

Abstract
Translation of successful target and compound validation studies into clinically effective therapies is a major challenge, with potential for costly clinical trial failures. This situation holds true for the epilepsies—complex diseases with different causes and symptoms. Although the availability of predictive animal models has led to the development of effective antiseizure therapies that are routinely used in clinical practice, showing that translation can be successful, several important unmet therapeutic needs still exist. Available treatments do not fully control seizures in a third of patients with epilepsy, and produce substantial side-effects. No treatment can prevent the development of epilepsy in at-risk patients or cure patients with epilepsy. And no specific treatment for epilepsy-associated comorbidities exists. To meet these demands, a redesign of translational approaches is urgently needed.
Introduction
Preclinical research has enabled the discovery of valuable drugs for the symptomatic suppression of seizures in patients with epilepsy. However, seizures are not adequately controlled in a third of cases, no disease-modifying therapies exist, and comorbidities are a major burden on quality of life. The introduction of new drugs into clinical practice over the past two decades has not substantially changed this situation.1 There is an urgent demand to address the unmet clinical needs of patients. In particular, we need treatments for drug-resistant seizures and for epilepsy syndromes with few or poor treatment options; treatments with improved tolerability; disease-modifying treatments that prevent or attenuate epileptogenesis; and treatments to prevent or ameliorate the common comorbidities that contribute to disability in people with epilepsy. New therapies should also address the special needs of certain subpopulations, including age-specific and gender-specific treatments. Preclinical development in these treatment areas is complex because of heterogeneity in presentation and cause, and might need to be formulated with a specific seizure, epilepsy syndrome, or comorbidity in mind.2
Work in other areas of neurology, such as stroke,3 Alzheimer’s disease,4 spinal cord injury,5 and amyotrophic lateral sclerosis,6 has indicated problems in the design of preclinical studies that probably contribute to poor translation of positive preclinical data to the clinic. Additional challenges for translation in epilepsy include gaps in understanding of the pathophysiology of most human epilepsies, and difficulty in the differentiation of mechanisms involved in ictogenesis, epileptogenesis, or comorbidities in a particular animal model, and from animal models to human epilepsies.
Awareness is increasing of the pressing need to improve the reliability and validity of preclinical studies, to aid the translation of preclinical findings into clinically testable and relevant interventions, and to reduce risk in the therapy discovery process by improving our ability to predict the efficacy, tolerability, and effect of potential new therapies on the quality of life of individuals with epilepsies. Several publications and workshops have drawn attention to the technical and methodological issues that need to be addressed to optimise study design, conduct, reporting, and validation of data across preclinical antiepilepsy and antiepileptogenic therapy development studies.2,6–16 In this Personal View, we aim to provide a framework for the development of guidelines to improve and standardise epilepsy therapy development studies. We draw together previously published recommendations in a single document, addressing specific issues associated with the validation of antiseizure and disease-modifying treatments, and with the discovery and validation of epilepsy biomarkers, and present our views about the prospects for future advances in therapy development. We focus on the new-therapy development process with animal models, from target identification to initial clinical trials. We do not discuss early proof-of-concept studies leading to target or compound identification—ie, the entry studies for translational development. A detailed account of this aspect of therapy development has been published.17 Table 1 gives key definitions used in this Personal View.
Table 1.
Definitions of key terms
| Definition | |
|---|---|
| Epilepsy | Conceptual definition: a disorder of the brain characterised by an enduring predisposition to generate unprovoked epileptic seizures and by the neurobiological, cognitive, psychological, and social consequences of this condition18* Operational definition: (i) at least two unprovoked (or reflex) seizures occurring more than 24 h apart; (ii) one unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk after two unprovoked seizures (at least 60%) occurring over the next 10 years; and (iii) diagnosis of an epilepsy syndrome19* |
| Epileptogenesis | The development and extension of tissue capable of generating spontaneous seizures, resulting in (1) development of an epileptic condition and/or (2) progression of the epilepsy after it is established20 |
| Ictogenesis | The acute neurobiological processes that result in a seizure |
| Epileptogenic abnormality | The pathophysiological substrate(s) responsible for the initiation and/or maintenance of epilepsy |
| Epilepsy comorbidity | A medical or psychiatric condition that occurs in association with epilepsy at frequencies that are substantially greater than those observed in an appropriately matched group without epilepsy A comorbidity might be a cause of epilepsy, a consequence of epilepsy, or its treatments, or a separate condition that is associated with epilepsy because there are common causes for the epilepsy and the comorbidity7 |
| Drug resistance | In patients: the failure of adequate trials of two tolerated and appropriately chosen and used antiseizure medication schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom21* In animal models: persistent seizure activity that does not respond to monotherapy with at least two appropriate antiseizure medications22 |
| Cure | The complete and permanent reversal of epilepsy, such that no seizures occur after treatment withdrawal2 |
| Epilepsy biomarker | An objectively measurable characteristic of a biological process that reliably identifies the development, presence, severity, progression, or localisation of an epileptogenic abnormality23 |
| Epilepsy surrogate endpoint | A laboratory measurement or physical sign that is used in therapeutic trials as a substitute for a clinically meaningful endpoint (epileptic seizures) and is expected to predict the effect of the therapy24 |
| Therapy | Symptomatic therapy or treatment: includes antiseizure drugs or in general antiseizure treatments and anticomorbidity treatments2 Disease-modifying therapy or treatment: includes antiepileptogenic and comorbidity-modifying treatments |
International League Against Epilepsy (ILAE) definition. With the exception of the ILAE definitions, the table gives working definitions used for the purpose of this Personal View.
