

Abstract
Immunoglobulin G4-related disease is a recently recognized systemic disease that can affect any organ or tissue in the body, including the kidneys. IgG4-related kidney disease (IgG4-RKD) is an important part of immunoglobulin G4-related disease. The most common renal manifestation of IgG4-RKD is tubulointerstitial nephritis and glomerular lesions. There, however, is few case of IgG4-RKD mimicking malignant ureter tumor leading to severe hydronephrosis. We herein report an unusual case of IgG4-RKD mimicking malignancy.
A 66-year-old Asian man presented to the nephrologist with soreness of loins, anorexia, and acute kidney injury in 2010. His renal function spontaneously improved after 2 weeks’ hemodialysis without systemic steroid therapy. Four years later, he presented to the urologist with severe left hydronephrosis because of marked thickness of the left ureter wall. As a ureteral malignancy could not be ruled out, laparoscopic nephroureterectomy was performed.IgG4-related kidney disease was confirmed by the histologic examination. Then, repeat laboratory test showed almost complete recovery of renal function after initiation of steroidal therapy.
This case highlights the rare possibility of IgG4-RKD mimicking malignant ureter tumor. Nephrologist and pathologists should be aware of the possibility that hydronephrosis with ureter obstruction may be involved in IgG4-RKD.
INTRODUCTION
An immunoglobulin G4-related disease (IgG4-RD) is a newly discovered multisystem disease characterized by IgG4 positive plasma cell infiltration in various organs, and, often but not always, elevated serum IgG4 concentrations. Immunoglobulin G4-related disease was first described by Hamano et al, who described elevated serum IgG4 concentrations in patients with autoimmune pancreatitis).1 Then it was reported in other organs.2–4 The affected common anatomic site is considered to be pancreas, salivary gland, liver, lung, periorbital tissues, thyroid gland, breast, prostate, and kidney. The histologic characteristics are inflammatory infiltration consisting of predominantly lymphocyte, IgG4 plasma cell without cytologic atypia, and a storiform pattern fibrosis.4–7 Immunoglobulin G4-related kidney disease (IgG4-RKD) signifies any form of renal involvement by IgG4-RD. The most common renal involvement by IgG4-RD is tubulointerstitial nephritis, and glomerular disease, in particular membranous glomerulonephritis. Only a limited number of IgG4-RD, however, had been reported arising in the renal pelvis and ureter. In this article, we report an unusual case of IgG4-related disease arising in the renal pelvis and ureter.
CASE PRESENTATION
A 66-year-old Chinese farmer with no significant previous medical history, presented to the nephrologist with soreness of loins, anorexia, and acute kidney injury in 2010. He was apyrexial and had mild abdominal tenderness. His biochemical test results were compatible with acute renal failure (blood urea nitrogen [BUN]: 17.9 mmol/l; creatinine, 729umol/l). Erythrocyte sedimentation rate (18 mm/h) and C-reactive protein (40 mg/dL) were mildly increased. Whole blood counts showed anemia (hemoglobin, 79 g/l). Urine examination showed neither red blood cells nor proteinuria. Antinuclear levels of C3 and C4, and antineutrophil cytoplasmic antibodies as well as other laboratory test results were within the normal range. A subsequent abdominal ultrasound scan examination revealed bilateral mild hydronephrosis. No lesions were observed in pancreas, retroperitoneum, or biliary tract. The initial clinical diagnosis was acute renal failure of uncertain etiology. The patient refused the kidney biopsy and started on hemodialysis because of consecutive oliguria for 7days. Systemic steroid therapy was not performed. Within 2 week, he had improvement in renal function so that dialysis could be discontinued. Then, he was discharged home.
In September 2014, he developed chronic kidney diseases with a peak serum creatinine of 146.0 μmol/L (baseline 3 months prior was 135 μmol/L). Renal ultrasonography revealed left moderate hydronephrosis. In November 2014, he presented to the urologist with left severe hydronephrosis associated with marked thickness of the left ureter wall and retroperitoneal fibrosis discovered by a computed tomography scan (Figures 1 and 2). Although urinary cytology was negative for malignancy, carcinoma of ureter was suspected. In imaging test, it was very difficult to distinguish malignant ureter tumor from benign lesion. Ultimately, we made a preoperative diagnosis as suspicious of malignant ureter tumor and then performed laparoscopic nephroureterectomy.
