

Summary
RUNX1 (previously termed AML1) is a frequent target of human leukaemia-associated gene aberrations, and it encodes the DNA-binding subunit of the Core-Binding Factor transcription factor complex. RUNX1 expression is essential for the initiation of definitive haematopoiesis, for steady-state thrombopoiesis, and for normal lymphocytes development. Recent studies revealed that protein arginine methyltransferase 1 (PRMT1), which accounts for the majority of the type I PRMT activity in cells, methylates two arginine residues in RUNX1 (R206 and R210), and these modifications inhibit corepressor-binding to RUNX1 thereby enhancing its transcriptional activity. In order to elucidate the biological significance of these methylations, we established novel knock-in mouse lines with non-methylable, double arginine-to-lysine (RTAMR-to-KTAMK) mutations in RUNX1. Homozygous Runx1KTAMK/KTAMK mice are born alive and appear normal during adulthood. However, Runx1KTAMK/KTAMK mice showed a reduction in CD3+ T lymphoid cells and a decrease in CD4+ T cells in peripheral lymphoid organs, in comparison to their wild-type littermates, leading to a reduction in the CD4+ to CD8+ T-cell ratio. These findings suggest that arginine-methylation of RUNX1 in the RTAMR-motif is dispensable for the development of definitive haematopoiesis and for steady-state platelet production, however this modification affects the role of RUNX1 in the maintenance of the peripheral CD4+ T-cell population.
Keywords: RUNX1, methylation, lymphocyte differentiation, transgenic mice models, transcription factor, leukaemia
Introduction
Runt-related transcription factor 1 (RUNX1; previously termed Acute Myeloid Leukaemia 1: AML1) was originally cloned from the breakpoint of chromosome 21 found in t(8;21)(q22;q22) of cases classified as French-American-British (FAB) subtype acute myeloid leukaemia (AML)-M2 (Miyoshi et al, 1991; Erickson et al, 1992; Miyoshi et al, 1993) and now recognized as one of the most frequently mutated genes associated with human leukaemia (Rabbitts, 1994; Look, 1997). RUNX1 encodes the DNA-binding subunit of the hetero-dimeric transcription factor complex, Core-Binding Factor (CBF) (Ogawa et al, 1993; Wang et al, 1993), and is a Class II leukaemia-associated mutation-target gene (Gilliland, 2001). RUNX1 serves as a transcriptional activator as well as a repressor to its target genes, depending on the cellular context, mediated through its interaction with co-factors: transcriptional co-activator(s) and/or co-repressor(s) (Frank et al, 1995; Lutterbach and Hiebert, 2000; Okuda et al, 2001; Speck, 2001).
RUNX1 plays an essential role in the generation of definitive haematopoietic stem cells (HSC) from the haemogenic endothelium of the dorsal aorta during mid-gestation, through a mechanism called endothelial-haematopoietic cell transition: thus the constitutional disruption of this gene blocks this process, resulting in the complete lack of fetal liver definitive haematopoiesis in mice (Okuda et al, 1996; Wang et al, 1996; North et al, 1999; North et al, 2002). In addition, observation of haploinsufficient mice and conditional Runx1-knockout experiments in adult animals, revealed that this transcription factor also plays important roles in steady state haematopoiesis: Loss of RUNX1 in adult animals leads to a slight increase of circulating and bone marrow myeloid precursor cells, due to altered balance between self-renewing and differentiation of the progenitor cells (Ichikawa et al, 2004; Sun and Downing, 2004; Growney et al, 2005; Putz et al, 2006). Similarly, abnormalities of megakaryocytic maturation with reduction of circulating platelets were observed in mutant mice following the MX-Cre- and polyinosinic/polycytidylic acid (pIpC)-induced disruption of the floxed-Runx1 gene of whole haematopoietic system (Ichikawa et al, 2004; Growney et al, 2005; Putz et al, 2006). In addition, B- and T-lymphocyte development was impaired in mice with conditional disruption (Taniuchi et al, 2002; Egawa et al, 2007; He et al, 2008; Setoguchi et al, 2008; Seo et al, 2012), pointing to the cell lineages where RUNX1 has essential biological roles in adult haematopoiesis (Ichikawa et al, 2004; Growney et al, 2005; Putz et al, 2006).
These biological functions of RUNX1 appear to correlate with its trans-activation activity because re-expression of transcriptionally-active RUNX1 is necessary and sufficient for rescuing all haematopoietic defects found in the Runx1-knockout animals or embryonic stem (ES)/primary cells, while transcriptionally inactive forms of this molecule did not rescue these defects (Okuda et al, 2000; Goyama et al, 2004; Nishimura et al, 2004; Fukushima-Nakase et al, 2005; Dowdy et al, 2013).
How the activity of RUNX1 is regulated remains obscure. Apparently, the expression level of the RUNX1 protein is important: Up-regulation of RUNX1 transcription, induced by mechanical sheer stress in the ventral endothelium of the dorsal aorta, is important to generate HSC during mid-gestation (Adamo et al, 2009; North et al, 2009). Translational regulation through its internal ribosomal entry site (IRES) element also plays a role (Nagamachi et al, 2010). Nonetheless, mechanisms through which the activity of the RUNX1 protein is regulated remain to be fully revealed.
Several research groups have recently shown that RUNX1 protein is processed with post-translational modifications, including phosphorylation (Tanaka et al, 1996; Zhang et al, 2004; Aikawa et al, 2006; Biggs et al, 2006; Wee et al, 2008; Zhang et al, 2008; Huang et al, 2012), acetylation (Yamaguchi et al, 2004; Nguyen et al, 2005; Wang et al, 2011), ubiquitination (Huang et al, 2001; Shang et al, 2009) and methylation (Chakraborty et al, 2003; Zhao et al, 2008). Importantly, these modifications are influential in the transcriptional-activating ability of RUNX1. Phosphorylation of serine/threonine residues by several kinases, including extracellular-signal kinases (ERKs), homeodomain-interacting protein kinases (HIPs) and cyclin-dependent kinases (CDKs), are reported to be key modifications that regulate RUNX1 transcription-activity. Similarly, some tyrosine residues in RUNX1 are phosphorylated by Src kinase. In addition, several arginine residues in RUNX1 have been recently reported as methylated (Zhao et al, 2008; Vu et al, 2013). Two arginine residues just 3′ of its runt-domain, R206 and R210, are methylated by protein arginine methyl-transferase 1 (PRMT1), and this methylation dissociates the transcription co-repressor, SIN3A, from RUNX1. Thus PRMT1 serves as a transcriptional co-activator for RUNX1 (Zhao et al, 2008). A second methylation event in RUNX1, at R223, by CARM1 (aka PRMT4), triggers the binding of a multi-protein repressor complex that allows RUNX1 to repress the expression of micro-RNA 223 (miR-223), that negatively regulates translation of a number of myeloid target-genes which functions to keep undifferentiated phenotype of haematopoietic progenitor cells (Vu et al, 2013). Alteration of RUNX1’s biochemical properties by these post-translational modifications regulates its activity in multiple in vitro and in vivo assays. However, the biological significance of these modifications on the function of RUNX1 during fetal or adult haematopoiesis remains to be elucidated (Table S1).
In this study, we focused on the R206 and R210 sites of arginine-methylation as an initial step towards a comprehensive understanding of how RUNX1 is regulated through these modifications, as both residues have been identified to be actually methylated in vivo, which increased RUNX1 transcriptional activity to a number of target gene promoters (Zhao et al, 2008). In order to elucidate biological significance of these methylations, we performed in vitro culture experiments using ES cell clones that contain non-methylatable, double arginine-to-lysine (KTAMK) mutations of RUNX1 at these residues. In addition, we generated genome manipulated mouse lines that express the mutant RUNX1 KTAMK molecule. Our results indicate that arginine methylation of RUNX1 is dispensable for RUNX1 function in definitive haematopoiesis and steady-state platelet production, but plays an indispensable role in the proper maintenance of the peripheral T cell population.
Materials and methods
The flow chart representing the methodology and strategy applied in this study is shown in Figure S1.
Construction of the mutant knock-in targeting vectors
We used the full-length mouse cDNA for the Runx1 gene, a mouse equivalent to the human AML1b isoform, as the backbone for mutagenesis to construct cDNAs with arginine-to-lysine substitution(s) at amino acid residue 206 (R206K mutant), R210K mutant and R206K/R210K-double mutant (KTAMK-double mutant) (Fig 1A and S2) by means of polymerase chain reaction (PCR)-based site-directed mutagenesis (PrimeSTAR Mutagenesis Kit, Takara Bio Inc., Shiga, Japan). The oligonucleotide primer pair used for the generating R206K-mutant was 5′-CTGCGGAAGACGGCCATGAGGGTCAGC-3′ (sense) and 5′-GGCCGTCTTCCGCAGCTGCTCCAATTC-3′ (antisense), where the underlined nucleotides indicate the mutated residues to introduce codon substitution(s). Similarly, the representative primer pairs for the R210K-mutant and the R206K/R210K-double mutant were 5′- GCCATGAAGGTCAGCCCGCACCACCC -3′ (sense) and 5′- GCTGACCTTCATGGCCGTGCGCCGCA -3′ (antisense), and 5′-GAAGACGGCCATGAAGGTCAGCCCGCACCACCC-3′ (sense) and 5′-TTCATGGCCGTCTTCCGCAGCTGCTCCAATTC-3′ (antisense), respectively. According to the manufacturer’s instructions, inverse-PCR reactions were performed with one of the primer pairs described above and the circular form of a pBluescript II plasmid vector (Stratagene, La Jolla, CA, USA) containing mouse Runx1 cDNA as the template, being supplemented with the reaction premix of the kit. The oligonucleotide primers extended during PCR reaction without primer replacement. The resultant double-strand PCR product was expected to undergo spontaneous circularization by annealing its overhangs. An aliquot of the circularized PCR products was used to transform competent E.coli cells, followed by the isolation of the recombinant plasmid clones. The integrity of the synthesized molecules was confirmed by sequencing both strands for the whole length of the cDNA clones (Fig S2).
