

Abstract
Evidence suggests that an imbalance between oxidation and antioxidation is involved in the pathogenesis of acute lung injury/acute respiratory distress syndrome (ALI/ARDS). Activation of AMP-activated protein kinase (AMPK) has been shown to inhibit the occurrence of ALI/ARDS. However, it is unknown whether activation of AMPK benefits ALI/ARDS by restoration of the oxidant and antioxidant balance, and which mechanisms are responsible for this process. The present study aimed to address these issues. Lipopolysaccharide (LPS) induced pronounced pathological changes of ALI in mice; these were accompanied by elevated production of malondialdehyde (MDA) and decreased activity of superoxide dismutase (SOD) compared with control mice. Prior treatment of mice with the AMPK agonist metformin significantly suppressed the LPS-induced development of ALI, reduced the elevation of MDA and increased the activity of SOD. Further analysis indicated that activation of AMPK also stimulated the protein expression of peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) and superoxide dismutase 1 (SOD1). This study suggests that activation of AMPK by metformin inhibits oxidative stress by upregulation of PGC1α and SOD1, thereby suppressing the development of ALI/ARDS, and has potential value in the clinical treatment of such conditions.
Keywords: acute lung injury, AMP-activated protein kinase, peroxisome proliferator-activated receptor γ coactivator 1α, superoxide dismutase 1
Introduction
Acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (ARDS) comprise diffuse heterogeneous lung injury characterized by dyspnea, severe hypoxemia and non-cardiogenic pulmonary edema, with significant morbidity and mortality (1). Studies have suggested that an imbalance between oxidant and antioxidant systems is involved in the pathogenesis of ALI/ARDS (2,3). In the context of ALI/ARDS, the generation of reactive oxygen species (ROS) is increased (2,3). There are numerous potential sources of ROS, including infiltrated leukocytes (neutrophils), parenchymal cells (epithelial and endothelial cells), circulating oxidant-generating enzymes (xanthine oxidase), and inhaled gases with high oxygen concentrations that are frequently used during mechanical ventilation (2,3). Concurrently, the expression levels and activity of ROS scavengers, including superoxide dismutase (SOD), are decreased significantly in ALI/ARDS (3,4). Other antioxidant defenses, specifically, urate, glutathione and ascorbate, are also reduced in the distal airspaces in patients with ALI (5), resulting the overproduction of ROS and leading to an imbalance between oxidation and antioxidation. Increased ROS production has the potential to induce DNA damage, lipid peroxidation and the activation of nuclear factor (NF)-κB and activator protein 1 (AP-1) leading to an expansive inflammatory reaction (2,3). Therefore, reducing oxidation or restoring the balance of oxidant and anti-oxidant systems may be a new strategy in the management of ALI/ARDS.
AMP-activated protein kinase (AMPK) is a serine/threonine protein kinase composed of a catalytic α subunit and regulatory β and γ subunits. Pathological changes, including glucose deprivation, hypoxia, ischemia and heat shock, causing cellular depletion of ATP or elevation of AMP induce AMPK activation. Studies have demonstrated that AMPK is also activated by metformin (6), berberine (7) and resveratrol (8), independent of energy crisis (9). AMPK has also been shown to negatively modulate inflammatory reactions. For example, the activation of AMPK has been found to protect endothelial cells by inhibition of the NF-κB pathway (10). A further study has indicated that the activation of AMPK attenuated neutrophil proinflammatory activity and decreased the severity of ALI in a lipopolysaccharide (LPS)-induced mouse model of ALI (11), yet its detailed molecular mechanisms have not yet been determined.
In the present study, LPS was used to induce a mouse model of ALI, and the effects of the activation of AMPK by metformin on oxidant production and the inhibition of ALI were examined. In addition, further investigations were conducted to clarify the potential molecular mechanisms.