FIGURE 1.
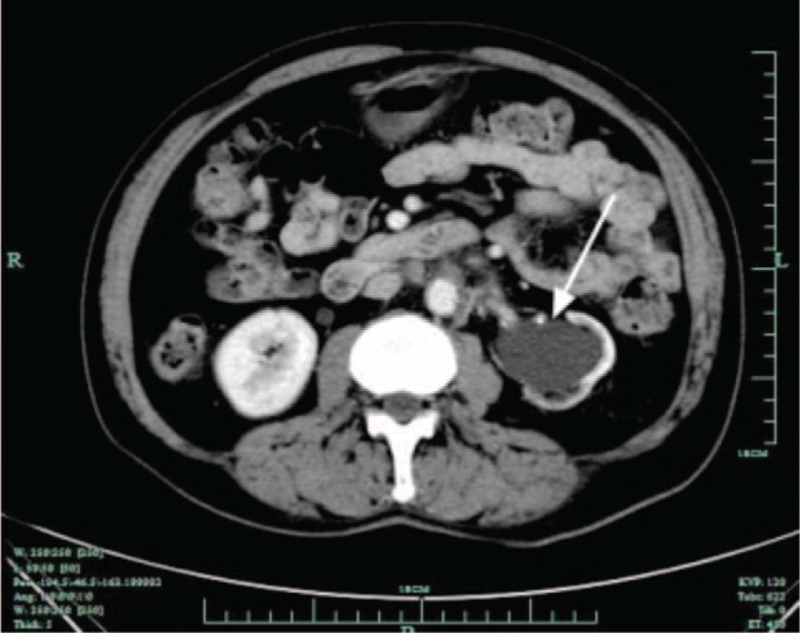
Computed tomography reveals left hydronephrosis (arrow).
FIGURE 2.
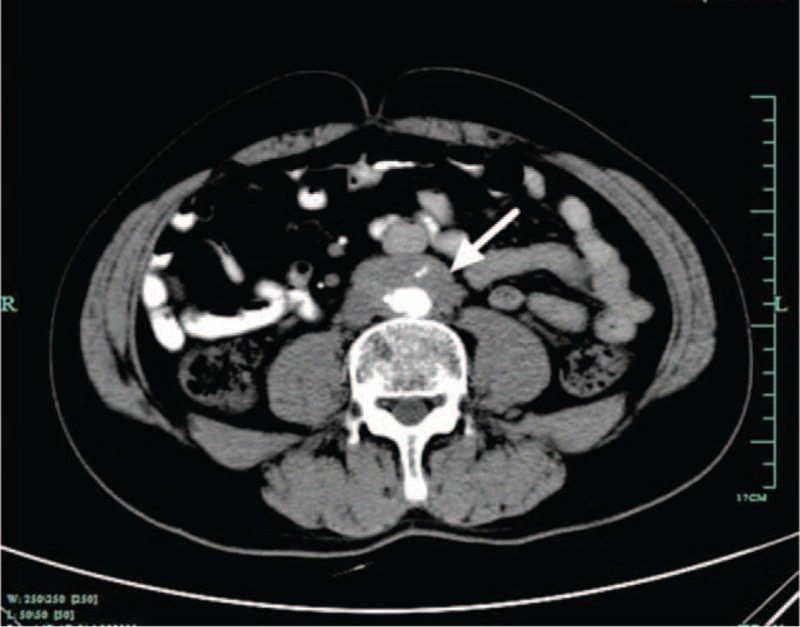
Contrast-enhanced arterial phase computed tomography image shows soft-tissue mass surrounding aorta (arrow), indicating retroperitoneal fibrosis.