Fig 1. Arginine-to-lysine mutations at R206 and R210 of RUNX1 attenuate itstranscriptional-activating ability.
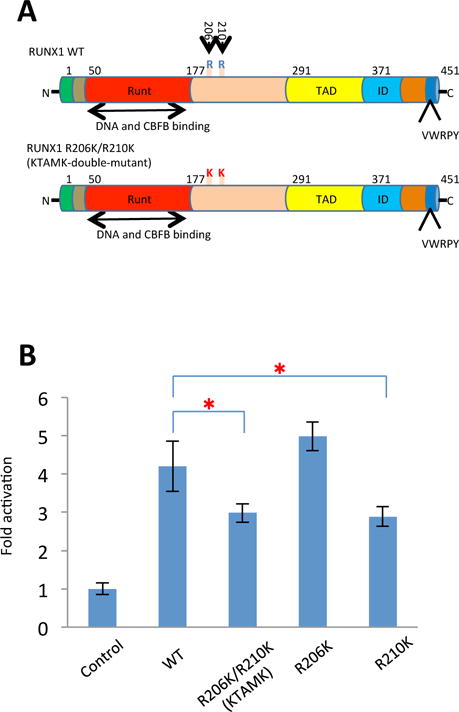
(A) Structure of wild type RUNX1 protein and arginine-to-lysine (RTAMR-to-KTAMK) mutant RUNX1 protein. Numerals indicate the positions of amino acid residues of the molecules. Runt indicates the Runt domain; TAD, trans-activation domain; ID, auto-inhibitory domain; VWRPY, VWRPY-motif; R, arginine (R) residue stretch; K, lysine (K) residue stretch. (B) Biochemical activities of RUNX1 (wild-type, WT) or the mutant molecules (R206K, R210K and KTAMK) were examined by means of reporter assay experiments. Each of the pRc effector constructs and pM-CSF-R-luc was cotransfected into HeLa cells with CBF-β and phRL-TK control vectors. Height of columns represents increase in relative luciferase activity on the CSF1R M-CSF receptor gene promoter (see “Materials and methods”). Bars indicate standard deviations of triplicate experiments. RUNX1 KTAMK and R210K mutants showed impaired trans-activating activity compared with wild-type RUNX1 (*P<0.05 t test).
To produce replacement-type vectors for generating knock-in alleles, a murine genomic DNA fragment of 12 kb encompassing exon 4 of the mouse Runx1 gene, which corresponds to the middle of the Runt domain (Okuda, et al 1996), was cloned into a pBluescript II plasmid vector, followed by an insertion of a poly-A-less diphtheria toxin-A suicide cassette at the utmost 3′ end of the construct for negative selection. Next, a SacII-restricted fragment from either the wild-type or the KTAMK mutant Runx1 cDNA, followed by a DNA fragment of the polyadenylation signal from the rabbit globin gene, was inserted into the corresponding SacII site within exon 4 of the Runx1 genomic DNA fragment so as to create a knocked-in exon containing the entire cDNA sequence of mouse Runx1 with the designed mutations downstream from exon 4. To complete the construction, a puromycin-resistance cassette for positive selection was inserted just downstream of the rabbit globin-polyadenylation signal sequence. The vectors, hereinafter referred to as pRES-constructs, were linearized at a unique ScaI site within the cloning vector just prior to being electroporated into ES cells (Fig S3A and S4A).
Luciferase reporter assay
Eukaryotic expression vectors, which transiently expressed the engineered RUNX1 molecules, were prepared by inserting the cDNAs into the pRc/CMV vector (Invitrogen, Carlsbad, CA, USA). The pRc vector constructs of the RUNX1-related molecules to be studied and the firefly luciferase reporter constructs, CSF1R-luc42 (a gift from Dr. Dong-Er Zhang, University of California San Diego) (Zhang et al, 1994) were transfected into HeLa cells (Riken Cell Bank, Tsukuba, Japan) with the calcium precipitation method. A control plasmid, phRL-TK (Promega, Madison, WI, USA), was cotransfected at a fixed ratio to provide internal transfection efficiency. After a 48-h culture, the cells were lysed and subjected to the double-luciferase reaction with specific substrates (Promega), according to the manufacturer’s instructions. Firefly luciferase activity relative to Renilla luciferase activity was measured with a luminometer (Berthorld Detection Systems, Portzheim, Germany).
Introducing Runx1 mutation alleles into murine ES cells by use of a knock-in approach
An aliquot (20 μg) of the linearized targeting vector for the pRES-constructs was transfected by electroporation into 9 × 106 E14-strain mouse ES cells of either the Runx1-deficient or the wild-type genotype. Runx1-deficient genotype ES cells had previously undergone homologous recombination for both Runx1 alleles at exon 4, with a neomycin-resistance cassette insertion for one allele and a hygromycin B-resistance cassette insertion for the other, as previously described (Okuda et al, 1996; Okuda et al, 2000) (Fig S3A). The resultant clones were selected in the presence of 1.3 mg/ml puromycin (Sigma, St. Louis, MO, USA), as described in the “Construction of the mutant knock-in targeting vectors” section, this vector contains a puromycin-resistance cassette (Fig S3A, and Fig S4A). Puromycin-resistant clones for the targeted integration were screened for homologous recombination by means of serial Southern blot analyses under conventional conditions with 5′ and 3′ outside probes as previously described (Okuda et al, 2000; Nishimura et al, 2004). Single-copy integration of the vector DNA to the clones was then confirmed by Southern blot analysis with an internal probe corresponding to the puromycin N-acetyltransferase sequence. Knock-in clones transfected into ES cells of the Runx1-deficient genotype (Fig S3B) were used for in vitro experiments on haematopoietic differentiation, whereas those introduced into wild-type ES cells (Fig S4B) were used for the generation of germ-line mice. ES cell clones were kept under conditions that preserved their undifferentiated phenotype until they were subjected to the experiments.
In vitro haematopoietic differentiation of ES cells clones
The procedure for the in vitro differentiation of embryoid bodies (EBs) was based on a previously described method (Okuda et al, 2000). ES cell clones were adapted to grow in media that contained 1000 u/ml murine recombinant leukaemia inhibitory factor. Triplicated cell suspensions of 3 × 102 adapted ES cells were subjected to the semisolid culture consisting of 1 ml of Iscove’s modified Dulbecco’s medium containing 1.2% methylcellulose, 15% fetal calf serum, 2 mM glutamine and 450 μM monothioglycerol. The cultures were supplemented with 2 units of human erythropoietin (EPO) per ml and 50 ng of human granulocyte colony–stimulating factor (G-CSF) per ml (both purchased from Kirin Brewery Co., Tokyo, Japan) and 10 ng of murine granulocyte-macrophage colony-stimulating factor (GM-CSF) per ml, 20 ng of murine stem cell factor (SCF, also termed KITL) per ml, and 5 ng of murine interleukin-3 (IL3) per ml (all from Genzyme, Cambridge, MA, USA) which allowed for haematopoietic differentiation of EBs. On day 14 of culture, haematopoietic differentiation of EBs was scored for the presence of differentiated haematopoietic components by visual examination with an inverted microscope (CK40, Olympus, Tokyo, Japan) equipped with a digital camera (C-5050 Zoom, Olympus) for the presence of surrounding macrophages. The morphology of the component cells was confirmed by microscopic examination using May-Grünwald-Giemsa staining of preparations generated by cytospin (Shandon Cytospin 3, Thermo Fisher Scientific, Cheshire, UK) of single dispersed embryoid bodies.
Generation of chimera and germline mice
Chimeric mice were generated as described (Okuda et al, 1996, Nishimura et al, 2004, Fukushima-Nakase et al, 2005). Briefly, mutant Runx1 ES cell clones with undifferentiated morphology, normal ploidy and a single vector insertion were injected into the E3.5 blastocysts obtained from the C57BL/6-strain by means of an inverted microscope (IX-70; Olympus) equipped with micromanipulators (Narishige, Tokyo, Japan). The manipulated blastocysts were then transferred into the uterines of pseudopregnant mother mice of the ICR strain 2.5- to 3-days after intercourse. The resultant chimera male mice were then crossed with C57BL/6-strain female mice to obtain ES-cell–derived offspring. The chimerism of the subsequently born mice was evaluated according to coat color, followed by screening by means of Southern blot analysis of tail biopsy specimens of agouti offspring for transmission of the knock-in allele. Germ-line transmission of the mutated alleles into F1 agouti mice and their progeny was also monitored with Southern blot analysis. The phenotype found in KTAMK-homozygous mice was analysed and compared with that of their wild-type siblings. When a difference was found between the two types of mice, the findings were further confirmed by comparing them with those for homozygous knock-in mice of wild-type Runx1 cDNA, which were published elsewhere (Nishimura et al, 2004, Fukushima-Nakase et al, 2005). All procedures of the animal experiments performed in this study were approved by the Committee for Animal Research, Kyoto Prefectural University of Medicine.
Flow cytometric analysis
Flow cytometric analysis was performed using conventional methods described elsewhere (Nishimura et al, 2004). The dissected thymus or spleen was mechanically disaggregated into single cells in RPMI 1640 medium (Invitrogen) containing 5% fetal bovine serum (FBS), which were then examined for live cells by using the trypan blue dye-exclusion test. We used samples with the viability of over 95% for the analysis. Next, appropriate aliquots of the cells were resuspended in phosphate-buffered saline (PBS) containing 3% FBS and 0.05% sodium azide. This was followed by incubation with each of the appropriately diluted monoclonal antibodies (MoAbs) on ice for 60 min. The following fluorochrome-conjugated antibodies were used: anti-CD3e conjugated with phycoerythrin (PE), anti-CD45R/B220 with fluorescein isothiocyanate (FITC), anti-CD4 with PE, anti-CD8a with FITC, anti-Ly6G (Gr-1) with PE, and anti-CD11b (Mac-1) with FITC (all from BD Biosciences, San Jose, CA, USA). After two washes, samples were analysed on a FACSCanto™ flow cytometer (BD Biosciences). For each analysis, a gate was placed on the appropriate region of thymocytes, splenocytes, or peripheral blood on the forward-scattered light (FSC)–side-scattered light (SSC) plot. Isotype-matched antibodies conjugated with the appropriate fluorochrome (BD Biosciences) at the same protein concentrations were used as negative controls for all experiments.