Animal models of epilepsy
Since the 1930s new epilepsy therapies have advanced into clinical practice based on tests of their effects in the prevention of chemically (eg, pentylenetetrazol) or electrically (eg, maximal electroshock [MES]) induced seizures, or in the slowing of kindling progression, using single-dose or repeat-administration protocols. These tests were generally done by companies with a product in the pipeline or as part of a longstanding effort to discover new therapies through screening—such as the National Institute for Neurological Diseases and Stroke (NINDS) Anticonvulsant Screening Program (ASP; figure 1A). Recent reviews have implied that, despite the introduction to the clinic of new antiseizure drugs based on these screens, the proportion of patients with treatment-resistant epilepsy has not significantly decreased.1 A few models (such as drug-resistant kindling or post-status epilepticus models, and 6 Hz models) have been introduced with a hope of better discriminating the drugs that will work in patients with treatment-resistant epilepsy (figure 1B), but their ability to discriminate has not yet been validated.
Figure 1. Antiseizure drug screening.
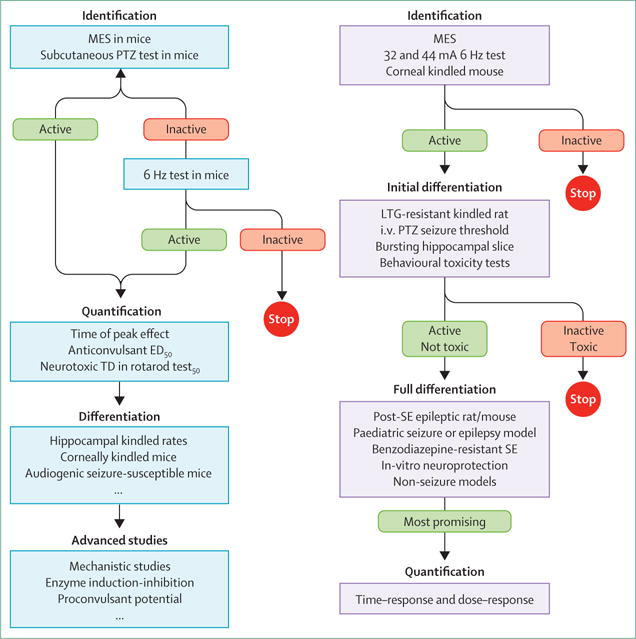
Classic Anticonvulsant Screening Program (ASP) testing protocol (A). Proposed testing protocol of the ASP, based on recommendations of the working group that reviewed the programme in 2011 (B).25 The inclusion of the corneal kindled mouse at the front end provides a chronic seizure model that was missing from the original screening mechanism. LTG=lamotrigine. MES=maximal electroshock. PTZ=pentylenetetrazol. ED50=median effective dose. TD50=median toxic dose. SE=status epilepticus.
We propose the following categorisation of animal models, which will help to guide their selection depending on the target area for therapy discovery. We use the terms acute or chronic to indicate the absence or presence of persisting changes in epileptogenicity, based on either evidence for higher propensity to manifest induced seizures in provocation tests (eg, flurothyl or pentylenetetrazol) or the documentation of spontaneous seizures.