Histologic examination revealed lymphoplasmacytic infiltrate with storiform fibrosis in renal cortex and ureter wall. Infiltrating plasma and lymphocytes cells did not show significant cytologic atypia. Also, there was no evidence of malignancy in ureter wall. Immunohistochemically, the IgG4 positive plasma cells infiltrated exceeding 10 cells per 1 high-power field. More than 50% of plasma cells positive for IgG were positive for IgG4 (Figure 3). By electron microscopy, electron-dense deposits were seen in tubular basement membrane (Figure 4). Immunoglobulin immunofluorescence revealed strong accumulation in the tubular basement membrane and Bowman capsule (Figure 5). Based on pathologic results after surgery, we analyzed stored preoperative serum retrospectively. As a result, the patients had polyclonal gammopathy with elevated levels of serum IgG (1910 mg/dl), but serum IgE was normal (76 IU/mL). Hypocomplementemia was found. The serum level of IgG4 was also elevated (913 mg/dL). Finally, we diagnosed IgG4-related kidney disease with renal and ureter lesion according to a diagnostic criteria proposed in the literature.8 After the operation, the patient received steroidal therapy. Oral prednisolone at initial dose of 60 mg/day was administrated after surgery. Within 2 week, he had improvement in renal function. The serum levels of creatinine returned to 94 μmol/l three months after the initiation of therapy and the serum level of IgG4 returned to 53 mg/dL (normal value ≤135 mg/dL). The dose of prednisolone was gradually tapered and he has been treated with 10 mg daily since March 2015, without further symptoms. His renal function and IgG4 levels were normal during follow-up of 10 months.
FIGURE 3.
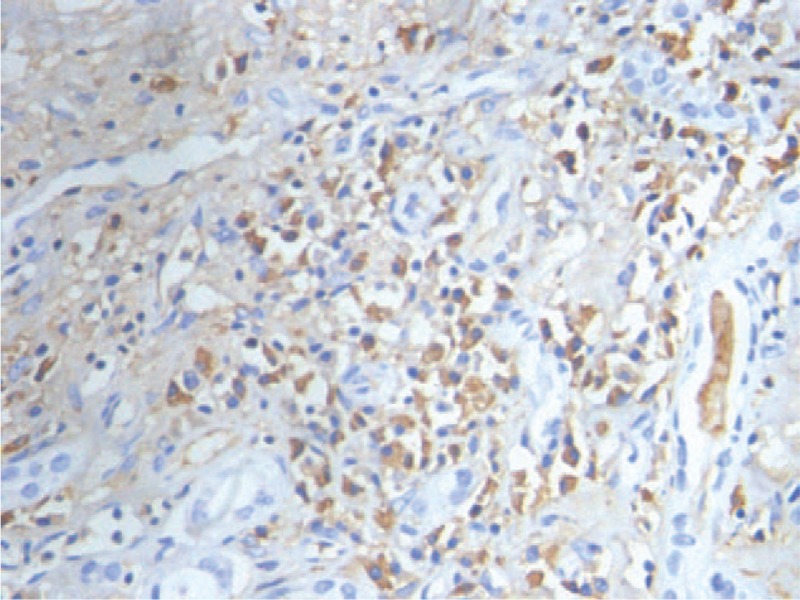
Immunohistochemistry of the inflammatory cells in renal cortex shows IgG-positive plasma cells, with 50% to 60% of these cells also being positive for IgG4 (brown) (×400).
FIGURE 4.
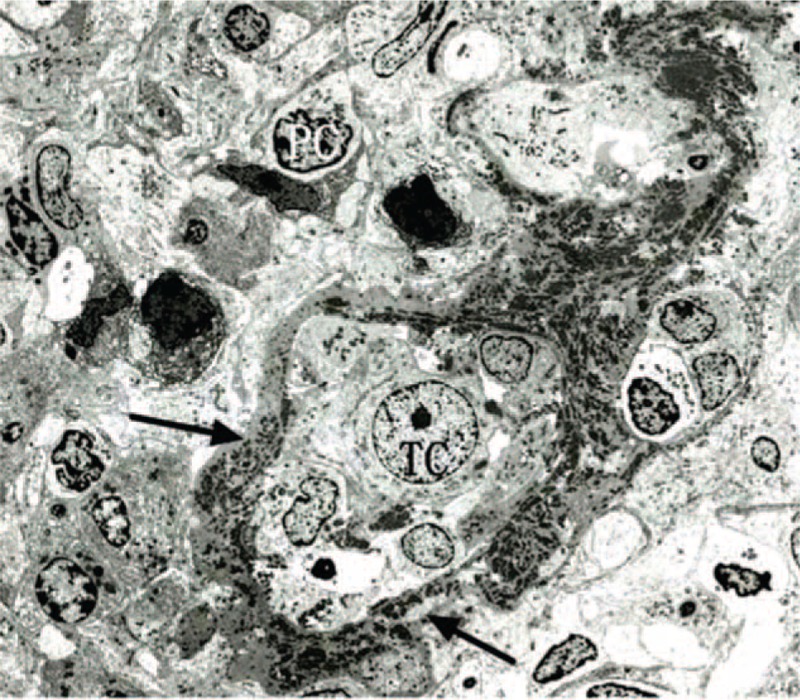
Granular electron-dense deposits (arrow) are accumulated in the thickened tubular basement membrane. TC = tubular cell, PC = plasma cell (×100).