Materials and methods
Animals and reagents
BALB/c mice were provided by the Experimental Animal Center of the Medical College of Xi'an Jiaotong University (Xi'an, China). The animal protocol was approved by the Institutional Animal Ethics Committee of Xi'an Jiaotong University. Malondialdehyde (MDA) and SOD assay kits were obtained from Jiancheng Bioengineering Institute (Nanjing, China). LPS from Escherichia coli 055:B5 (Sigma-Aldrich, St. Louis, MO, USA) was used to induce ALI.
Generation of the ALI model
Male BALB/c mice (6–8 weeks old, 20–25 g) were randomly divided into three groups with 8 mice per group. Control mice received intratracheal instillation of sterile phosphate-buffered saline (PBS) alone. Models of ALI were induced by the intratracheal instillation of 50 µl LPS (100 µg) for 24 h as previously described (12). Mice of the third group were pre-treated with metformin (250 mg/kg) by intraperitoneal injection at 0.5 h prior to stimulation with LPS. All the mice were maintained in an animal holding room under special pathogen-free conditions with free access to food and water.
Harvesting of bronchoalveolar lavage fluid (BALF)
Animals were sacrificed at 24 h after LPS exposure by exsanguination under anesthesia with 80 mg/kg ketamine (K2573, Sigma-Aldrich). Bronchoalveolar lavage (BAL) was performed by intratracheal injection. The lungs were lavaged three times with ice-cold sterile PBS at a volume of 0.8 ml/wash and the average fluid recovery was >85%. The BALF was centrifuged at 800xg for 10 min at 4°C to pelletize cells. The cell pellets were resuspended in PBS. Total cells were counted under light microscopy using a hemocytometer and neutrophils were determined as described previously (12).
Histology
Histopathological evaluation was performed on mice that were not subjected to BAL. Lungs were inflated and fixed with 10% buffered formalin for 48 h and embedded in paraffin. Tissue sections (4 µm thick) were cut and stained with hematoxylin and eosin (H&E) according to the regular staining method for histological analysis.
MDA content assay
In order to determine the lipid peroxidation level, MDA contents in the lung tissues were examined using an assay kit. Briefly, the lung tissue samples were homogenized in cool normal saline (lung tissue to normal saline ratio, 1:10). The homogenate was then assessed according to the manufacturer's protocol. The concentration of MDA was measured by absorbance at 523 nm and was expressed in units of nmol/mg protein.
SOD activity assay
To examine the oxidation resistance of the lung tissue samples from each group, the activity of SOD in the lung tissue was determined using an assay kit following the manufacturer's protocol. The activity of SOD was quantized by absorbance at 550 nm and was expressed in units of U/mg protein.
Western blotting
Lungs were removed and homogenized in ice-cold radioimmunoprecipitation assay lysis buffer (Sigma-Aldrich) with phosphatase inhibitor and protease inhibitor. Lysates were centrifuged at 14,000 × g for 20 min at 4°C, and the supernatant was collected as the total protein. Protein was separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a trans-blot nitrocellulose membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Polyclonal antibodies against total-AMPK (2532; 1:500), phospho-AMPK (2535; 1:500; Cell Signaling Technology, Inc., Danvers, MA, USA), peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α; NBP1-04676; 1:800; Novus Biologicals, LLC, Littleton, CO, USA), SOD1 (P00441; 1:1,000; Bioworld Technology, Inc., St. Louis Park, MN, USA) and glyceraldehyde 3-phosphate dehydrogenase (9545; 1:5,000; GAPDH; Sigma-Aldrich) were used following the manufacturer's protocols. Horseradish peroxidase-conjugated goat anti-rabbit IgG (1:5,000) was used as the secondary antibody (A9169; Sigma-Aldrich). Reactions were developed with SuperSignal West Pico Chemiluminescent Substrate (A9169; Pierce; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and exposure to autoradiographic film. Signaling was quantified from scanned films using Scion Image software version 4.03 (Scion Corporation, Frederick, MD, USA).
Statistical analysis
Values are presented as the mean ± standard deviation. Data were analyzed using one way analysis of variance followed by Tukey's post hoc test. P<0.05 was considered to indicate a significant difference between groups.