Acute seizure models
Acute seizure models are models of induced seizures with no evidence of persisting changes in seizure threshold or spontaneous seizures. Induction protocols might include single or repetitive exposure to seizure-provoking protocols (including chemoconvulsants, electrical stimulation, hypoxia, hyperthermia), provided that persisting epileptogenic propensity is not an important element for the model to be effective. The advantage of these models is that they are high-throughput and their use in screening has been proven to select drugs that can reduce or stop seizures in the clinic. The disadvantages are that acute seizure models typically will not select drugs (or therapies) that affect or prevent the underlying epilepsy or associated comorbidities; cannot discriminate drugs on the basis of their relative capability to treat seizures; could miss potentially efficacious therapies;26 and might not predict certain adverse or toxic effects noted in human beings.8
Chronic models with high propensity for induced seizures or epileptogenesis
Chronic models with a high propensity for induced seizures or epileptogenesis show a persisting decrease in seizure threshold in provocation tests, but no evidence yet of spontaneous seizures. Such models can be induced (eg, kindling) or genetic. These models have several advantages: testing of propensity for provoked seizures yields faster results and is less technically demanding and labour intensive than is documentation of spontaneous seizures by long-term video-EEG; they offer an alternative for the development of treatments that can reduce the propensity to develop seizures, including, in some cases, drug-resistant induced seizures; and they might be useful in the testing of anticomorbidity therapies (if documented in these models). The disadvantages are that these models cannot test the effects on spontaneous seizures and that a higher propensity to induce seizures might not be an accurate marker of the epileptic state.
Chronic models of epilepsy
Chronic models of epilepsy are models of epileptogenesis with documented spontaneous seizures in long-term video-EEG studies. These seizures can be induced (ie, post-status epilepticus models of epilepsy) or genetic (eg, tuberous sclerosis models; genetic absence epilepsy rats from Strasbourg [GAERS]). The advantage of chronic models over other models is that they might better represent the human disorder, model the development of epileptogenesis including drug-sensitive and drug-resistant spontaneous seizures (enabling testing of antiepileptogenic drugs), and enable better testing of potential for adverse events in the populations of interest.22,8 The disadvantage is that a specific insult (eg, stroke, status epilepticus) might not produce results that are generalisable to epilepsy resulting from other types of injury (eg, traumatic brain injury). Moreover, most human epilepsies do not result from a known insult, and therefore these models might not be fully representative. No therapy has been brought to clinic solely on the basis of efficacy in a chronic model, but this might be due to the fast turnover of screening in the acute models or models of epileptogenesis, or to the limited use of chronic models of epilepsy in therapy development so far. An exception might be the use of mTOR inhibitors in epilepsy due to tuberous sclerosis, which was supported by results of studies in mouse models of tuberous sclerosis before introduction to clinical testing.27,28 The use of chronic models offers promise in meeting some of the treatment gaps described, but it is too soon to confirm whether promising therapeutic leads from such models will result in clinically relevant disease-modifying treatments or symptomatic treatments for drug-resistant epilepsies.
In our opinion, animal models pose several challenging questions. An important issue is brain development. Clinically, epilepsy is highly prevalent in infancy and childhood, and neurotransmitter distribution, metabolic pathways, myelination, and other factors that are probably key in epileptogenesis are substantially different in infants and children compared with adults.29 Both acute target validation and later-life consequences need to be explored for unique opportunities for the infant and paediatric populations with epilepsy.
For each class of epilepsy models, the timecourse of the cascade of events that follow the triggering insult must be established. In this way, discrete targets can be identified that might be useful only for immediate, subacute, or delayed administration, and biomarkers might be found for different steps in the process. Chronic models might therefore be suitable to screen for therapies targeting different stages of epileptogenesis or the associated comorbidities. Finally, the identification of these specific changes will need to be validated between animal and human. Importantly, an effort should be made to use clinically feasible diagnostic methods in animal models—such as imaging, serum markers, EEG, and behavioural tests—to enable translation to human beings.
Another important issue, in our view, is the question of how to select a model. Different options exist for animal model selection for preclinical therapy studies. Why would one model be selected over another? Standard screens (figure 1) are very useful, because they enable comparisons of the potency of the drug relative to other compounds (which is most useful when comparing compounds with similar mechanisms of action), are high-throughput, and have been validated in the clinic. Tests in models of different epilepsy syndromes, such as the genetic absence models, are typically done to establish spectrum of activity and potential for use in specific human syndromes for drugs that are already selected for development, or to predict the potential of a drug to exacerbate seizures in those syndromes. Conversely, certain therapeutic interventions might only be expected to be effective in the epileptic brain, and need to be tested in chronic models. An example would be a drug that blocks proconvulsant inflammatory pathways. If these proconvulsant inflammatory pathways are activated postictally or late in epileptogenesis, such an intervention would not be expected to prevent acutely induced seizures. By contrast, treatments that prevent induced seizures in otherwise naive brains might not have the same effect on spontaneous seizures manifesting during the chronic epileptic state. The acute experiments will continue to be important in the discovery of drugs but, to make further progress, the model of therapeutic testing in epilepsy will have to shift to also include chronic models (genetic or acquired) in which an epileptogenic alteration (eg, a genetic mutation) or insult (eg, stroke, status epilepticus) ultimately leads to recurrent, spontaneous seizures. We emphasise that a shift to inclusion of chronic epilepsy models is in process, and a crucial need exists to address proper methods for chronic trials in epilepsy so that these studies will be successful and lead to effective new therapies. Table 2 outlines recommendations adapted for the early preclinical epilepsy research studies.8
Table 2.