Statistical analysis
All statistical analyses were made by using Student’s t test. P values < 0.05 were considered statistically significant.
Results
Arginine-to-lysine mutations at R206 and R210 of Runx1 attenuate its transcriptional-activating ability
R206 and R210 are target residues that are methylated by PRMT1 (Zhao et al, 2008). These arginine residues are located just C-terminal to the Runt domain of RUNX1 (Fig 1A), and the R210 residue is highly conserved among human and mouse RUNX family proteins. As the initial step to analyse the biological significance of these methylations, we introduced an arginine-to-lysine mutation at codon 206 (R206K-mutant), codon 210 (R210K-mutant), and at both sites (R206K and R210K, the KTAMK-mutant) in the mouse Runx1 cDNA (Fig 1A and Fig S2). Lysine residues have a basic side chain, as do arginine residues, thus lysine can mimic an arginine-like basic amino acid residue but it cannot be methylated by PRMTs. Methylation at these arginine residues has been reported to result in the dissociation of SIN3A, from RUNX1, thus enhancing its transcriptional activating activity. When these arginine-mutants were exogenously expressed in mammalian cell lines, they showed similar or modestly reduced trans-activating activity, compared to wild-type RUNX1, on one known target promoter, the CSF1R-promoter (Fig 1B). While the R206K-mutant showed the same transcriptional-activity as wild-type RUNX1, the KTAMK-double-mutant showed a modest reduction of transcriptional-activating ability, which is compatible with the original report (Zhao et al, 2008) (Fig 1B). Thus, we focused on the KTAMK-double-mutant, and performed the following experiments.
Introducing KTAMK-mutation into Runx1 locus of mouse ES cells by use of knock-in strategy
With the KTAMK-mutant cDNA, we constructed replacement-type vectors for generating knock-in alleles in order to introduce the targeted base pair substitutions at the murine Runx1 gene locus, allowing the mutant RUNX1 protein to be expressed under the control of the endogenous RUNX1 promoter. Our vector-construction strategy is depicted in the scheme shown in Figures S3A and S4A. Multiple ES cell clones that underwent homologous recombination for either allele were successfully isolated for the KTAMK (R206K and R210K)-double knock-in mutations. Homologous recombinant ES cell clones for the wild-type genotype were used to generate chimeric mice (Fig S4B), while homologous ES cell clones with the Runx1−/− genotype were used for the in vitro assays and haematopoietic rescue experiments described below (Fig S3B).
KTAMK-mutant Runx1 retains its ability to rescue the in vitro haematopoietic defects observed in Runx1-deficient mouse ES cells
RUNX1 is essential for the early development of definitive haematopoiesis, but not for primitive erythropoiesis, as conventional knockout of this gene resulted in mid-gestational death due to complete block of definitive haematopoiesis. This haematopoietic defect can be replicated in vitro, using a murine ES cell differentiation system. Wild-type ES cells can develop haematopoietic cells, including myeloid cells and definitive erythroid cells, via embryoid-formation in semisolid culture conditions supplemented with the proper combination of colony-stimulating factors. In contrast, Runx1-deficient ES cells lose their ability to develop haematopoietic components of definitive origin. As previously described (Okuda et al, 2000), when wild-type Runx1 is introduced into Runx1-deficient ES cells using a knock-in strategy, the ES cells regain their ability to develop blood cells in vitro.
To evaluate if the KTAMK-mutant retains this rescue activity we subjected the ES-cell clones expressing the KTAMK-mutant protein from the disrupted Runx1-locus to in vitro culture and analysed whether haematopoietic differentiation was restored. We found that expression of the KTAMK-mutant cDNA from the knock-in allele in the Runx1-deficient ES cell clones restored their ability to develop haematopoietic cells in culture, similar to the Runx1-deficient ES cells with knocked-in wild-type Runx1 (Fig 2A). In addition, examination of cytospin preparations by May-Grünwald-Giemsa staining showed that the cells that make up the colonies grown from the KTAMK-mutant knock-in clones were indistinguishable from those generated from the heterozygous ES clones (Fig 2B). Thus, the modest impairment in trans-activating ability of the double-arginine-to-lysine mutant form of RUNX1 does not impair the in vitro haematopoietic function of wild-type RUNX1. This may be expected given the absence of a detectable haematopoietic phenotype for Runx1+/− ES cell clones, or haematopoietic stem progenitor cells (HSPC).
Fig 2. KTAMK-mutant RUNX1 retains its ability to rescue the in vitro haematopoietic defects observed in Runx1-defficient mouse ES cells.
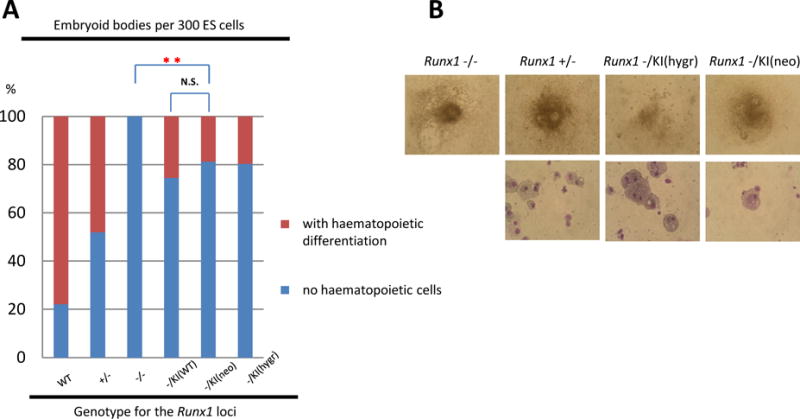
(A) Results of embryoid body (EB) differentiation. Incidence of haematopoietic differentiation of day-14 EBs derived from embryonic stem (ES) cell clones was determined in a representative experiment using the following genotypes: wild-type, heterozygous for Runx1-disruption (+/−), homozygous for the disruption (−/−), one knock-in allele for wild-type Runx1 cDNA (−/KI(WT)), one knock-in allele for KTAMK-mutant at the hygromycin allele (−/KI(hygr)), and one knock-in allele for KTAMK-mutant at the neomycin allele (−/KI(neo)). Red sections of columns indicate the proportion of grown embryoid bodies (EBs) with haematopoietic differentiation and blue sections represent the proportion of those without haematopoietic elements. ES clones of KTAMK-mutant knock-in allele developed into haematopoietic cells in vitro as did control clones. Statistically significant differences are shown as P values (**P<0.001 t test). N.S., not statistically different. (B) Appearance of representative day-14 EBs derived from each of the ES cell clones (top row). Morphology of the haematopoietic cells developed from ES cells of KTAMK-mutant knock-in clones showed no remarkable abnormalities of component cells examined with May-Grünwald-Giemsa staining (bottom row). Original magnifications: top row, ×20; bottom row, ×132.
Generation of germline mice bearing the knock-in allele of the KTAMK mutant of Runx1
To further evaluate the biological activity of this mutant in the context of an intact animal, we introduced this mutant cDNA into the Runx1 locus of wild-type mouse ES cells by means of a targeted-insertion strategy (Fig S4A, B). Through chimera mice production by conventional blastocyst-injection of these mutant ES cell clones, germline mutant mouse lines were successfully established. Heterozygous mice were healthy and fertile. Genotyping for the live pups generated from heterozygotes-crossing revealed that this KTAMK-double-mutant allele segregated according to the Mendelian ratio (Table S2). Homozygous Runx1KTAMK/KTAMK mice were born alive and became adults, circumventing the mid-embryonic death that was originally found for conventional Runx1-deficient mice: both males and females proved to be fertile, as was the case for the knock-in animal with wild-type Runx1 cDNA, as previously reported (Nishimura et al, 2004; Fukushima-Nakase et al, 2005). Thus, the knock-in procedure generated mice that expressed exogenous KTAMK-mutant Runx1 cDNA: it appeared that these arginine-methylation events are not essential for early haematopoietic development, based on the in vivo and in vitro experiments we have conducted.
Genetically modified mice of the Runx1KTAMK/KTAMK genotype showed minimal abnormality in myeloid haematopoiesis
We previously reported that knock-in mice expressing the wild-type Runx1 cDNA have minimal abnormalities so far as steady state myeloid haematopoiesis is concerned (Nishimura et al, 2004; Fukushima-Nakase et al, 2005). Thus, we analysed Runx1KTAMK/KTAMK knock-in mice for their haematopoietic phenotypic behaviour. There were no significant differences in peripheral blood cell counts among mice of the Runx1KTAMK/KTAMK genotypes, in comparison to their wild-type littermates (Fig 3A, Table I). In addition, the morphological appearance of the circulating blood cells, including neutrophils, monocytes, lymphocytes, platelets and erythrocytes, of these mutant mice, was indistinguishable from that of their wild-type littermates (data not shown). Furthermore, microscopic examination of the haematoxylin-eosin (HE) stained bone marrow sections of these mutant mice showed no consistent changes in the architecture or cellularity in comparison to their wild-type littermates (Fig S5B).
Fig 3. Findings for the peripheral blood of germline mice carrying the knock-in Runx1 KTAMK mutant allele.