Results
Metformin inhibits LPS-induced ALI
The results of the present study demonstrated that LPS induced significant ALI in mice. Histological analysis revealed substantial hemorrhage, infiltration by inflammatory cells and damage of epithelial and endothelial cells in mice with LPS-induced ALI in comparison with the control (Fig. 1A). Furthermore, the number of total inflammatory cells and neutrophils in the BALF was increased in the mice that received LPS stimulation (P<0.01 vs. control; Fig. 1B and C). The aforementioned alterations were substantially ameliorated by prior treatment of the mice with metformin (Fig. 1), suggesting that metformin effectively inhibits the development of LPS-induced ALI in mice.
Figure 1.

Metformin (Met) inhibits LPS-induced ALI in mice. (A) Representative photomicrographs (magnification, ×200) of hematoxylin and eosin stained lung tissues from control (Con) mice and LPS-induced ALI mice models with or without prior treatment with Met. (B) Total number of inflammatory cells and (C) number of neutrophils in the BALF (n=3 per group). *P<0.01 vs. the Con group; #P<0.01 vs. the LPS-induced ALI model (LPS group). LPS, lipopolysaccharide; ALI, acute lung injury; BALF, bronchoalveolar lavage fluid.
Metformin attenuates the LPS-induced reduction of AMPK phosphorylation
To investigate the mechanisms by which metformin inhibits LPS-induced ALI, the phosphorylation of AMPK was examined using western blotting. As shown in Fig. 2, the phosphorylation level of AMPK declined to 28.3% of the control value in LPS-treated mice (P<0.05 vs. control). However, pretreatment of the mice with metformin (250 mg/kg) at 0.5 h prior to LPS instillation significantly increased AMPK phosphorylation, which exhibited a 1.86-fold increase compared with that in the LPS group (P<0.05 vs. LPS). These results suggest that metformin attenuates the impairment of AMPK activity in LPS-induced ALI, which might be associated with the development of ALI.
Figure 2.
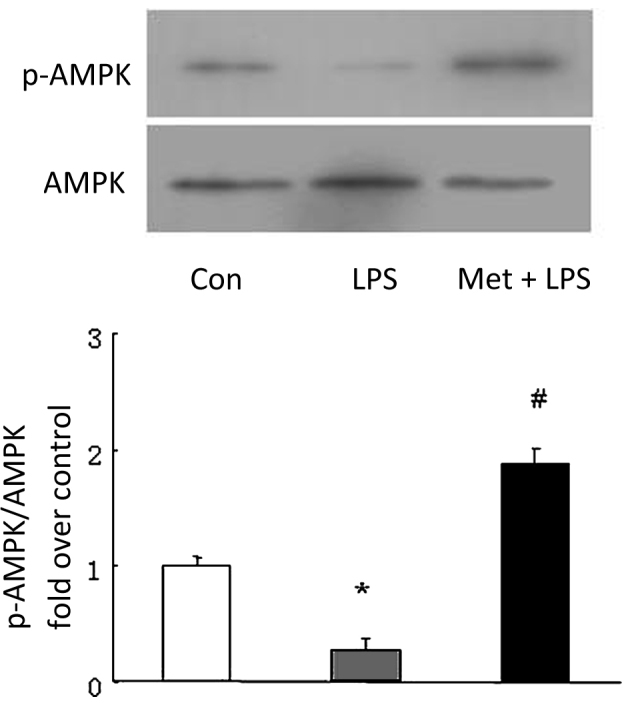
Effect of metformin (Met) on the phosphorylation of AMPK. Mice were pre-treated with Met (250 mg/kg) intraperitoneally for 30 min prior to stimulation with LPS. Phosphorylation of AMPK was examined using western blotting (n=3 per group). *P<0.05 vs. the control (Con) group; #P<0.05 vs. the LPS-induced acute lung injury model (LPS group). AMPK, AMP-activated protein kinase; p-AMPK, phospho-AMPK; LPS, lipopolysaccharide.