FIGURE 5.
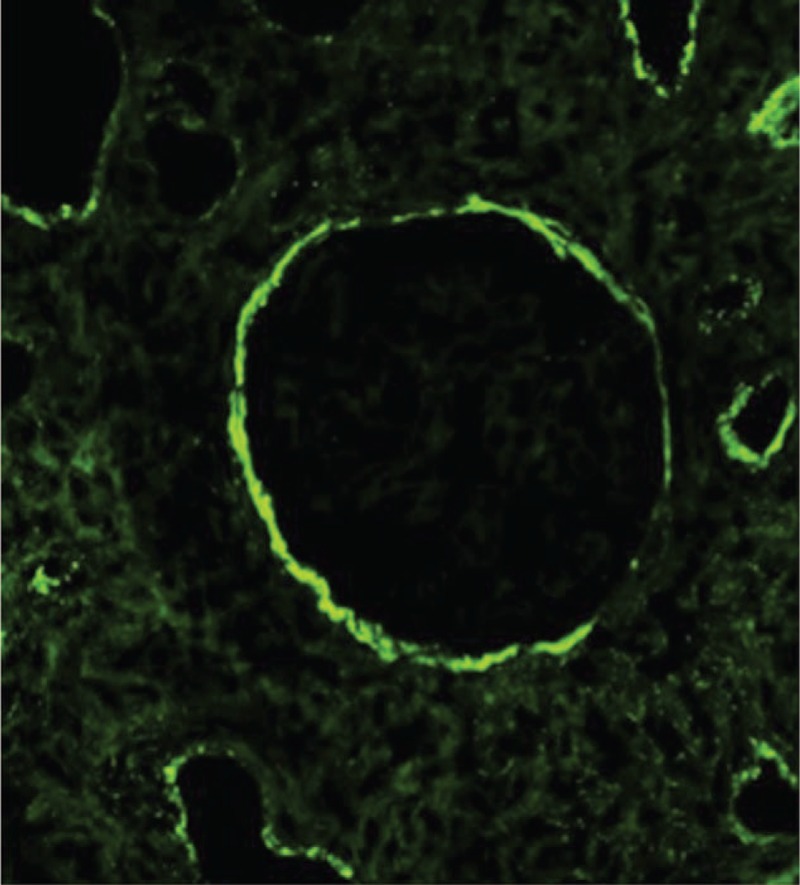
IgG immunofluorescence reveal strong accumulation in the tubular basement membrane and Bowman capsule. Interstitial deposits are also observed.
DISCUSSION
Immunoglobulin G4-related disease is a recently recognized systemic inflammatory disorder characterized by dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, mass-forming lesions, storiform fibrosis and often, raised serum IgG4 levels.3 It was firstly described as a cause of recurrent pancreatitis, but may also affect lymph nodes, salivary glands, and renal interstitium.9,10 It almost affects all other organ systems. The multiorganic involvement supports the concept that IgG4-RD is a systemic disease. Renal involvement is believed to be relatively uncommon, but it may present with a variety of biochemical, clinical, and radiologic abnormalities. The most common renal manifestation is tubulointerstitial nephritis. Although there are no internationally accepted diagnostic criteria for IgG4-RKD, our case patient's clinical manifestations and laboratory test results (elevated serum IgG4 levels, retroperitoneal fibrosis, dense lymphoplasmacytic infiltrate, storiform-type fibrosis) conformed to the diagnostic criteria of IgG4-RKD, which was proposed by Kawano et al.8 In the current case, development of bilateral mild hydronephrosis, however, lead to acute kidney injury, which could be spontaneous remission is an uncommon presentation of IgG4-RD. To our knowledge, a limited number of this kind of presentation of IgG4-RD has been reported.