Recommendations practices to improve the design and reporting of preclinical epilepsy research studies
| Recommendations | |
|---|---|
| Rationale |
|
| Experimental design |
|
| Treatment delivery |
|
| Outcome assessment |
|
| Data collection, analysis, and reporting |
|
| Interpretation |
|
| Publication issues |
|
Finally, the predictive validity of animal models deserves some attention. The availability of animal models that can predict treatment responses in specific epilepsy syndromes or seizures is expected to reduce risk in therapy discovery. However, no specific criteria exist that define the predictive validity of epilepsy models, and the formulation of such criteria is likely to face several challenges. For example, how do we define the borders between a model with poor predictive validity versus a treatment-refractory animal model? A new model in which many of the syndrome-appropriate drugs do not work could be a good model of a treatment-resistant human state or, alternatively, could be a poorly predictive model for therapy development. The same conceptual challenge also holds for human trials; the response of a person with treatment-refractory epilepsy is poorly predictive of the response of individuals with non-refractory epilepsies. Most importantly, in animal models of epilepsy syndromes with very few or imperfect available treatments—eg, infantile spasms or Dravet syndrome—predictive validity of a model becomes less meaningful and might hinder the development of therapies for drug-resistant populations. Going forward, the identification of the mechanism by which targeted treatments work and their validation in treatment-responsive and treatment-refractory populations, in both animal models and relevant patient cohorts, could offer a guide to predict clinical success.
Discovery of antiseizure and disease-modifying treatments
Antiseizure treatments
As we have indicated, the need is pressing for new therapies to treat seizures in many diverse paediatric and adult patients who do not respond well to existing drugs. Antiseizure treatments can be selected in a non-specific way (irrespective of potential success in any specific syndrome) or they can be selected to target syndromes or patient populations with unmet needs, or to target the population of patients who have not responded to other treatments (therapy resistant).
The testing proposed by the Anticonvulsant Screening Program would still rely heavily on acute models for initial candidate selection (figure 1B), but would then use other animal models to characterise compounds. However, very little is known about the pharmacological profiles of these other models,22 which have not been validated for their ability to predict clinical efficacy in patients who are pharmacoresistant. Antiseizure therapy targeted at specific syndromes can be identified using genetic models; these models are exemplified by GAERS, which model human absence epilepsies. The advantage is that GAERS provide an example of spontaneous, naturally occurring seizures and, in this sense, most closely resemble human epilepsy; they have provided the best prediction of efficacy in specific human syndromes. The use of transgenic approaches also enables the generation of animals that carry genetic mutations that result in human epilepsy (eg, in SCN1A), with the advantage that therapies effective in these models might target specific human epilepsy mechanisms. The disadvantage of both spontaneous and transgenic epilepsy models is that they only represent a few syndromes, and therefore treatments effective in these animals are not necessarily relevant to most patients.