Metformin increases SOD activity and decreases MDA production
To determine the mechanisms by which the metformin-stimulated AMPK activation inhibits LPS-induced ALI, the production of MDA and activity of SOD were determined. The results shown in Fig. 3 reveal that LPS instillation caused a 2.58-fold increase over control in MDA content and a 2.16-fold reduction in SOD activity in lung tissues compared with those in the control group (P<0.05). However, pre-treatment with metformin markedly reduced LPS-induced MDA production and increased SOD activity in mice with LPS-induced ALI (Fig. 2), suggesting that metformin restores the balance between oxidation and antioxidation.
Figure 3.

Effects of metformin (Met) on MDA production and SOD activity. (A) Levels of the lipid peroxidation product MDA in lung tissues from all groups of mice. (B) SOD activity was determined in lung tissues from all groups of mice (n=3 per group). *P<0.05 vs. the control (Con) group; #P<0.05 vs. the LPS-induced ALI model (LPS group). MDA, malondialdehyde; SOD, superoxide dismutase; LPS, lipopolysaccharide; ALI, acute lung injury.
Metformin induces SOD1 expression
SOD1 is a most important enzyme that catalyzes the elimination of oxygen free radicals produced by infiltrated leukocytes and parenchymal cells in the antioxidative system (13). To examine whether metformin induces the expression of SOD1 and benefits the lungs, the protein levels of SOD1 were determined in the lung tissues. The results shown in Fig. 4 indicate that LPS reduced the SOD1 protein level to 0.28-fold of that in the control group (P<0.05 vs. control mice). Prior treatment of the mice with metformin attenuated the LPS-induced reduction in SOD1 level; the SOD1 protein level was raised to 2.86-fold compared with that in untreated mice with LPS-induced ALI (P<0.05), suggesting that metformin induces SOD1 expression in the pulmonary system.
Figure 4.
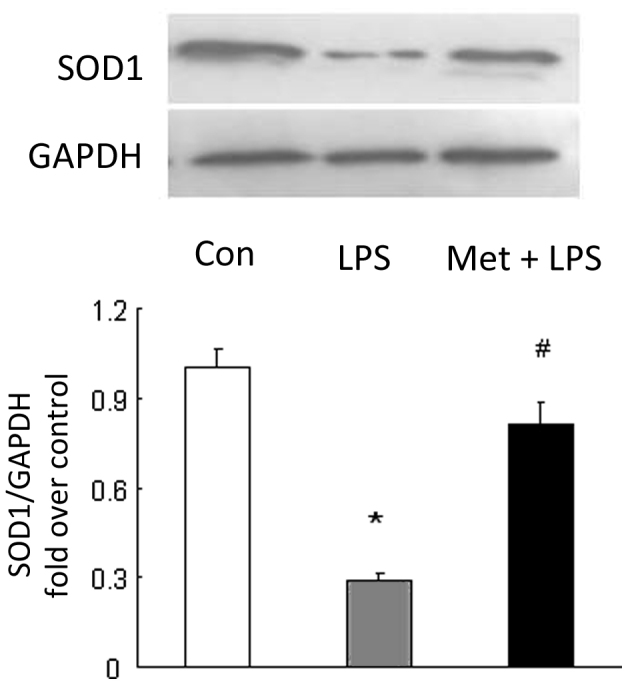
Metformin (Met) induces the expression of SOD1. Metformin was administered at 0.5 h prior to LPS injection in mice. Expression of SOD1 in lung tissue was determined using immunoblotting. GAPDH was used as loading control. Representative western blots and quantification of bands are shown (n=3 per group). *P<0.05 vs. the control (Con) group; #P<0.05 vs. the LPS-induced ALI model (LPS group). SOD1, superoxide dismutase 1; LPS, lipopolysaccharide; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; ALI, acute lung injury.