Previously, imaging abnormality and unexplained renal dysfunction were believed to be the 2 major clinical presentations. In 1 study, approximately half of IgG4-RKD patients manifested renal dysfunction, with renal lesions detected by imaging evaluation for IgG4-RD.8 In some study, more than half of patients presented with acute or progressive renal failure, which required renal biopsy.11–13 Multiple or solitary, round or wedge-shaped, parenchymal low-density lesions are common on computed tomography. In our current case, we, however, did not find parenchymal low-density lesions in the renal; and, the first clinical signs and symptoms of our current case were bilateral mild hydronephrosis and acute kidney injury, which could spontaneous resolution. It was consistent with prior literature.14–16
We found left hydronephrosis with marked thickness of the left ureter wall after a 4-year history of spontaneous remission. Although the thickness of ureter wall was not typical as ureteral cancer, we could not make a diagnosis of a benign tumor by image examinations. The current case is unusual because IgG4-positive plasma cell infiltration and fibrosis were arising in the renal pelvis and ureter.
IgG4-related disease arising in the renal pelvis with thickness of the renal pelvic wall had been previously reported.17 In 2011, Abe et al18 reported a case of IgG4-related fibrosis presented as a unilateral ureteral mass in a 39-year-old man. To our knowledge, there are few medical literatures,19 which report the IgG4-related disease that develop a lymphoproliferative disorder involving the ureter wall and resembled malignant ureter tumor both symptomatically and radiologically. It is important to consider IgG4-related disease in the differential diagnosis of a unilateral malignant ureter tumor. Therefore, this is a rare case of IgG4-RKD mimicking malignant ureter tumor. Finally, nephrologist and pathologists should be aware of the possibility that hydronephrosis because of thickness of ureter wall may be involved in IgG4-RKD.
Recently, the international consensus guidance statement on the management and treatment of IgG4-RD has been published.20 There, however, is no definitive treatment strategy for IgG4-RD, especially in IgG4-RKD. Some important current approach to treatment of the disease are discussed, which includes “Watch-and-wait” strategy, systemic glucocorticoids, rituximab, and steroid-sparing immunomodulators.4
Although spontaneous remission is possible in IgG4-RD, most patients require systemic glucocorticoids to suppress renal inflammation.21 In the current case, we adopted systemic glucocorticoids for the treatment of IgG4-RKD and the renal function improved rapidly. Some literature, however, reported that 25% to 50% of patients relapse on low maintenance dose or after discontinuation of glucocorticoids. Furthermore, larger studies are needed to understand more about clinical manifestations, and approaches to diagnosis and treatment of IgG4-RKD.
CONCLUSIONS
In summary, IgG4-RD may present as hydronephrosis arising in the renal pelvis and ureter, mimicking a ureteral tumor. Although IgG4-RD may have many “faces” with multiorgan involvement, there are few reports of this kind renal manifestation. Thus, it is of great importance to take into account the possibility that hydronephrosis and thickness of ureter wall may be involved in IgG4-RKD. Early recognition of this disease is important, particularly because most patients respond well to glucocorticoids. Large clinical trials are needed for studying clinical manifestations, approaches to diagnosis, and treatment of patients with IgG4-RKD.
Footnotes
Abbreviations: BUN = blood urea nitrogen, CT = computed tomography, IgG4 = immunoglobulin G4, IgG4-RKD = IgG4-related kidney disease.
Lie Jin, Jun Xin and Chui-fen Wu are co-corresponding author
W-HL, LJ, C-FW planned the article and contributed to data collection, discussing content, writing, and reviewing the article. C-XS, M-FM, C-YZ, JX conceived the study and participated in its design, study supervision, and helping to writing the article.
