles to the bud tip. While the exact mechanism for bem3 activity is unclear, it highlights the involvement of Rho GTPases in vesicle trafficking. In animal cells, Rho GTPases not only associate with the plasma membrane but also localize to numerous intracellular compartments where they regulate endosome recycling, clathrin-dependent and -independent endocytosis, and Golgi compartment-to-ER transport. How Rho GTPases control membrane trafficking is ill defined; however, actin reorganization appears to be crucial (58).
Collectively, our results support a model in which CirA is essential for pathogenesis and the formation of the CCV and targets RhoA. The localization of ectopically expressed CirA to the plasma membrane suggests that CirA may reside on the CCV during infection. Our data suggest that CirA may engage RhoA on the CCV and stimulate its GTPase activity. The toxic nature of CirA when ectopically expressed in yeast and the cytopathic effects in mammalian cells suggest that regulation of this effector is tightly controlled, as C. burnetii is described as a stealth pathogen invading and replicating undetected by the host cells. The importance of spatiotemporal regulation of toxic effectors has been highlighted byusing an example in L. pneumophila where the toxicity of SidE is suppressed by another effector SidJ (59). It is likely that functions of CirA are tightly controlled during C. burnetii infection to prevent the toxic effects seen when this effector is overexpressed. The spatiotemporal regulation of CirA and the consequences during infection are under investigation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ted Hackstadt and Paul de Figueiredo for helpful discussions and for critical review of the manuscript. We also thank Paul Beare for providing reagents.
This work was supported by National Institute of Allergy and Infectious Diseases, National Institutes of Health, grants AI088430 (J.E.S.), K02AI085403 (Z.-Q.L.), and AI090142 (J.E.S. and Z.-Q.L.) and by Department of Defense, Defense Threat Reduction Agency, grant HDTRA1-13-1-0003 (J.E.S.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01554-15.
REFERENCES
- 1.van Schaik EJ, Chen C, Mertens K, Weber MM, Samuel JE. 2013. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat Rev Microbiol 11:561–573. doi: 10.1038/nrmicro3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Hoek W, Schneeberger PM, Oomen T, Wegdam-Blans MC, Dijkstra F, Notermans DW, Bijlmer HA, Groeneveld K, Wijkmans CJ, Rietveld A, Kampschreur LM, van Duynhoven Y. 2012. Shifting priorities in the aftermath of a Q fever epidemic in 2007 to 2009 in The Netherlands: from acute to chronic infection. Euro Surveill 17:20059. [PubMed] [Google Scholar]
- 3.Campoy EM, Zoppino FCM, Colombo MI. 2011. The early secretory pathway contributes to the growth of the Coxiella-replicative niche. Infect Immun 79:402–413. doi: 10.1128/IAI.00688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romano PS, Gutierrez MG, Berón W, Rabinovitch M, Colombo MI. 2007. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell Microbiol 9:891–909. doi: 10.1111/j.1462-5822.2006.00838.x. [DOI] [PubMed] [Google Scholar]
- 5.Howe D, Melnicáková J, Barák I, Heinzen RA. 2003. Maturation of the Coxiella burnetii parasitophorous vacuole requires bacterial protein synthesis but not replication. Cell Microbiol 5:469–480. doi: 10.1046/j.1462-5822.2003.00293.x. [DOI] [PubMed] [Google Scholar]
- 6.Lührmann A, Nogueira CV, Carey KL, Roy CR. 2010. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc Natl Acad Sci U S A 107:18997–19001. doi: 10.1073/pnas.1004380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winchell CG, Graham JG, Kurten RC, Voth DE. 2014. Coxiella burnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect Immun 82:2229–2238. doi: 10.1128/IAI.01236-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voth DE, Heinzen RA. 2009. Sustained activation of Akt and Erk1/2 is required for Coxiella burnetii antiapoptotic activity. Infect Immun 77:205–213. doi: 10.1128/IAI.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hussain SK, Broederdorf LJ, Sharma UM, Voth DE. 2010. Host kinase activity is required for Coxiella burnetii parasitophorous vacuole formation. Front Microbiol 1:137. doi: 10.3389/fmicb.2010.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahapatra S, Ayoubi P, Shaw EI. 2010. Coxiella burnetii Nine Mile II proteins modulate gene expression of monocytic host cells during infection. BMC Microbiol 10:244–257. doi: 10.1186/1471-2180-10-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Omsland A, Beare PA, Hill J, Cockrell DC, Howe D, Hansen B, Samuel JE, Heinzen RA. 2011. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl Environ Microbiol 77:3720–3725. doi: 10.1128/AEM.02826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beare PA, Larson CL, Gilk SD, Heinzen RA. 2012. Two systems for targeted gene deletion in Coxiella burnetii. Appl Environ Microbiol 78:4580–4589. doi: 10.1128/AEM.00881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beare PA, Howe D, Cockrell DC, Omsland A, Hansen B, Heinzen RA. 2009. Characterization of a Coxiella burnetii ftsZ mutant generated by Himar1 transposon mutagenesis. J Bacteriol 191:1369–1381. doi: 10.1128/JB.01580-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVb secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2:e00175-11. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carey KL, Newton HJ, Lührmann A, Roy CR. 2011. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog 7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, Tan Y, Luo Z-Q, Samuel JE. 2010. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc Natl Acad Sci U S A 107:21755–21760. doi: 10.1073/pnas.1010485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larson CL, Beare PA, Howe D, Heinzen RA. 2013. Coxiella burnetii effector protein subverts clathrin-mediated vesicular trafficking for pathogen vacuole biogenesis. Proc Natl Acad Sci U S A 110:e4770–e4779. doi: 10.1073/pnas.1309195110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber MM, Chen C, Rowin K, Mertens K, Galvan G, Zhi H, Dealing CM, Roman VA, Banga S, Tan Y, Luo Z-Q, Samuel JE. 2013. Identification of Coxiella burnetii type IV secretion substrates required for intracellular replication and Coxiella-containing vacuole formation. J Bacteriol 195:3914–3924. doi: 10.1128/JB.00071-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan X, Lührmann A, Satoh A, Laskowski-Arce MA, Roy CR. 2008. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science 320:1651–1654. doi: 10.1126/science.1158160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maturana P, Graham JG, Sharma UM, Voth DE. 2013. Refining the plasmid-encoded type IV secretion system substrate repertoire of Coxiella burnetii. J Bacteriol 195:3269–3276. doi: 10.1128/JB.00180-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voth DE, Beare PA, Howe D, Sharma UM, Samoilis G, Cockrell DC, Omsland A, Heinzen RA. 2011. The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV secretion system substrates. J Bacteriol 193:1493–1503. doi: 10.1128/JB.01359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larson CL, Beare PA, Voth DE, Howe D, Cockrell DC, Bastidas RJ, Valdivia RH, Heinzen RA. 2015. Coxiella burnetii effector proteins that localize to the parasitophorous vacuole membrane promote intracellular replication. Infect Immun 83:661–670. doi: 10.1128/IAI.02763-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newton HJ, Kohler LJ, McDonough JA, Temoche-Diaz M, Crabill E, Hartland EL, Roy CR. 2014. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog 10:e1004286. doi: 10.1371/journal.ppat.1004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klingenbeck L, Eckart RA, Berens C, Lührmann A. 2012. The Coxiella burnetii type IV secretion system substrate CaeB inhibits intrinsic apoptosis at the mitochondrial level. Cell Microbiol 15:675–687. doi: 10.1111/cmi.12066. [DOI] [PubMed] [Google Scholar]
- 25.Andoh M, Zhang G, Russell-Lodrigue KE, Shive HR, Weeks BR, Samuel JE. 2007. T cells are essential for bacterial clearance, and gamma interferon, tumor necrosis factor alpha, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect Immun 75:3245–3255. doi: 10.1128/IAI.01767-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan Y, Arnold RJ, Luo Z. 2011. Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proc Natl Acad Sci U S A 108:21212–21217. doi: 10.1073/pnas.1114023109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mumberg D, Müller R, Funk M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 28.Self AJ, Hall A. 1995. Measurement of intrinsic nucleotide exchange and GTP hydrolysis rates. Methods Enzymol 256:67–76. doi: 10.1016/0076-6879(95)56010-6. [DOI] [PubMed] [Google Scholar]
- 29.Dong N, Liu L, Shao F. 2010. A bacterial effector targets host DH-PH domain RhoGEFs and antagonizes macrophage phagocytosis. EMBO J 29:1363–1376. doi: 10.1038/emboj.2010.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Black DS, Bliska JB. 2000. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol Microbiol 37:515–527. [DOI] [PubMed] [Google Scholar]
- 31.Newton HJ, McDonough JA, Roy CR. 2013. Effector protein translocation by the Coxiella burnetii Dot/Icm type IV secretion system requires endocytic maturation of the pathogen-occupied vacuole. PLoS One 8:e54566. doi: 10.1371/journal.pone.0054566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cirillo SLG, Lum J, Cirillo JD. 2000. Identification of novel loci involved in entry by Legionella pneumophila. Microbiology 146:1345–1359. doi: 10.1099/00221287-146-6-1345. [DOI] [PubMed] [Google Scholar]
- 33.Chi X, Wang S, Huang Y, Stamnes M, Chen J-L. 2013. Roles of Rho GTPases in intracellular transport and cellular transformation. Int J Mol Sci 14:7089–7108. doi: 10.3390/ijms14047089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aguilera M, Salinas R, Rosales E, Carminati S, Colombo MI, Berón W. 2009. Actin dynamics and Rho GTPases regulate the size and formation of parasitophorous vacuoles containing Coxiella burnetii. Infect Immun 77:4609–4620. doi: 10.1128/IAI.00301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valdivia RH. 2004. Modeling the function of bacterial virulence factors in Saccharomyces cerevisiae. Eukaryot Cell 3:827–834. doi: 10.1128/EC.3.4.827-834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo Z-Q. 2010. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog 6:e1000822. doi: 10.1371/journal.ppat.1000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banga S, Gao P, Shen X, Fiscus V, Zong W-X, Chen L, Luo Z-Q. 2007. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc Natl Acad Sci U S A 104:5121–5126. doi: 10.1073/pnas.0611030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Z, Stephenson R, Qiu J, Zheng S, Luo Z-Q. 2014. A Legionella effector modulates host cytoskeletal structure by inhibiting actin polymerization. Microbes Infect 16:225–236. doi: 10.1016/j.micinf.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Viner R, Chetrit D, Ehrlich M, Segal G. 2012. Identification of two Legionella pneumophila effectors that manipulate host phospholipids biosynthesis. PLoS Pathog 8:e1002988. doi: 10.1371/journal.ppat.1002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu Y, Galán JE. 1999. A Salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature 401:293–297. doi: 10.1038/45829. [DOI] [PubMed] [Google Scholar]
- 41.Sheahan K-L, Satchell KJF. 2007. Inactivation of small Rho GTPases by the multifunctional RTX toxin from Vibrio cholerae. Cell Microbiol 9:1324–1335. doi: 10.1111/j.1462-5822.2006.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lemichez E, Aktories K. 2013. Hijacking of Rho GTPases during bacterial infection. Exp Cell Res 319:2329–2336. doi: 10.1016/j.yexcr.2013.04.021. [DOI] [PubMed] [Google Scholar]
- 43.Arbeloa A, Garnett J, Lillington J, Bulgin RR, Berger CN, Lea SM, Matthews S, Frankel G. 2010. EspM2 is a RhoA guanine nucleotide exchange factor. Cell Microbiol 12:654–664. doi: 10.1111/j.1462-5822.2009.01423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cassel D, Pfeuffer T. 1978. Mechanism of cholera toxin action: covalent modification of the guanyl nucleotide-binding protein of the adenylate cyclase system. Proc Natl Acad Sci U S A 75:2669–2673. doi: 10.1073/pnas.75.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murata T, Delprato A, Ingmundson A, Toomre DK, Lambright DG, Roy CR. 2006. The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat Cell Biol 8:971–977. doi: 10.1038/ncb1463. [DOI] [PubMed] [Google Scholar]
- 46.Lerm M, Selzer J, Hoffmeyer A, Rapp UR, Aktories K, Schmidt G. 1999. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of c-Jun N-terminal kinase in HeLa cells. Infect Immun 67:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teichert M, Tatge H, Schoentaube J, Just I, Gerhard R. 2006. Application of mutated Clostridium difficile toxin A for determination of glucosyltransferase-dependent effects. Infect Immun 74:6006–6010. doi: 10.1128/IAI.00545-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orth JHC, Preuss I, Fester I, Schlosser A, Wilson BA, Aktories K. 2009. Pasteurella multocida toxin activation of heterotrimeric G proteins by deamidation. Proc Natl Acad Sci U S A 106:7179–7184. doi: 10.1073/pnas.0900160106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haglund CM, Welch MD. 2011. Pathogens and polymers: microbe-host interactions illuminate the cytoskeleton. J Cell Biol 195:7–17. doi: 10.1083/jcb.201103148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lutter EI, Barger AC, Nair V, Hackstadt T. 2013. Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Rep 3:1921–1931. doi: 10.1016/j.celrep.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar Y, Valdivia RH. 2008. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hardt WD, Chen LM, Schuebel KE, Bustelo XR, Galán JE. 1998. S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93:815–826. doi: 10.1016/S0092-8674(00)81442-7. [DOI] [PubMed] [Google Scholar]
- 53.Stender S, Friebel A, Linder S, Rohde M, Mirold S, Hardt WD. 2000. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol Microbiol 36:1206–1221. [DOI] [PubMed] [Google Scholar]
- 54.Terebiznik MR, Vieira OV, Marcus SL, Slade A, Yip CM, Trimble WS, Meyer T, Finlay BB, Grinstein S. 2002. Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat Cell Biol 4:766–773. doi: 10.1038/ncb854. [DOI] [PubMed] [Google Scholar]
- 55.Kjeken R, Egeberg M, Habermann A, Kuehnel M, Peyron P, Floetenmeyer M, Walther P, Jahraus A, Kuznetsov SA, Griffiths G. 2004. Fusion between phagosomes, early and late endosomes: a role for actin in fusion between late, but not early endocytic organelles. Mol Biol Cell 15:345–358. doi: 10.1091/mbc.E03-05-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ridley AJ. 2006. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol 16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 57.Mukherjee D, Sen A, Aguilar RC. 2014. RhoGTPase-binding proteins, the exocyst complex and polarized vesicle trafficking. Small GTPases 5:e28453. doi: 10.4161/sgtp.28453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yalovsky S, Bloch D, Sorek N, Kost B. 2008. Regulation of membrane trafficking, cytoskeleton dynamics, and cell polarity by ROP/RAC GTPases. Plant Physiol 147:1527–1543. doi: 10.1104/pp.108.122150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jeong KC, Sexton JA, Vogel JP. 2015. Spatiotemporal regulation of a Legionella pneumophila T4SS substrate by the metaeffector SidJ. PLoS Pathog 11:e1004695. doi: 10.1371/journal.ppat.1004695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.