Metformin increases PGC1α production
To further clarify the mechanisms of induction of SOD1 by metformin, the level of PGC1α, a transcriptional factor shown to upregulate SOD1 in non-pulmonary tissues was examined (14). As shown in Fig. 5, the protein level of PGC1α was 0.36-fold that of the control in the LPS-induced mice model of ALI (P<0.05 vs. control), while mice treated with metformin prior to the induction of ALI displayed a 1.74-fold increase in PGC1α protein level in the lung compared with untreated LPS model mice (P<0.05 vs. LPS treated mice), indicating that metformin upregulates the expression of PGC1α in the lung.
Figure 5.
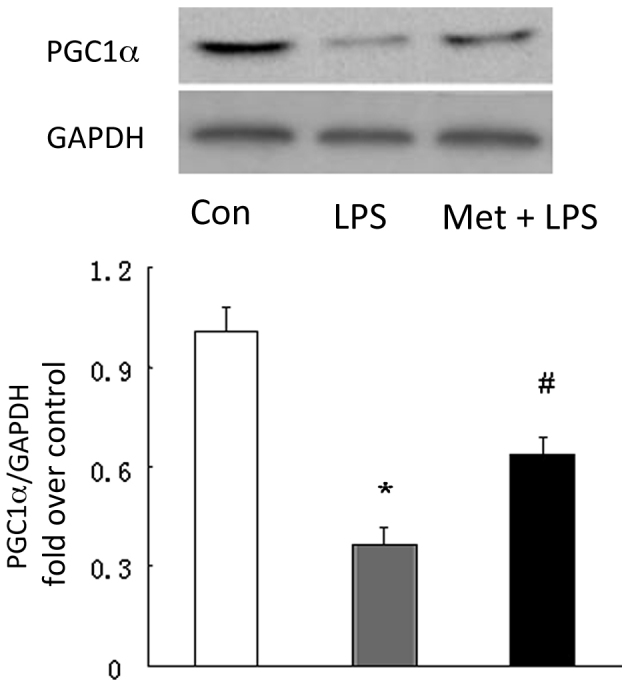
Metformin (Met) upregulates the expression of PGC1α. Equal amounts of protein were loaded and probed using specific PGC1α and GAPDH (loading control) antibodies. Representative western blots and quantification of bands are shown (n=3 per group). *P<0.05 vs. the control (Con) group; #P<0.05 vs. the LPS-induced ALI model (LPS group). PGC1α, peroxisome proliferator-activated receptor γ coactivator 1α; LPS, lipopolysaccharide; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; ALI, acute lung injury.
Discussion
The present study demonstrates that the activation of AMPK by metformin suppresses the development of LPS-induced ALI; this effect is coupled with the upregulation of PGC-1α upregulation and the subsequent induction of SOD1. The present study also reveals a novel molecular mechanism by which activation of AMPK benefits LPS-induced ALI. This study further confirms that the strategy of activation of AMPK might have potential value in the treatment of ALI/ARDS.
A disruption of oxidant-antioxidant balance is likely to be important in the pathogenesis of ALI/ARDS (15). In ALI/ARDS, increased ROS production induces lipid peroxidation, DNA damage and protein inactivation, and stimulates the expression of proinflammatory transcriptional factors, such as NF-κB and AP-1, which in turn upregulate the expression of adhesion molecules, chemokines and inflammatory cytokines (for example, tumor necrosis factor-α), leading to cell injury and death (15). The present study suggests that the production of MDA, which indicates the level of lipid peroxidation, was significantly elevated while the activity of SOD was decreased in the LPS-induced mouse model of ALI, and this was accompanied by the infiltration of a large number of neutrophils into the pulmonary tissue. These results further confirm the involvement of oxidative stress in the pathogenesis of ALI.
The activation of AMPK using pharmacological ligands has been demonstrated to induce anti-inflammatory and immunosuppressive effects in a variety of cell types, and thus is potentially useful in the treatment of various diseases (7,8,16,17). A study has shown that activation of the AMPK signaling pathway by 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside and berberine ameliorates the LPS-induced mice model of ALI (11). However, the mechanisms responsible for AMPK activation exerting an inhibitory effect on ALI remain largely unknown. In the present study, it was found that the activation of AMPK by metformin suppressed the development of LPS-induced ALI, which was accompanied by significantly increased activity and expression of SOD1 and reduced concentration of MDA in lung tissues, counteracting the changes induced in the LPS-induced ALI model. These indicate that the activation of AMPK by metformin rebuilds the balance between oxidation and antioxidation has a beneficial effect on LPS-induced ALI.