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012; 366:539–551. [DOI] [PubMed] [Google Scholar]
- 2.Zhang H, Li P, Wu D, et al. Serum IgG subclasses in autoimmune diseases. Medicine 2015; 94:e387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu KH, Chan TM, Tsai PH, et al. Diagnostic performance of serum IgG4 Levels in patients with IgG4-related disease. Medicine 2015; 94:e1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vasaitis L. IgG4-related disease: a relatively new concept for clinicians. Eur J Inter Med. Oct 16 2015. [DOI] [PubMed] [Google Scholar]
- 5.Glass LR, Freitag SK. Management of orbital IgG4-related disease. Curr Opin Ophthalmol 2015; 26:491–497. [DOI] [PubMed] [Google Scholar]
- 6.Pradhan D, Pattnaik N, Silowash R, et al. IgG4-related kidney disease: a review. Pathol Res Pract 2015; 211:707–711. [DOI] [PubMed] [Google Scholar]
- 7.Prucha M, Kolombo I, Stadler P. Ormond's disease: IgG4-related disease. Prague Med Rep 2015; 116:181–192. [DOI] [PubMed] [Google Scholar]
- 8.Kawano M, Saeki T, Nakashima H, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol 2011; 15:615–626. [DOI] [PubMed] [Google Scholar]
- 9.Inoue D, Yoshida K, Yoneda N, et al. IgG4-related disease: dataset of 235 consecutive patients. Medicine 2015; 94:e680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandez-Codina A, Martinez-Valle F, Pinilla B, et al. IgG4-related disease: results from a Multicenter Spanish Registry. Medicine 2015; 94:e1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandez Lorente L, Lopez Alvarez D, Garcia Lopez V, et al. IgG4-related disease: description of a case with pulmonary lesions, mediastinal lymphadenopathies and rapidly progressive renal failure. Nefrologia: publicacion oficial de la Sociedad Espanola Nefrologia 2015; 35:218–223. [DOI] [PubMed] [Google Scholar]
- 12.Maeta S, Munemura C, Ishida C, et al. Case report: acute renal failure due to IgG4-related retroperitoneal fibrosis. Nihon Naika Gakkai Zasshi 2012; 101:1079–1081. [DOI] [PubMed] [Google Scholar]
- 13.Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol 2011; 22:1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramasamy VB, Trefor R, Rajamani K, et al. Lesson of the month 2: IgG4-related renal mass with spontaneous resolution. Clin Med (Lond) 2015; 15:396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohshima K, Sato Y, Yoshino T. A case of IgG4-related dacryoadenitis that regressed without systemic steroid administration. J Clin Exp Hematop 2013; 53:53–56. [DOI] [PubMed] [Google Scholar]
- 16.Miura H, Miyachi Y. IgG4-related retroperitoneal fibrosis and sclerosing cholangitis independent of autoimmune pancreatitis. A recurrent case after a 5-year history of spontaneous remission. JOP 2009; 10:432–437. [PubMed] [Google Scholar]
- 17.Kuroda N, Nakamura S, Miyazaki K, et al. Chronic sclerosing pyelitis with an increased number of IgG4-positive plasma cells. Med Mol Morphol 2009; 42:236–238. [DOI] [PubMed] [Google Scholar]
- 18.Abe H, Morikawa T, Araki A, et al. IgG4-related periureteral fibrosis presenting as a unilateral ureteral mass. Pathol Res Pract 2011; 207:712–714. [DOI] [PubMed] [Google Scholar]
- 19.Hamano H, Kawa S, Ochi Y, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet 2002; 359:1403–1404. [DOI] [PubMed] [Google Scholar]
- 20.Khosroshahi A, Wallace ZS, Crowe JL, et al. International consensus guidance statement on the management and treatment of igg4-related disease. Arthritis Rheumatol 2015; 67:1688–1699. [DOI] [PubMed] [Google Scholar]
- 21.Saeki T, Saito A, Hiura T, et al. Lymphoplasmacytic infiltration of multiple organs with immunoreactivity for IgG4: IgG4-related systemic disease. Inter Med (Tokyo, Japan) 2006; 45:163–167. [DOI] [PubMed] [Google Scholar]