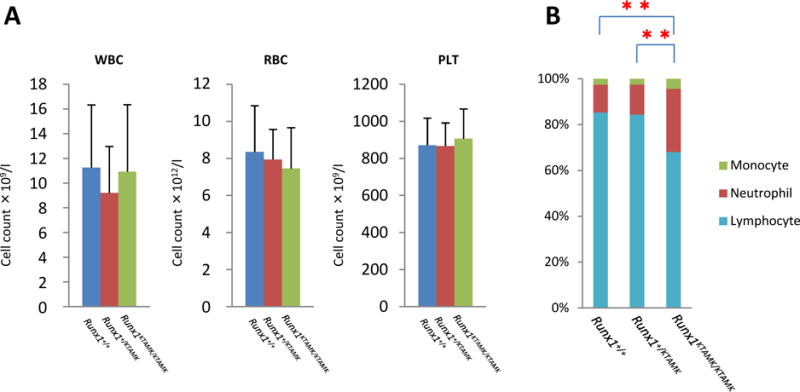
(A) Peripheral blood cell counts for the mutant mice compared with those for the matched-sibling control mice are indicated by the height of the columns. Lines above the columns signify standard deviations. RBC: red blood cell count; WBC: white blood cell count; PLT: platelet counts. (B) Bar graphs showing differences in peripheral leukocytes from knock-in and control mice. Lymphoid population in peripheral blood of Runx1KTAMK/KTAMK mice is reduced (**P<0.001 t test). Light blue: lymphocytes; red: neutrophils; green: monocytes.
Table I.
Peripheral blood analysis of Runx1+/+, Runx1+/KTAMK and Runx1KTAMK/KTAMK mice
| Haematological parameter |
Runx1+/+ (n = 34) |
Runx1+/KTAMK (n = 49) |
Runx1KTAMK/KTAMK (n = 43) |
|---|---|---|---|
| RBC (×1012/l) | 8.72 ± 1.46 | 8.67 ± 1.25 | 9.07 ± 1.6 |
| Hb (g/l) | 158.9 ±22.0 | 159.6 ±19.6 | 163.2 ± 20.9 |
| HCT (%) | 45.62 ± 5.90 | 46.04 ± 5.53 | 47.33 ± 6.58 |
| WBC (×109/l) | 11.26 ± 5.08 | 9.20 ± 3.78 | 10.94 ± 5.42 |
| Differential of WBC | |||
| Lymphocytes (%) | 85.27 ± 4.69 | 84.32 ± 4.97 | 67.83 ± 10.0** |
| (×109/l) | 9.67 ± 4.35 | 7.79 ± 3.34 | 7.58 ± 4.16* |
| Neutrophils (%) | 12.06 ±4.89 | 13.06 ±4.54 | 27.52 ± 9.98** |
| (×109/l) | 1.32 ± 0.76 | 1.20 ± 0.61 | 2.90 ± 1.56** |
| Monocytes (%) | 2.55 ± 1.50 | 2.45 ± 1.56 | 4.36 ± 2.58** |
| (×109/l) | 0.30 ±0.29 | 0.23 ± 0.23 | 0.43 ± 0.28 |
| Platelet count (×109/l) | 836.03 ±248.58 | 795.12 ±162.27 | 745.71 ± 219.52 |
+ indicates wild-type allele for the Runx1 gene locus; KTAMK, knock-in allele with KTAMK mutant cDNA of Runx1 gene.
Results are listed as the average for a group ± standard deviations.
RBC indicates red blood cell count; Hb, haemoglobin; HCT, haematocrit; WBC, white blood cell count.
P<0.05 and
P<0.001 t test
Blood samples were collected when the mice were 3 to 22 weeks old.
In contrast, we observed a slight decrease in the number of lymphocytes and a slight increase in the number of neutrophils and monocytes in the KTAMK-homozygous mice, and this decrease of the lymphocyte was statistically significant (Table I, Fig 3B), while knock-in mice expressing the wild-type Runx1 cDNA showed no such difference in their complete blood counts (Nishimura et al, 2004, Fukushima-Nakase et al, 2005). Thus, KTAMK-mutant alleles can largely overcome the haematopoietic defect resulting from the loss of Runx1, whereas lymphoid and/or myeloid haematopoiesis appeared to be modestly influenced by these knock-in mutant alleles.
It has been reported that conditionally knocked-out adult mice for Runx1 were viable with minimal abnormality for the myeloid haematopoiesis in the beginning of their life but tend to develop myeloid hyper-proliferation caused by an imbalance of haematopoietic stem cells and myeloid progenitor cells (Ichikawa et al, 2004; Growney et al, 2005; Putz et al, 2006). Histological findings of their bone marrow and/or spleen were reported to show high cellularity of myeloid components compatible to myeloproliferative characteristics at 7 to 22 weeks after pIpC-induced conditional disruption of Runx1. In contrast, the RUNX1-KTAMK mutant knock-in mice showed no such findings by following up observations through the age of 22 weeks (Fig 3A, Fig S5B, and data not shown). KTAMK-mutant mice had no splenomegaly, or architectural abnormality by histological examinations (Table II, and Fig S5A, B). In addition, cells with myeloid markers, including Mac1 and Gr-1, were not increased in the spleen of Runx1KTAMK/KTAMK mice, compared to their wild-type littermates, based upon flow cytometric analysis and immunohistochemistry for Gr-1 positive cell population (Fig 4A, B, C, D and Tables S3 and S4). This contrasts sharply from the adult conditional knock-out animals that suffer from splenomegaly and myeloproliferative phenotype in later life (Ichikawa et al, 2004; Growney et al, 2005; Putz et al, 2006).
Table II.
Size and body-weight ratios of organs of Runx1+/+, Runx1+/KTAMK and Runx1KTAMK/KTAMK mice
|
Runx1+/+ (n = 9) |
Runx1+/KTAMK (n = 6) |
Runx1KTAMK/KTAMK (n = 12) |
|
|---|---|---|---|
| Body weight (g) | 28.5 ± 6.25 | 26.9 ± 5.55 | 28.0 ± 4.73 |
| Thymus (mg) | 47.9 ± 6.92 | 39.5 ± 6.19 | 38.8 ± 10.9 |
| Tw (mg)/Bw (g) | 1.74 ± 0.43 | 1.53 ± 0.46 | 1.40 ± 0.36 |
| Spleen (mg) | 113.0 ± 40.6 | 86.7 ± 15.1 | 90.1 ± 20.0 |
| Sw (mg)/Bw (g) | 4.01 ± 1.68 | 3.32 ± 0.78 | 3.26 ± 0.71 |
| Kidney (mg) | 206.9 ± 68.2 | 181.0 ± 36.7 | 195.1 ± 35.8 |
| Kw (mg)/Bw (g) | 7.14 ± 0.89 | 6.77 ± 0.96 | 7.00 ± 0.90 |
Results are listed as the average for a group ± standard deviations.
Bw indicates body weight; Tw, thymus weight; Sw, spleen weight; and Kw, kidney weight.
No statistical difference was observed between any genotype-groups.
Fig 4. Findings for myeloid populations in spleen of Runx1+/+ and Runx1KTAMK/KTAMK mice.
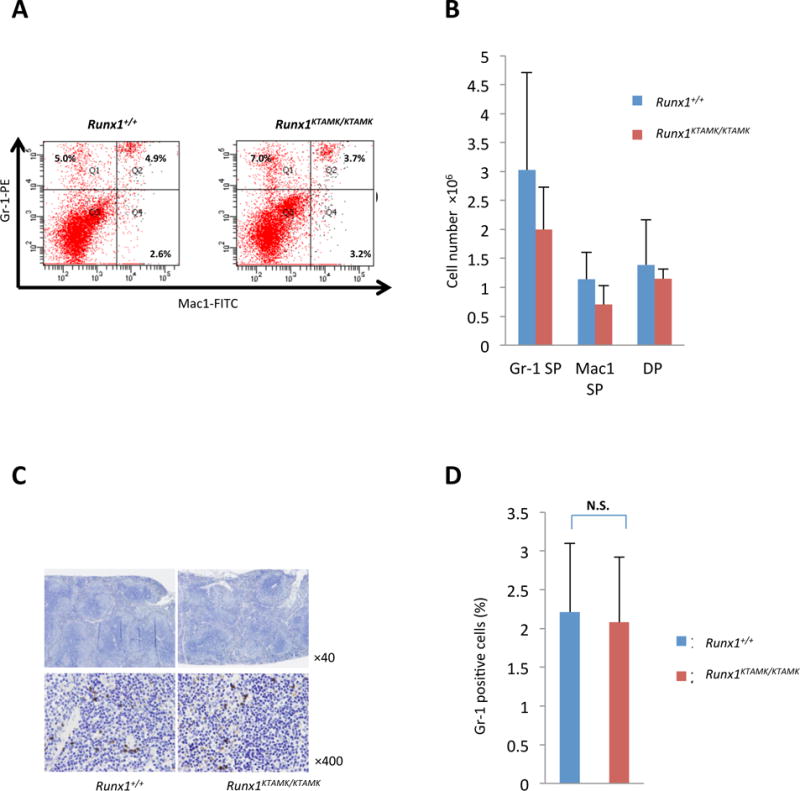
(A) Results of flow cytometric analysis of Mac1 and Gr-1 expression for splenocytes. Numbers in graph quadrants indicate fractions of the cells by percentile. Four mice for each group were analysed (Table S3) and representative results are shown. (B) Columns with bars for standard deviation indicate absolute numbers of Gr-1, Mac1 and double positive (DP) lymphocytes per spleen from littermate mice of each genotype. (C) Immunohistochemistry analysis of spleen of Runx1+/+ and Runx1KTAMK/KTAMK mice. Spleen tissue sections stained with antibody for Gr-1; original magnification ×40 and ×400. Three mice for each group were analysed, and representative results are shown. (D) Counts of the frequency of Gr-1 positive cells per high-power field (×400) in 20 fields. Bar graph shows the average positive ratio from 3 independent pairs of each group of mice. Runx1+/+ mice (2.21 ± 0.89 %) and Runx1KTAMK/KTAMK mice (2.08 ± 0.84 %) (P=0.86 t test). SP, single positive; N.S., not statistically different. Data are also shown as a table (Table S4).