We anticipate that, with better predictive screening methods and improved study design and reporting, seizure control in patients who are pharmacoresistant will be improved. Because epilepsy is a spectrum disorder, more attention needs to be paid to non-seizure components, such as neuropsychiatric disorders, and the extent to which drugs ameliorate symptoms other than seizures. Because seizures activate so many elements of neuronal function that are important for normal cognition and behaviour, further development of treatments that isolate seizure-only mechanisms without interfering with other brain functions would fill a treatment gap. Also, the development of successful therapies is more likely to be achieved when the complex network interactions that result in seizure generation, propagation, and termination are better understood, from basic research into the mechanisms of epilepsy. Therefore, it is paramount that more funding be appropriated to bring epilepsy therapy development in line with funding spent on other, less common neurological disorders.30
Disease-modifying treatments
Disease-modifying treatments are, by necessity, assessed using chronic models. Disease-modifying treatments can be either antiepileptogenic (ie, therapy prevents or alleviates the development of epilepsy or its progression) or comorbidity modifying (table 1). Results of several preclinical proof-of-concept studies have provided evidence for positive treatment effects on acquired epileptogenesis with two causes: status epilepticus and traumatic brain injury.31 Evidence shows that treatments, including some antiseizure therapies, can modulate epileptogenesis and alleviate the severity of comorbidities in some genetic animal models.32,33 None of these positive experimental results has advanced to an established antiepileptogenic treatment in the clinic, but several clinical trials are in progress and recruiting patients, in which epileptogenesis is a primary or secondary outcome measure. One trial uses low-dose adrenocorticotropic hormone in infants to prevent the development of a paediatric epilepsy (West’s syndrome; NCT01367964), and most of the others are investigating the effect of treatment with standard antiseizure drugs such as levetiracetam (NCT01463033) and topiramate (NCT00598923) to prevent seizures and epilepsy after a brain insult, particularly after traumatic brain injury (www.clinicaltrials.gov). Paradoxically, none of these treatments has shown efficacy in preclinical models of post-traumatic epilepsy; in these cases, an attractive mechanism of action was deemed to outweigh the need for preclinical efficacy testing before clinical trials (whether this is appropriate has not been established). In summary, few clinical trials are investigating treatments targeting neurobiological processes that have been implicated in animal models as being involved in epileptogenesis.
In many experimental models, treatments have been initiated after a known epileptogenic insult (genetic or acquired), and the development and severity of epilepsy or the severity of behavioural or cognitive impairment have been used as outcome measures. Several treatments have affected epilepsy development in a single model, but only rapamycin has shown favourable effects on epileptogenesis triggered by several factors (genetic risk factors, status epilepticus, traumatic brain injury), although it also failed in some preclinical studies. Preclinical experience with mTOR inhibitors is an excellent example of how successful a treatment can be if clear evidence exists for target relevance and engagement, and how random the treatment’s success rate might be if used with no knowledge of target relevance or modification. Whether a magic bullet will be found that will alleviate different types of epileptogenesis, or whether such a goal needs to be personalised for different indiviuals or epilepsy syndromes or is even feasible, is unclear.
How do we decide which disease-modifying treatments identified in proof-of-concept studies should proceed to a preclinical study? One criterion should be that we have a thorough understanding of the nature of epilepsy in the model used for proof-of-concept studies, which is crucial for data interpretation. For example, what is the speed of epileptogenesis and inter-animal variability? If the target is known, evidence of target engagement should exist.
Table 3 summarises the requirements for an adequately powered disease-modifying preclinical trial combating epileptogenesis. The preclinical antiepileptogenesis study should show a reduction in seizure frequency (reported as seizures or recording period) or an increase in the percentage of seizure-free animals, or both.20 One could also report the percentage of animals with a greater than 50% reduction in seizure frequency—a common endpoint in clinical trials. To obtain reliable data, seizures should be monitored when seizure frequency has stabilised in the particular model, and be of sufficient duration to account for the usual fluctuations in seizure frequency in individual animals. Information on seizure duration and type would be clinically important. Further, assessment with long-term video-EEG should be undertaken to enable seizure characterisation. This assessment generates a need for rapid analysis of large amounts of data, which, with the available methods, is a major bottleneck. Another practical problem is the delivery of treatments at effective levels for long periods.
Table 3.
Recommended features of a preclinical disease-modifying monotherapy trial in adult rodents
| Recommendations | |
|---|---|
| Study design |
|
| Animals |
|
| Experimental model |
|
| Number of studies |
|
| Timing, dosing, and duration of treatment (if target is known) |
|
| Potential outcome measures |
|
| Statistics |
|
| Reporting |
|
See also reference 20 for an expanded explanation of these recommendations. With reference to the choice of animal, the rat was deemed to be the primary species to use because it has some advantages over mice (eg, the larger size of the brain and the body is advantageous for EEG and MRI, and makes repeated blood sampling possible). Efficacy in more than one species adds value, and genetically modified mice (or rats) could offer some advantages.