The induction of PGC-1α has been shown to confer protective against oxidative stress with AMPK activation in non-pulmonary systems (16,18). Therefore, it is worthwhile to examine the expression level of PGC-1α in LPS-induced ALI. The present study demonstrated that ALI model mice exhibited reduced levels of PGC-1α, and the application of metformin attenuated the decline of PGC-1α in the ALI model. This was accompanied by a reduction in the level of MDA production, suggesting that the induction of PGC-1α mediates the protective effects of AMPK activation on lung tissues.
PGC-1α is a transcriptional coactivator that acts to regulate lipid catabolism, mitochondrial number and function. PGC-1α has been identified to be a central regulator of energy metabolism, and is expressed at high levels in tissues with high metabolic rates (19). A previous study suggests that PGC-1α is required for the induction of numerous ROS-detoxifying enzymes, including SOD1, SOD2 and glutathione peroxidase 1, and cells lacking PGC-1α have an increased sensitivity to oxidative stress (14). Overexpression of PGC-1α in vitro and in vivo significantly upregulates the expression of antioxidant enzymes, including SOD and catalase, thereby reducing ROS levels, suppressing oxidative damage and protecting cells and tissues (20,21). Consistent with the pattern of changes of PGC-1α levels, the expression of SOD1 was markedly raised in metformin-treated ALI model mice, whereas the level was reduced in the ALI model compared with that in the control. Thus, it is proposed that the protective effect of metformin on LPS-induced ALI is mediated via the AMPK/PGC-1α/SOD1 axis.
Metformin is an in vitro synthetic AMPK agonist which has been commonly used clinically to treat type 2 diabetes for many years (22). The wide clinical experience and safety record of metformin suggest that it may be an optimal therapeutic approach for human ALI/ARDS. The present study also provides novel insights into the mechanism of metformin in the regulation of the balance between oxidation and antioxidation, and the subsequent amelioration or prevention of the development of ALI/ARDS.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 81070045 and No. 81330002).
References
- 1.Walkey AJ, Summer R, Ho V, Alkana P. Acute respiratory distress syndrome: Epidemiology and management approaches. Clin Epidemiol. 2012;4:159–169. doi: 10.2147/CLEP.S28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosanna DP, Salvatore C. Reactive oxygen species, inflammation and lung diseases. Curr Pharm Des. 2012;18:3889–3900. doi: 10.2174/138161212802083716. [DOI] [PubMed] [Google Scholar]
- 3.Park HS, Kim SR, Lee YC. Impact of oxidative stress on lung diseases. Respirology. 2009;14:27–38. doi: 10.1111/j.1440-1843.2008.01447.x. [DOI] [PubMed] [Google Scholar]
- 4.Crimi E, Sica V, Williams-Ignarro S, Zhang H, Slutsky AS, Ignarro LJ, Napoli C. The role of oxidative stress in adult critical care. Free Radic Biol Med. 2006;40:398–406. doi: 10.1016/j.freeradbiomed.2005.10.054. [DOI] [PubMed] [Google Scholar]
- 5.Bowler RP, Velsor LW, Duda B, Chan ED, Abraham E, Ware LB, Matthay MA, Day BJ. Pulmonary edema fluid antioxidants are depressed in acute lung injury. Crit Care Med. 2003;31:2309–2315. doi: 10.1097/01.CCM.0000085090.06078.8C. [DOI] [PubMed] [Google Scholar]
- 6.Xu JN, Zeng C, Zhou Y, Peng C, Zhou YF, Xue Q. Metformin inhibits StAR expression in human endometriotic stromal cells via AMPK-mediated disruption of CREB-CRTC2 complex formation. J Clin Endocrinol Metab. 2014;99:2795–2803. doi: 10.1210/jc.2014-1593. [DOI] [PubMed] [Google Scholar]