Runx1KTAMK/KTAMK mice showed a reduction in the peripheral CD4 single positive cell population
Given that there was significant lymphoid depletion in the peripheral blood of KTAMK-mutant mice, as above (Table I, Fig 3B), we analysed the lymphoid tissues of these mice. There were no significant differences in the size of the thymus (per body-weight) (Table II) or the macroscopic appearance of the thymus among the Runx1KTAMK/KTAMK genotypes, compared to their wild-type littermates (Fig S5A). Histological findings for the thymus of Runx1KTAMK/KTAMK mice, obtained from microscopic examination of haematoxylin-eosin stained sections, were also not morphologically different from their wild-type littermates (Fig S5B). In addition, flow cytometric analysis showed that the proportions of CD4- and CD8-expressing thymocytes in Runx1KTAMK/KTAMK mice were indistinguishable from those found in their wild-type littermates (Fig 5A, B, Table S5).
Fig 5. Findings for lymphoid populations in thymus and spleen of Runx1+/+ and Runx1KTAMK/KTAMK mice.
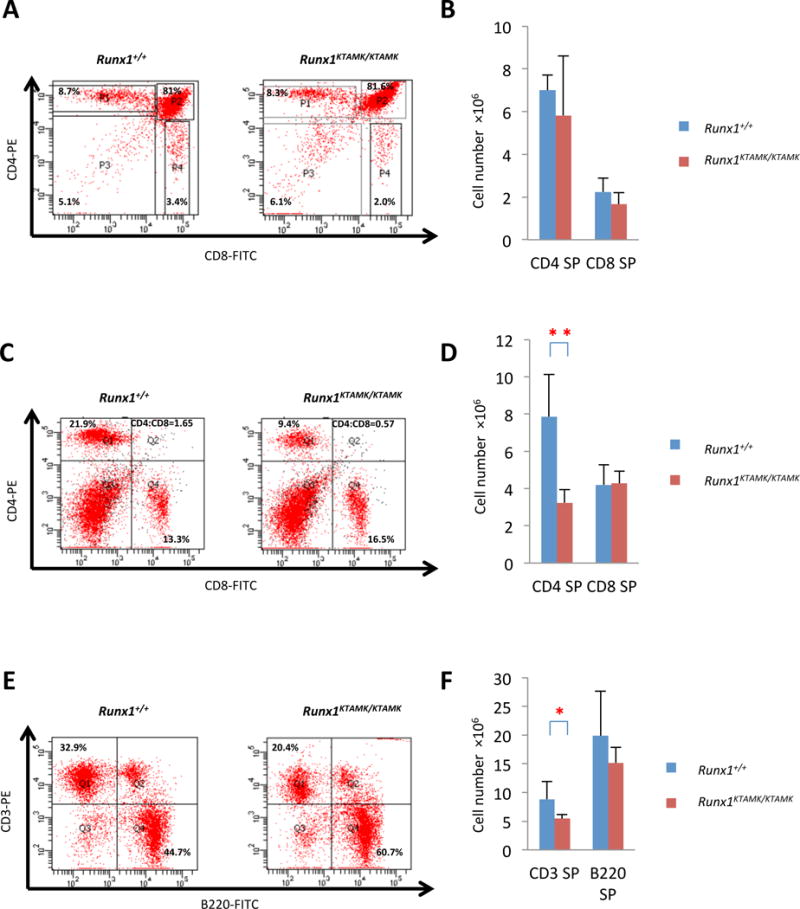
(A) Results of flow cytometric analysis of CD4 and CD8 expression for thymocytes. Four mice for each group were analysed, and representative data are shown. Numbers in graph quadrants indicate fractions of the cells by percentile. (B) Columns with bars for standard deviation indicate absolute numbers of CD4 or CD8 positive lymphocytes per thymocyte from littermate mice of each genotype. Data are also shown as a table (Table S5). (C) Results of flow cytometric analysis of CD4 and CD8 expression for splenocytes. Numbers in graph quadrants indicate fractions of the cells by percentile and CD4/8 ratio. Seven mice for each group were analysed, and representative data are shown. (D) Columns with bars for standard deviation indicate absolute numbers of CD4 or CD8 positive lymphocytes per spleen from littermate mice of each genotype. Statistically significant differences are shown as P values (**P<0.001 t test). Data are also shown as a table (Table S6). (E) Results of flow cytometric analysis of CD3 and B220 expression for splenocytes. Numbers in graph quadrants indicate fractions of the cells by percentile. Seven mice for each group were analysed, and representative data are shown. (F) Columns with bars for standard deviation indicate absolute numbers of CD3 or B220 positive lymphocytes per spleen from littermate mice of each genotype. Statistically significant differences are shown as P values (*P<0.01 t test). Data are also shown as a table (Table S7). SP, single positive
In contrast, Runx1KTAMK/KTAMK mice showed a reduction in CD3-expressing T lymphoid populations, mainly caused by a decrease in CD4 expressing T cells, with no change in CD8-expressing T cells in the spleen. This results in a reduction in the CD4+ to CD8+ T-cell ratio in comparison to their wild-type littermates (Fig 5C, D, E, F, Tables S6, S7). In addition, CD4/CD8 ratio reduction was also detected in peripheral blood in the KTAMK-mutant animals (Fig S6, and Table S8). Importantly, this reduction in the peripheral T-cell population was not observed for the wild-type Runx1 cDNA knock-in mice that we previously generated (Nishimura et al, 2004; Fukushima-Nakase et al, 2005) using the same targeting strategy. Thus it appears that this difference depends on, and is specific to, the mutations at the two sites of arginine methylation in RUNX1.
Discussion
In the present study, in order to analyse the biological significance of the arginine methylation at R206 and R210 residues we performed a number of genetic experiments. Compatible with the previous report that methylation of these residues are important for dissociating the transcriptional-corepressor SIN3A from RUNX1 (Zhao et al, 2008), we confirmed that arginine-to-lysine mutations at these residues resulted in a modest attenuation of its transactivating-activity on heterologous promoter constructs in vitro: this reduction in transactivation-activity was maximum when both R206 and R210 residues were mutated to lysine. Yet, the KTAMK-mutant retained its biological activity to rescue the haematopoietic defect of Runx1-deficient ES cells, when introduced into the cells, by means of knock-in strategy, as was the case for the wild–type Runx1 cDNA. In addition, mutant mice that are homozygous for the KTAMK-knock-in allele, develop definitive haematopoiesis and circumvent the embryonic death that observed in conventional Runx1-knockout (Okuda et al, 1996; Wang et al, 1996). Born mutant-mice showed no detectable abnormalities in the myeloid, megakaryocyte/platelet, or B-lymphocyte lineages in contrast to the conditional knockout mice. However, the KTAMK-knock-in mice showed modest but significant depletion of CD4+ T-lymphocyte in the peripheral blood and spleen. Thus, our results clearly indicate that the methylation of these arginine residues is of biological importance and that it does function in peripheral CD4+ T-cell population maintenance.
RUNX1 is known to play pivotal roles at multiple steps in T cell development: T-lymphocytes start their development when fetal liver lymphoid progenitor cells migrate into the thymus, and begin to differentiate through a series of stages, which are characterized by expression of two prominent cell surface markers: CD4 and CD8. Thymocytes that achieve successful rearrangement of the T-cell receptor gene are positively selected to survive and become CD4/CD8-double-positive cells. Double-positive thymocytes then lose either CD4 or CD8 on their surface to further differentiate into either CD8-sigle positive cytotoxic cells or to CD4-single-positive helper cells (Shortman and Wu, 1996). ThPOK and RUNX3 serve as lineage-specific master transcription factors for CD4+ cell differentiation and CD8+ cell proliferation, respectively (He et al, 2008; Setoguchi et al, 2008; Hassan et al, 2011). These single-positive cells are then released from the thymus to peripheral lymphatic tissues and they are maintained in the peripheral lymphoid tissues, including the lymph nodes and the spleen, where they play discrete roles to orchestrate the immune response of the entire body. It has been elucidated that RUNX1 is necessary for the transition from double-negative stage to double-positive stage of thymocytes as revealed by conditional knockout mice that use Lck-promoter-mediated Cre excision of RUNX1 (Taniuchi et al, 2002). RUNX1 also appears to function at the double-positive stage because transgenic mice with CD2-promoter-mediated expression of RUNX1a (also termed AML1a – a truncated form of RUNX1 that has trans-dominant repressing effects) showed increased apoptotic sensitivity of thymocytes at the double-positive stage (Abe et al, 2005). Of interest is another conditional knockout mouse in which floxed-Runx1 was excised by CD4-promoter mediated expression of Cre recombinase (Egawa et al, 2007). In comparison to the lck-mediated knockout, CD4/CD8-double positive thymocytes were unaffected in this mouse line and these cells proceeded to further differentiate into either CD4+ or CD8+ single positive T-cells even in the absence of RUNX1, indicating that RUNX1 is not required for the differentiation once the thymocytes reach the differentiation potential-stage in the thymus. However, the mutant mice showed depletion of CD4+ cells in the peripheral blood and spleen, with a reduced CD4/CD8 ratio. Although the magnitude of the phenotype was somewhat milder, this phenotype appears to be the same as that caused by the KTAMK-double mutation in mice: The arginine methylation of RUNX1 by PRMT1 is dispensable for T-cell development inside the thymus but this modification appears to be necessary for the maintenance of the peripheral CD4+ T-cell population.
Although it is beyond the scope of the present study, future research should explore whether subpopulations of the CD4+ cells are affected by this mutation. It would be intriguing to see, for example, whether regulatory T cell subsets are impaired or not in the animals, through multicolour flow cytometry, using additional surface markers, including IL2RA (CD25), IL7R (CD127) and FOXP3.