The final challenges will be in moving from a positive preclinical disease-modifying study to a clinical trial. In most of the available preclinical antiepileptogenesis studies, treatment was started after the insult, before epilepsy onset, and animals were followed to establish seizure development and seizure frequency as endpoints. Similar studies in people would require many patients and follow-up periods of years to decades, which make the studies unfeasible. In consideration of which disease to model, and how to design a preclinical disease-modifying trial, it is important to keep a focus on the ability to intervene at a similar timepoint in the human disease, and the number needed to treat to prevent or reverse one case of epilepsy. The necessity of treating many patients, many of whom would not be affected even without treatment, might reduce motivation to enrol in a trial, especially if the intervention produced potential adverse effects.26 An alternative clinically relevant, and potentially more feasible, study design would be to initiate the antiepileptogenesis treatment after epilepsy diagnosis, and record its effects on progression of epileptogenesis and comorbidities. Another challenge relates to the establishment of preclinical endpoints that can be reliably assessed in the clinic—for example, seizure number, the major endpoint in preclinical anti epileptogenesis studies, is difficult to assess reliably in human beings.34,35
In view of the profound effect of behavioural and cognitive epilepsy comorbidities on quality of life, the absence of effective treatments for them, and the likelihood that the molecular mechanisms of epileptogenesis and comorbidities overlap, we advocate that the designs of preclinical studies enable the detection of effects on both epileptogenesis and comorbidities.6,7 Age-specific effects of therapies will also have to be considered. Most of the proof-of-concept testing of disease-modifying treatments has been done only in adult animals.
Another concern is that we are modelling only a few clinically relevant initial hits, and the fact that timing of the initial hit can be defined only in a subpopulation of patients might complicate the translation of preclinical studies in which the therapeutic time window is strictly associated with occurrence of the insult. Also, whether the treatment could be combined with surgical removal of the focus (if known) and novel devices, or both, should be considered.
Identification and use of biomarkers
Identification of reliable epilepsy biomarkers would greatly ease antiepileptogenic and antiseizure therapy discovery if the biomarkers could be used to devise more cost-effective and rapid-throughput approaches to screening. Biomarkers would also greatly reduce the cost of clinical trials to validate antiepileptogenic and antiseizure therapies by enriching subject populations, and by acting as surrogate endpoints to document remission, prevention, or cure without the need to wait for seizures to occur. Unfortunately, no validated biomarkers exist that can be used to reliably measure aspects of epilepsy in the same way that the blood glycosylated haemoglobin test is used as a biomarker of diabetes.
Reliable epilepsy biomarkers could revolutionise diagnosis and treatment. Biomarkers that predict the development of an epilepsy condition before it manifests would enable testing of antiepileptogenic interventions. Biomarkers that reliably indicate the presence of an epilepsy disorder would enable diagnosis and treatment after a single seizure without the need to wait for further seizures, and could distinguish epilepsy from disorders with nonepileptic seizures without the need to monitor with expensive video-EEG. Biomarkers that indicate the severity of an epilepsy disorder could be used to rapidly tailor antiseizure interventions to individual patients, avoiding the time-consuming and potentially dangerous trial-and-error approach. Biomarkers of disease progression or pharmacoresistance would enable more timely referral for aggressive alternative therapies such as surgery. Biomarkers that localise epileptogenic brain tissue could be used to establish the extent of surgical resection without the need for expensive, and sometimes invasive, pre-surgical assessments. Figure 2 shows how epilepsy biomarkers, measured at different timepoints, could be used to indicate processes of epileptogenesis and the propensity to generate epileptic seizures (ictogenesis). Identification of reliable biomarkers might also elucidate underlying fundamental mechanisms that could be targets for the development of new antiepileptogenic and antiseizure therapies. Moreover, if the biomarkers were present in both animals and human beings, they could provide a link between preclinical and clinical studies.
Figure 2. Epilepsy biomarkers.