- 7.Jeong HW, Hsu KC, Lee JW, Ham M, Huh JY, Shin HJ, Kim WS, Kim JB. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am J Physiol Endocrinol Metab. 2009;296:E955–E964. doi: 10.1152/ajpendo.90599.2008. [DOI] [PubMed] [Google Scholar]
- 8.Han Y, Jiang C, Tang J, Wang C, Wu P, Zhang G, Liu W, Jamangulova N, Wu X, Song X. Resveratrol reduces morphine tolerance by inhibiting microglial activation via AMPK signalling. Eur J Pain. 2014;18:1458–1470. doi: 10.1002/ejp.511. [DOI] [PubMed] [Google Scholar]
- 9.Yee SW, Chen L, Giacomini KM. The role of ATM in response to metformin treatment and activation of AMPK. Nat Genet. 2012;44:359–360. doi: 10.1038/ng.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su RY, Chao Y, Chen TY, Huang DY, Lin WW. 5-Aminoimidazole-4-carboxamide riboside sensitizes TRAIL- and TNF{alpha}-induced cytotoxicity in colon cancer cells through AMP-activated protein kinase signaling. Mol Cancer Ther. 2007;6:1562–1571. doi: 10.1158/1535-7163.MCT-06-0800. [DOI] [PubMed] [Google Scholar]
- 11.Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, Tsuruta Y, Abraham E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L497–L504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang G, Liu L, Zhang Y, Han D, Liu J, Xu J, Xie X, Wu Y, Zhang D, Ke R, et al. Activation of PPARγ attenuates LPS-induced acute lung injury by inhibition of HMGB1-RAGE levels. Eur J Pharmacol. 2014;726:27–32. doi: 10.1016/j.ejphar.2014.01.030. [DOI] [PubMed] [Google Scholar]
- 13.Fukai T, Ushio-Fukai M. Superoxide dismutases: Role in redox signaling, vascular function and diseases. Antioxid Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 15.Tasaka S, Amaya F, Hashimoto S, Ishizaka A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid Redox Signal. 2008;10:739–753. doi: 10.1089/ars.2007.1940. [DOI] [PubMed] [Google Scholar]
- 16.Kim MY, Lim JH, Youn HH, Hong YA, Yang KS, Park HS, Chung S, Ko SH, Shin SJ, Choi BS, et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1 α axis in db/db mice. Diabetologia. 2013;56:204–217. doi: 10.1007/s00125-012-2747-2. [DOI] [PubMed] [Google Scholar]
- 17.Tamaki N, Cristina Orihuela-Campos R, Inagaki Y, Fukui M, Nagata T, Ito HO. Resveratrol improves oxidative stress and prevents the progression of periodontitis via the activation of the Sirt1/AMPK and the Nrf2/antioxidant defense pathways in a rat periodontitis model. Free Radic Biol Med. 2014;75:222–229. doi: 10.1016/j.freeradbiomed.2014.07.034. [DOI] [PubMed] [Google Scholar]
- 18.Marmolino D, Manto M, Acquaviva F, Vergara P, Ravella A, Monticelli A, Pandolfo M. PGC-1alpha down-regulation affects the antioxidant response in Friedreich's ataxia. PLoS One. 2010;5:e10025. doi: 10.1371/journal.pone.0010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, Lin JD. PGC-1 coactivators in the control of energy metabolism. Acta Biochim Biophys Sin (Shanghai) 2011;43:248–257. doi: 10.1093/abbs/gmr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 21.Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1α rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu SC, Tu YK, Chien MN, Chien KL. Effect of antidiabetic agents added to metformin on glycaemic control, hypoglycaemia and weight change in patients with type 2 diabetes: A network meta-analysis. Diabetes Obes Metab. 2012;14:810–820. doi: 10.1111/j.1463-1326.2012.01606.x. [DOI] [PubMed] [Google Scholar]