In the Cd4-mediated Runx1 disruption mentioned above, cell kinetic analysis using BrdU-labelling suggested that the decrease in peripheral CD4+ T-cells was due to accelerated cell death, rather than to impaired homeostatic proliferation. This phenomenon is likely to be mediated by reduction of the expression of IL7-receptor (IL7R) (Egawa et al, 2007), as mice that have Cd4-mediated-knockout of the Il7r gene show a considerable reduction in number of peripheral CD4+ lymphocytes (Tani-ichi et al, 2013). Although it was documented that Runx1-disruption in CD4+ lymphocytes reduced transcription of the IL7R gene, it is still not clear whether this gene is a direct target for RUNX family transcription factors or not. The KTAMK-mutant mice generated in the present study will serve as an experimental tool to further analyse these underlying mechanisms.
Post-translational modification of target proteins by the protein arginine N-methyltransferase (PRMT) family of proteins play an important role in various biological processes, such as signal transduction, mRNA splicing, transcriptional control, DNA repair and protein translocation (Bedford and Clarke, 2009). PRMTs methylate arginine residues by transferring methyl groups from S-adenosyl-L-methionine to terminal guanidino nitrogen atoms. The PRMT family can be divided into two major groups. PRMT1 belongs to the type I PRMTs that also includes PRMT2, 3, 6, 8 and CARM1, which generate asymmetric dimethylarginine through the addition of a second methyl group to the same nitrogen. Type II PRMTs, including PRMT 5, 7 and 9, generate symmetric dimethylarginine through the addition of one methyl group to each of the two nitrogen (Nicholson et al, 2009). PRMT1 generally predominates the total cellular PRMT activities (Tang et al, 2000); it is mainly cytoplasmic, with only a fraction of PRMT1 located in the nucleus (Herrmann et al, 2005). PRMT1 methylates RUNX1 at several arginine residues and affects its transcriptional activity (Zhao et al, 2008; Shia et al, 2012), while CARM1also methylates RUNX1, at R233, affecting myeloid differentiation through the regulation of MIR223 and, probably, other target molecules (Vu et al, 2013). Recently PRMT1 was shown to function as a key regulator of signal transduction with an important role in T lymphocyte activation (Parry and Ward, 2010). Indeed, PRMT1 is highly expressed in T helper cells (Mowen et al, 2004). Thus, it seems to be natural that the effect of interfering with the RUNX1 and PRMT1 interaction manifests itself in CD4 single positive cells than in other linage cells.
As described in the introduction, RUNX1 is subjected to a variety of post-translational modifications, including phosphorylation, acetylation, ubiquitination and methylation. These modifications should change its conformation and co-factor binding: some of these modifications have already been documented to alter its biochemical properties. Although we saw the biological consequence derived from loss of RUNX1 methylation in the present study, in only a few cases have the biological significance of these modifications been revealed in the context of an entire animal study. For example, S249, S266, and S276 are phosphorylated by ERK, and S249 and S276 by HIPK2: both of these modifications increase transcription activating ability (Tanaka et al, 1996; Zhang et al, 2004; Aikawa et al, 2006). Although knock-in animals that carry serine-to-alanine substitutions at these residues of Runx1 allele (S249A/S266A mouse and S249A/S276A mouse) showed no notable phenotype (Tachibana et al, 2008), mutants that carry two more nearby S/T residues (T273A and S276A) as well as S249A/S266A substitutions showed abnormalities in T-cell development in the rescued primary cells from Runx1-deficient mice. The addition of one more non-phosphorable substitution at S435 resulted in the complete loss of RUNX1 biological activity in this experimental system (Yoshimi et al, 2012). These findings suggest that phosphorylation of these residues in RUNX1 is required at some time. Similarly, RUNX1 has a number of modification sites, including serine/threonine residues phosphorylated by CDKs, tyrosine residues phosphorylated by Src kinase and dephosphorylated by SHP2, arginine residues methylated by CARM1, and lysine residues acetylated by p300 or ubiquitinated by APC, etc. (Wang et al, 2009). Elucidation of the biological significance of these post-translational modifications should shed insight into the proper regulating mechanisms for this important transcription factor, and contribute to the consideration of RUNX1 as a possible molecular target for haematopoietic manipulation or leukaemia treatment.
In order to identify the biological consequence of the Runx1 mutation, we utilized a knock-in approach in this study. Using this method, the introduced knocked-in gene was expected to be expressed under the endogenous transcriptional and translational regulatory environment. Recently, alternatively-spliced forms of mouse Runx1 allele were newly isolated, and it was proposed that these isoforms, with alternative 3′-exon(s), have something to do with the half-life of the RUNX1 protein (Komeno et al, 2014). Splicing regulation also appears to be important for the regulation of RUNX1-activity, but those mutations remain to be tested for biological significance. Knock-out or knock-in approaches, like the methods utilized in this study, will be one of choices to analyse the importance of the newly identified exons.
Interference of RUNX1 activity, on the other hand, appears to contribute to leukaemic transformation. Most, if not all, known leukemogenic forms of this transcription factor caused by genomic mutations affect the transcriptional activity of the molecule. The RUNX1-RUNX1T1 (AML1-ETO/MTG8) protein generated by the t(8;21) chromosome translocation, for example, loses RUNX1the C-terminal transactivation domain of RUNX1 and fuses most of the RUNX1T1 transcriptional co-repressor domains to RUNX1, generating a chimeric polypeptide with transcription repressing and activating functions (Frank et al, 1995; Meyers et al, 1995; Yergeau et al, 1997; Kitabayashi et al, 1998; Lutterbach et al, 1998; Okuda et al, 1998). Similarly, congenital loss of one allele of RUNX1 and/or point mutations in RUNX1 are often encountered in sporadic or familial cases of acute myeloblastic leukaemia or myelodysplastic syndromes (Osato et al, 1999; Song et al, 1999; Preudhomme et al, 2000; Imai et al, 2000; Harada et al, 2003; Nakao et al, 2004). Hot spots of the RUNX1 mutations are clustered in the DNA-contacting residues within the Runt domain and result in loss-of-function or dominant-negative functions (Osato, 2004). Of interest is that the same missense mutation examined in the present study, R210K, has been reported in two cases: chronic myelomonocytic leukaemia (Kohlmann et al, 2010) and AML patients (McNerney et al, 2014). Although the KTAMK-mutant mice has shown no preleukaemic findings thus far, these clinical observations suggest the possibility that this missense mutation contributes to a multistep-leukaemogenic process.
To summarize, we have identified the biological function of RUNX1, which is mediated by its methylation at R206/R210, as being primarily the maintenance of peripheral CD4-single positive lymphocytes. Our present results, together with accumulating insights into the relationship between post-translational modification of RUNX1 and its biological role in the context of entire animals, will contribute to a more profound comprehension of the molecular mechanisms through which normal and malignant haematopoiesis are regulated. In addition, these studies will help us consider the RUNX1 molecule as a possible molecular target in the haematopoietic disorders.
Supplementary Material
Acknowledgments
We wish to thank all past and present members of Okuda Laboratory, Kyoto Prefectural University of Medicine, for their encouragement and helpful discussions. This work was supported by grants from the Ministry of Education, Sports, Culture, Science and Technology, Japan to TO, and NIH R01 grant CA166835 to SDN. TO also received a research grant from The Shimizu Foundation for the Promotion of Immunology Research, Kyoto, Japan.
Footnotes
Author’s contributions
SM performed the research, analysed the data, and wrote the manuscript; TY performed the research and analysed the data; XZ contributed to designing the research and analysed data; SDN designed the research, analysed data, edited and revised the manuscript; TM revised the manuscript; TO designed the research, performed the research, analysed the data and wrote, edited and revised the manuscript.
Conflict of interest
The authors declare no conflict of interest.