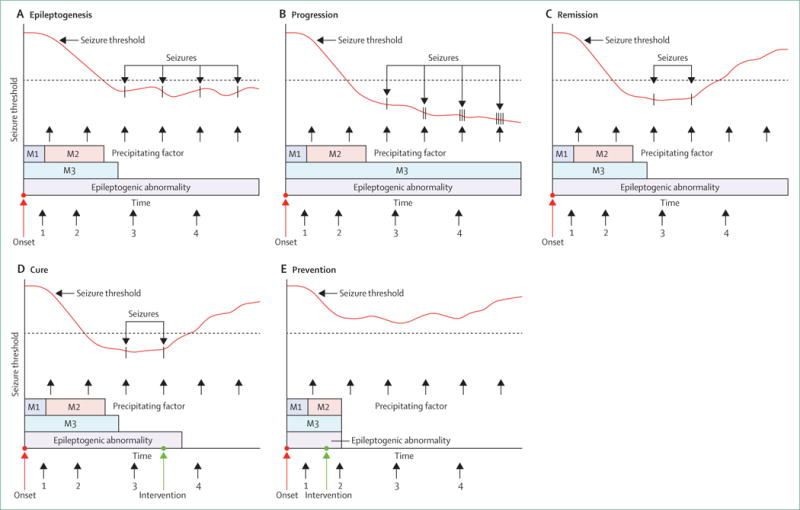
The bars at the bottom of each panel indicate epileptogenic mechanisms (M1, M2, and M3) involved in the development of an epilepsy condition. Some of these processes are time-limited and others can continue as the enduring epileptogenic abnormality responsible for the propensity to generate spontaneous seizures, in response to precipitating factors that might or might not be easily identified. The red line represents the seizure threshold, which decreases during epileptogenesis to a point at which spontaneous seizures can occur. Epileptogenesis (A). The cascade of epileptogenic mechanisms can be measured at points 1 and 2. These points and might enable staging of the epileptogenic process. The existence of an enduring epileptogenic abnormality can be measured at points 3 and 4, and other measures at these timepoints could indicate that the seizure threshold is at a level where spontaneous seizures can occur. Progression (B). The cascade of epileptogenic abnormalities at points 1 and 2 would be similar to that in part A, but measures at points 3 and 4 would show that a more pronounced epileptogenic abnormality persists. These timepoints would also indicate that the threshold continues to decrease, causing seizures to be more frequent and more severe. Remission (C). Following an intervention after point 3, a measurement at point 4 would show that the threshold is raised above the level at which spontaneous seizures occur; however, the epileptogenic abnormality persists. This persistence is in contradistinction to cure (D). In this case, a measurement taken at point 4 would show that the return of threshold to a normal level is due to the fact that the epileptogenic abnormality has gone. Prevention (E), in which an intervention between points 1 and 2 results in a complete elimination of the epileptogenic process. Measurements at points 3 and 4 would then show that the threshold never dips below a level at which spontaneous seizures would occur, and that an epileptogenic abnormality never developed. Reproduced from Engel and colleagues,23 by permission of John Wiley and Sons.
Findings from research into fundamental mechanisms of epileptogenesis and ictogenesis have identified several potential targets that could be used to develop biomarkers,36 and several potential epilepsy biomarkers are under investigation. Electrophysiological biomarkers include EEG transients recorded from the scalp and directly from the brain. The most promising are pathological high-frequency oscillations (pHFOs)—brief events with frequencies ranging from 80 to 600 Hz—which are only reliably identified when recorded with intracranial electrodes.37,38 Although pHFOs might soon be used to localise the epileptogenic region for resective surgical treatment and could aid drug discovery in animal models, noninvasive approaches to measure pHFOs will be necessary before they can enrich clinical trial populations, serve as surrogate endpoints, or be used for routine diagnostic purposes. Other aspects of EEG activity, as well as transcranial magnetic stimulation, are also being investigated as potential biomarkers of epileptogenic excitability.
Potential imaging biomarkers of epilepsy development include MRI-identified alterations in the hippocampus after prolonged febrile seizures, which might predict progression to mesial temporal lobe epilepsy with hippocampal sclerosis.39 Such a biomarker could permit intervention with an antiepileptogenic drug. Positron emission tomography with α-methyltryptophan (AMT-PET) provides another potential epilepsy biomarker, especially in patients with tuberous sclerosis who have epileptic seizures and multiple tubers40—the AMT tracer is selectively taken up by the specific tuber responsible for seizure generation.
Research is underway to identify molecular and cellular biomarkers from blood, CSF, or brain tissue that might provide important information about the epileptogenic process or the potential for ictogenesis.41,42 We anticipate that a single biomarker is unlikely to emerge and a panel of biomarkers will be necessary instead. In future, characteristic patterns of changes in genetic expression might serve as biomarkers of epileptogenesis and ictogenesis, and these could conceivably be measured in white blood cells, which would allow us to predict the development of epilepsy, diagnose epilepsy, and test the effectiveness of antiepileptogenic and antiseizure treatments with a simple finger stick.
General issues
Economics
Epilepsy therapeutics is perceived by many in industry as a saturated market. Many pharmaceutical companies no longer develop or license antiseizure drugs or other therapies because it is difficult to justify the huge financial investment needed to obtain approval for patient use of drugs that have the same efficacy as established treatments. Enthusiasm for innovation might be restored if some of the unmet needs we have noted (such as more effective therapies in the treatment-resistant population, therapies with better tolerability, or the development of targeted therapies against a specific seizure type or epilepsy syndrome in a patient population that could take advantage of orphan drug status) could be addressed. The implementation of new or modified regulatory statutes might encourage companies to pursue the development of new antiseizure drugs and novel therapeutic approaches. Desirable changes in regulation include increasing the life of patents, providing a mechanism for monotherapy licences for first-in-class compounds, orphan disease status for particular forms of epilepsy, and approval of a broad-spectrum epilepsy indication that does not specify age, seizure type, or adjunctive use restrictions.