References
- Abe N, Kohu K, Ohmori H, Hayashi K, Watanabe T, Hozumi K, Sato T, Habu S, Satake M. Reduction of Runx1 transcription factor activity up-regulates Fas and Bim expression and enhances the apoptotic sensitivity of double positive thymocytes. J Immunol. 2005;175:4475–4482. doi: 10.4049/jimmunol.175.7.4475. [DOI] [PubMed] [Google Scholar]
- Adamo L, Naveiras O, Wenzel PL, McKinney-Freeman S, Mack PJ, Gracia-Sancho J, Suchy-Dicey A, Yoshimoto M, Lensch MW, Yoder MC, Garcia-Cardena G, Daley GQ. Biomechanical forces promote embryonic haematopoiesis. Nature. 2009;459:1131–1135. doi: 10.1038/nature08073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikawa Y, Nguyen LA, Isono K, Takakura N, Tagata Y, Schmitz ML, Koseki H, Kitabayashi I. Roles of HIPK1 and HIPK2 in AML1- and p300-dependent transcription, hematopoiesis and blood vessel formation. Embo j. 2006;25:3955–3965. doi: 10.1038/sj.emboj.7601273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs JR, Peterson LF, Zhang Y, Kraft AS, Zhang DE. AML1/RUNX1 Phosphorylation by Cyclin-Dependent Kinases Regulates the Degradation of AML1/RUNX1 by the Anaphase-Promoting Complex. Molecular and Cellular Biology. 2006;26:7420–7429. doi: 10.1128/MCB.00597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Sinha KK, Senyuk V, Nucifora G. SUV39H1 interacts with AML1 and abrogates AML1 transactivity. AML1 is methylated in vivo. Oncogene. 2003;22:5229–5237. doi: 10.1038/sj.onc.1206600. [DOI] [PubMed] [Google Scholar]
- Dowdy CR, Frederick D, Zaidi SK, Colby JL, Lian JB, van Wijnen AJ, Gerstein RM, Stein JL, Stein GS. A germline point mutation in Runx1 uncouples its role in definitive hematopoiesis from differentiation. Exp Hematol. 2013;41:980–991.e981. doi: 10.1016/j.exphem.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egawa T, Tillman RE, Naoe Y, Taniuchi I, Littman DR. The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J Exp Med. 2007;204:1945–1957. doi: 10.1084/jem.20070133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson P, Gao J, Chang KS, Look T, Whisenant E, Raimondi S, Lasher R, Trujillo J, Rowley J, Drabkin H. Identification of breakpoints in t(8;21) acute myelogenous leukemia and isolation of a fusion transcript, AML1/ETO, with similarity to Drosophila segmentation gene, runt. Blood. 1992;80:1825–1831. [PubMed] [Google Scholar]
- Frank R, Zhang J, Uchida H, Meyers S, Hiebert SW, Nimer SD. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML1B. Oncogene. 1995;11:2667–2674. [PubMed] [Google Scholar]
- Fukushima-Nakase Y, Naoe Y, Taniuchi I, Hosoi H, Sugimoto T, Okuda T. Shared and distinct roles mediated through C-terminal subdomains of acute myeloid leukemia/Runt-related transcription factor molecules in murine development. Blood. 2005;105:4298–4307. doi: 10.1182/blood-2004-08-3372. [DOI] [PubMed] [Google Scholar]
- Gilliland DG. Hematologic malignancies. Current Opinion in Hematology. 2001;8:189–191. doi: 10.1097/00062752-200107000-00001. [DOI] [PubMed] [Google Scholar]
- Goyama S, Yamaguchi Y, Imai Y, Kawazu M, Nakagawa M, Asai T, Kumano K, Mitani K, Ogawa S, Chiba S, Kurokawa M, Hirai H. The transcriptionally active form of AML1 is required for hematopoietic rescue of the AML1-deficient embryonic para-aortic splanchnopleural (P-Sp) region. Blood. 2004;104:3558–3564. doi: 10.1182/blood-2004-04-1535. [DOI] [PubMed] [Google Scholar]
- Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, Curley DP, Kutok JL, Akashi K, Williams IR, Speck NA, Gilliland DG. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106:494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Harada Y, Tanaka H, Kimura A, Inaba T. Implications of somatic mutations in the AML1 gene in radiation-associated and therapy-related myelodysplastic syndrome/acute myeloid leukemia. Blood. 2003;101:673–680. doi: 10.1182/blood-2002-04-1010. [DOI] [PubMed] [Google Scholar]
- Hassan H, Sakaguchi S, Tenno M, Kopf A, Boucheron N, Carpenter AC, Egawa T, Taniuchi I, Ellmeier W. Cd8 enhancer E8I and Runx factors regulate CD8alpha expression in activated CD8+ T cells. Proc Natl Acad Sci U S A. 2011;108:18330–18335. doi: 10.1073/pnas.1105835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Park K, Wang H, He X, Zhang Y, Hua X, Li Y, Kappes DJ. CD4-CD8 lineage commitment is regulated by a silencer element at the ThPOK transcription-factor locus. Immunity. 2008;28:346–358. doi: 10.1016/j.immuni.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Herrmann F, Lee J, Bedford MT, Fackelmayer FO. Dynamics of Human Protein Arginine Methyltransferase 1(PRMT1) in Vivo. Journal of Biological Chemistry. 2005;280:38005–38010. doi: 10.1074/jbc.M502458200. [DOI] [PubMed] [Google Scholar]
- Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, Ito Y. Dimerization with PEBP2beta protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. Embo j. 2001;20:723–733. doi: 10.1093/emboj/20.4.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Woo AJ, Waldon Z, Schindler Y, Moran TB, Zhu HH, Feng GS, Steen H, Cantor AB. A Src family kinase-Shp2 axis controls RUNX1 activity in megakaryocyte and T-lymphocyte differentiation. Genes Dev. 2012;26:1587–1601. doi: 10.1101/gad.192054.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa M, Asai T, Saito T, Seo S, Yamazaki I, Yamagata T, Mitani K, Chiba S, Ogawa S, Kurokawa M, Hirai H. AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med. 2004;10:299–304. doi: 10.1038/nm997. [DOI] [PubMed] [Google Scholar]
- Imai Y, Kurokawa M, Izutsu K, Hangaishi A, Takeuchi K, Maki K, Ogawa S, Chiba S, Mitani K, Hirai H. Mutations of the AML1 gene in myelodysplastic syndrome and their functional implications in leukemogenesis. Blood. 2000;96:3154–3160. [PubMed] [Google Scholar]
- Kitabayashi I, Ida K, Morohoshi F, Yokoyama A, Mitsuhashi N, Shimizu K, Nomura N, Hayashi Y, Ohki M. The AML1-MTG8 leukemic fusion protein forms a complex with a novel member of the MTG8(ETO/CDR) family, MTGR1. Mol Cell Biol. 1998;18:846–858. doi: 10.1128/mcb.18.2.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B, Dicker F, Schnittger S, Dugas M, Kern W, Haferlach C, Haferlach T. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010;28:3858–3865. doi: 10.1200/JCO.2009.27.1361. [DOI] [PubMed] [Google Scholar]
- Komeno Y, Yan M, Matsuura S, Lam K, Lo MC, Huang YJ, Tenen DG, Downing JR, Zhang DE. Runx1 exon 6-related alternative splicing isoforms differentially regulate hematopoiesis in mice. Blood. 2014;123:3760–3769. doi: 10.1182/blood-2013-08-521252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- Lutterbach B, Hiebert SW. Role of the transcription factor AML-1 in acute leukemia and hematopoietic differentiation. Gene. 2000;245:223–235. doi: 10.1016/s0378-1119(00)00014-7. [DOI] [PubMed] [Google Scholar]
- Lutterbach B, Westendorf JJ, Linggi B, Patten A, Moniwa M, Davie JR, Huynh KD, Bardwell VJ, Lavinsky RM, Rosenfeld MG, Glass C, Seto E, Hiebert SW. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol. 1998;18:7176–7184. doi: 10.1128/mcb.18.12.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNerney ME, Brown CD, Peterson AL, Banerjee M, Larson RA, Anastasi J, Le Beau MM, White KP. The spectrum of somatic mutations in high-risk acute myeloid leukaemia with -7/del(7q) British Journal of Haematology. 2014;166:550–556. doi: 10.1111/bjh.12964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers S, Lenny N, Hiebert SW. The t(8;21) fusion protein interferes with AML-1B-dependent transcriptional activation. Mol Cell Biol. 1995;15:1974–1982. doi: 10.1128/mcb.15.4.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proceedings of the National Academy of Sciences. 1991;88:10431–10434. doi: 10.1073/pnas.88.23.10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Kozu T, Shimizu K, Enomoto K, Maseki N, Kaneko Y, Kamada N, Ohki M. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. Embo j. 1993;12:2715–2721. doi: 10.1002/j.1460-2075.1993.tb05933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowen KA, Schurter BT, Fathman JW, David M, Glimcher LH. Arginine methylation of NIP45 modulates cytokine gene expression in effector T lymphocytes. Mol Cell. 2004;15:559–571. doi: 10.1016/j.molcel.2004.06.042. [DOI] [PubMed] [Google Scholar]
- Nagamachi A, Htun PW, Ma F, Miyazaki K, Yamasaki N, Kanno M, Inaba T, Honda Z, Okuda T, Oda H, Tsuji K, Honda H. A 5′ untranslated region containing the IRES element in the Runx1 gene is required for angiogenesis, hematopoiesis and leukemogenesis in a knock-in mouse model. Dev Biol. 2010;345:226–236. doi: 10.1016/j.ydbio.2010.07.015. [DOI] [PubMed] [Google Scholar]
- Nakao M, Horiike S, Fukushima-Nakase Y, Nishimura M, Fujita Y, Taniwaki M, Okuda T. Novel loss-of-function mutations of the haematopoiesis-related transcription factor, acute myeloid leukaemia 1/runt-related transcription factor 1, detected in acute myeloblastic leukaemia and myelodysplastic syndrome. British Journal of Haematology. 2004;125:709–719. doi: 10.1111/j.1365-2141.2004.04966.x. [DOI] [PubMed] [Google Scholar]
- Nguyen LA, Pandolfi PP, Aikawa Y, Tagata Y, Ohki M, Kitabayashi I. Physical and functional link of the leukemia-associated factors AML1 and PML. Blood. 2005;105:292–300. doi: 10.1182/blood-2004-03-1185. [DOI] [PubMed] [Google Scholar]
- Nicholson TB, Chen T, Richard S. The physiological and pathophysiological role of PRMT1-mediated protein arginine methylation. Pharmacological Research. 2009;60:466–474. doi: 10.1016/j.phrs.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Fukushima-Nakase Y, Fujita Y, Nakao M, Toda S, Kitamura N, Abe T, Okuda T. VWRPY motif-dependent and -independent roles of AML1/Runx1 transcription factor in murine hematopoietic development. Blood. 2004;103:562–570. doi: 10.1182/blood-2003-06-2109. [DOI] [PubMed] [Google Scholar]
- North T, Gu TL, Stacy T, Wang Q, Howard L, Binder M, Marin-Padilla M, Speck NA. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development. 1999;126:2563–2575. doi: 10.1242/dev.126.11.2563. [DOI] [PubMed] [Google Scholar]
- North TE, de Bruijn MF, Stacy T, Talebian L, Lind E, Robin C, Binder M, Dzierzak E, Speck NA. Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity. 2002;16:661–672. doi: 10.1016/s1074-7613(02)00296-0. [DOI] [PubMed] [Google Scholar]
- North TE, Goessling W, Peeters M, Li P, Ceol C, Lord AM, Weber GJ, Harris J, Cutting CC, Huang P, Dzierzak E, Zon LI. Hematopoietic stem cell development is dependent on blood flow. Cell. 2009;137:736–748. doi: 10.1016/j.cell.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the Drosophila runt gene and the human AML1 gene. Proceedings of the National Academy of Sciences. 1993;90:6859–6863. doi: 10.1073/pnas.90.14.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, Van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- Okuda T, Cai Z, Yang S, Lenny N, Lyu CJ, Van Deursen JMA, Harada H, Downing JR. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood. 1998;91:3134–3143. [PubMed] [Google Scholar]
- Okuda T, Takeda K, Fujita Y, Nishimura M, Yagyu S, Yoshida M, Akira S, Downing JR, Tatsuo A. Biological characteristics of the leukemia-associated transcriptional factor AML1 disclosed by hematopoietic rescue of AML1-deficient embryonic stem cells by using a knock-in strategy. Molecular and Cellular Biology. 2000;20:319–328. doi: 10.1128/mcb.20.1.319-328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, Nishimura M, Nakao M, Fujita Y. RUNX1/AML1: A central player in hematopoiesis. International Journal of Hematology. 2001;74:252–257. doi: 10.1007/BF02982057. [DOI] [PubMed] [Google Scholar]
- Osato M. Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene. 2004;23:4284–4296. doi: 10.1038/sj.onc.1207779. [DOI] [PubMed] [Google Scholar]
- Osato M, Asou N, Abdalla E, Hoshino K, Yamasaki H, Okubo T, Suzushima H, Takatsuki K, Kanno T, Shigesada K, Ito Y. Biallelic and heterozygous point mutations in the runt domain of the AML1/PEBP2alphaB gene associated with myeloblastic leukemias. Blood. 1999;93:1817–1824. [PubMed] [Google Scholar]
- Parry RV, Ward SG. Protein arginine methylation: a new handle on T lymphocytes? Trends Immunol. 2010;31:164–169. doi: 10.1016/j.it.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Preudhomme C, Warot-Loze D, Roumier C, Grardel-Duflos N, Garand R, Lai JL, Dastugue N, Macintyre E, Denis C, Bauters F, Kerckaert JP, Cosson A, Fenaux P. High incidence of biallelic point mutations in the Runt domain of the AML1/PEBP2 alpha B gene in Mo acute myeloid leukemia and in myeloid malignancies with acquired trisomy 21. Blood. 2000;96:2862–2869. [PubMed] [Google Scholar]
- Putz G, Rosner A, Nuesslein I, Schmitz N, Buchholz F. AML1 deletion in adult mice causes splenomegaly and lymphomas. Oncogene. 2006;25:929–939. doi: 10.1038/sj.onc.1209136. [DOI] [PubMed] [Google Scholar]
- Rabbitts TH. Chromosomal translocations in human cancer. Nature. 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- Seo W, Ikawa T, Kawamoto H, Taniuchi I. Runx1-Cbfbeta facilitates early B lymphocyte development by regulating expression of Ebf1. J Exp Med. 2012a;209:1255–1262. doi: 10.1084/jem.20112745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoguchi R, Tachibana M, Naoe Y, Muroi S, Akiyama K, Tezuka C, Okuda T, Taniuchi I. Repression of the transcription factor Th-POK by Runx complexes in cytotoxic T cell development. Science. 2008;319:822–825. doi: 10.1126/science.1151844. [DOI] [PubMed] [Google Scholar]
- Shang Y, Zhao X, Xu X, Xin H, Li X, Zhai Y, He D, Jia B, Chen W, Chang Z. CHIP functions an E3 ubiquitin ligase of Runx1. Biochemical and Biophysical Research Communications. 2009;386:242–246. doi: 10.1016/j.bbrc.2009.06.043. [DOI] [PubMed] [Google Scholar]
- Shia WJ, Okumura AJ, Yan M, Sarkeshik A, Lo MC, Matsuura S, Komeno Y, Zhao X, Nimer SD, Yates JR, 3rd, Zhang DE. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood. 2012;119:4953–4962. doi: 10.1182/blood-2011-04-347476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortman K, Wu L. Early T lymphocyte progenitors. Annu Rev Immunol. 1996;14:29–47. doi: 10.1146/annurev.immunol.14.1.29. [DOI] [PubMed] [Google Scholar]
- Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, Ratajczak J, Resende IC, Haworth C, Hock R, Loh M, Felix C, Roy DC, Busque L, Kurnit D, Willman C, Gewirtz AM, Speck NA, Bushweller JH, Li FP, Gardiner K, Poncz M, Maris JM, Gilliland DG. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23:166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- Speck NA. Core binding factor and its role in normal hematopoietic development. Curr Opin Hematol. 2001;8:192–196. doi: 10.1097/00062752-200107000-00002. [DOI] [PubMed] [Google Scholar]
- Sun W, Downing JR. Haploinsufficiency of AML1 results in a decrease in the number of LTR-HSCs while simultaneously inducing an increase in more mature progenitors. Blood. 2004;104:3565–3572. doi: 10.1182/blood-2003-12-4349. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Tezuka C, Muroi S, Nishimoto S, Katsumoto T, Nakajima A, Kitabayashi I, Taniuchi I. Phosphorylation of Runx1 at Ser249, Ser266, and Ser276 is dispensable for bone marrow hematopoiesis and thymocyte differentiation. Biochemical and Biophysical Research Communications. 2008;368:536–542. doi: 10.1016/j.bbrc.2008.01.124. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kurokawa M, Ueki K, Tanaka K, Imai Y, Mitani K, Okazaki K, Sagata N, Yazaki Y, Shibata Y, Kadowaki T, Hirai H. The extracellular signal-regulated kinase pathway phosphorylates AML1, an acute myeloid leukemia gene product, and potentially regulates its transactivation ability. Molecular and Cellular Biology. 1996;16:3967–3979. doi: 10.1128/mcb.16.7.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, Clarke S, Herschman HR. PRMT1 Is the Predominant Type I Protein Arginine Methyltransferase in Mammalian Cells. Journal of Biological Chemistry. 2000;275:7723–7730. doi: 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- Tani-ichi S, Shimba A, Wagatsuma K, Miyachi H, Kitano S, Imai K, Hara T, Ikuta K. Interleukin-7 receptor controls development and maturation of late stages of thymocyte subpopulations. Proc Natl Acad Sci U S A. 2013;110:612–617. doi: 10.1073/pnas.1219242110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T, Ito Y, Littman DR. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- Vu Ly P, Perna F, Wang L, Voza F, Figueroa Maria E, Tempst P, Erdjument-Bromage H, Gao R, Chen S, Paietta E, Deblasio T, Melnick A, Liu Y, Zhao X, Nimer Stephen D. PRMT4 Blocks Myeloid Differentiation by Assembling a Methyl-RUNX1-Dependent Repressor Complex. Cell Rep. 2013;5:1625–1638. doi: 10.1016/j.celrep.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Huang G, Zhao X, Hatlen MA, Vu L, Liu F, Nimer SD. Post-translational modifications of Runx1 regulate its activity in the cell. Blood Cells, Molecules, and Diseases. 2009;43:30–34. doi: 10.1016/j.bcmd.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, Hatlen MA, Vu L, Liu F, Xu H, Asai T, Xu H, Deblasio T, Menendez S, Voza F, Jiang Y, Cole PA, Zhang J, Melnick A, Roeder RG, Nimer SD. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science. 2011;333:765–769. doi: 10.1126/science.1201662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proceedings of the National Academy of Sciences. 1996;93:3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang Q, Crute BE, Melnikova IN, Keller SR, Speck NA. Cloning and characterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor. Molecular and Cellular Biology. 1993;13:3324–3339. doi: 10.1128/mcb.13.6.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee HJ, Voon DC, Bae SC, Ito Y. PEBP2-beta/CBF-beta-dependent phosphorylation of RUNX1 and p300 by HIPK2: implications for leukemogenesis. Blood. 2008;112:3777–3787. doi: 10.1182/blood-2008-01-134122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Kurokawa M, Imai Y, Izutsu K, Asai T, Ichikawa M, Yamamoto G, Nitta E, Yamagata T, Sasaki K, Mitani K, Ogawa S, Chiba S, Hirai H. AML1 Is Functionally Regulated through p300-mediated Acetylation on Specific Lysine Residues. Journal of Biological Chemistry. 2004;279:15630–15638. doi: 10.1074/jbc.M400355200. [DOI] [PubMed] [Google Scholar]
- Yergeau DA, Hetherington CJ, Wang Q, Zhang P, Sharpe AH, Binder M, Marin-Padilla M, Tenen DG, Speck NA, Zhang DE. Embryonic lethality and impairment of haematopoiesis in mice heterozygous for an AML1-ETO fusion gene. Nat Genet. 1997;15:303–306. doi: 10.1038/ng0397-303. [DOI] [PubMed] [Google Scholar]
- Yoshimi M, Goyama S, Kawazu M, Nakagawa M, Ichikawa M, Imai Y, Kumano K, Asai T, Mulloy JC, Kraft AS, Takahashi T, Shirafuji N, Kurokawa M. Multiple phosphorylation sites are important for RUNX1 activity in early hematopoiesis and T-cell differentiation. Eur J Immunol. 2012;42:1044–1050. doi: 10.1002/eji.201040746. [DOI] [PubMed] [Google Scholar]
- Zhang DE, Fujioka K, Hetherington CJ, Shapiro LH, Chen HM, Look AT, Tenen DG. Identification of a region which directs the monocytic activity of the colony-stimulating factor 1 (macrophage colony-stimulating factor) receptor promoter and binds PEBP2/CBF (AML1) Mol Cell Biol. 1994;14:8085–8095. doi: 10.1128/mcb.14.12.8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Biggs JR, Kraft AS. Phorbol Ester Treatment of K562 Cells Regulates the Transcriptional Activity of AML1c through Phosphorylation. Journal of Biological Chemistry. 2004;279:53116–53125. doi: 10.1074/jbc.M405502200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Fried FB, Guo H, Friedman AD. Cyclin-dependent kinase phosphorylation of RUNX1/AML1 on 3 sites increases transactivation potency and stimulates cell proliferation. Blood. 2008;111:1193–1200. doi: 10.1182/blood-2007-08-109702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Jankovic V, Gural A, Huang G, Pardanani A, Menendez S, Zhang J, Dunne R, Xiao A, Erdjument-Bromage H. Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev. 2008;22:640–653. doi: 10.1101/gad.1632608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.