Multicentre preclinical trials
The organisation of multicentre preclinical studies modelled on phase 2 or 3 clinical trials might ease translation and de-risk clinical studies.43 An important explanation for the frequent failure for positive results from preclinical studies in animal models to translate into positive clinical trials in human beings is thought to be the paucity of methodological rigour in preclinical studies compared with phase 2 or 3 clinical trials.16,43–46 The pivotal phase 2 or 3 clinical trials required by regulatory agencies to show efficacy and safety of a potential new treatment have randomised, double-blind, controlled study design, pre-specified study endpoints, large numbers of participants (hundreds) established according to pre-study sample-size calculations, rigorous statistical analysis specified a priori, involvement of many centres, careful monitoring of data and study site, and mandatory study registration. These regulations minimise biases and the chance that false-positive results will be obtained and reported. By contrast, most preclinical studies involve small numbers of animals (as few as 4–5, and rarely more than 30 per group) that are not pre-specified on the basis of power analysis, are done in a single laboratory without rigorous blinding or statistical analysis, without data or site monitoring, and with a publication bias towards positive results. As a consequence, false-positive results are much more likely to be reported from preclinical studies than from phase 2 or 3 clinical studies.47 However, the decision to proceed with clinical studies for a potential new treatment is typically made on the basis of these results, often even without validation in a second laboratory. Industry reports anecdotally that more than 70% of compounds reported to be effective in academic laboratories do not replicate when tested in-house.45
In view of these challenges, it is not surprising that many of the potential treatments for neurological conditions identified in preclinical studies have not shown efficacy in clinical trials. Trials of neuroprotection for stroke or in neurodegenerative conditions exemplify the problem. In epilepsy, compounds that are reported to have antiseizure activity in preclinical studies have mostly had antiseizure effects in clinical trials. The success of these compounds probably rests on the wide availability of efficient and practical seizure models for drug testing, which means that, generally, the compounds that have been taken into clinical trials have been effective in several, different animal models, thus reducing the chances of a false-positive result. However, rigorous double-blind comparative preclinical studies have not been done to show that these new compounds have incremental efficacy over established antiseizure drugs, which is probably why they did not result in significant improvements in the overall proportion of patients with drug-resistant epilepsy.48
To address these issues, and thereby potentially improve the reliability of preclinical testing results to predict which treatments will show efficacy in the clinic, we advocate the implementation of a preclinical phase 2 multicentre drug trial model based on clinical phase 2 or 3 studies (figure 3).42,43 The goal is to improve the evidence from preclinical studies for treatments that have shown strong promise in early proof-of-concept (preclinical phase 1) studies. This improvement should reduce the risk for expensive clinical studies and therefore increase the appeal for funders (industry and government) to invest in their clinical development. Such an approach is already in the advanced stages of planning in other specialties, notably the MultiPART initiative in stroke research.
Figure 3. Different phases of preclinical therapy discovery with the proposal of optional preclinical trials.
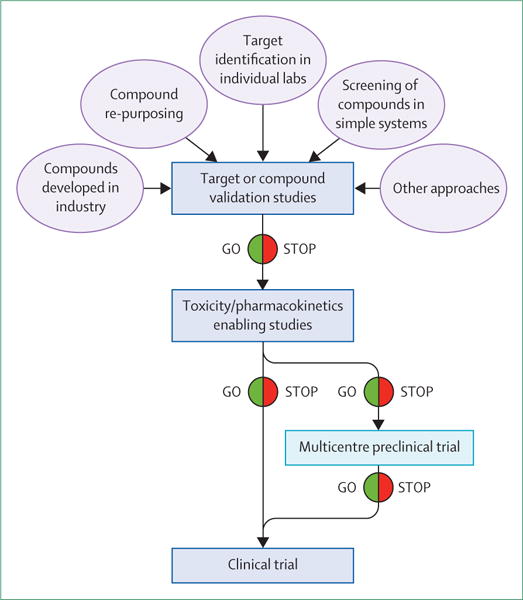
One option that could be considered to enable translation and de-risk clinical studies is the organisation of multicentre preclinical studies modelled on phase 2 or 3 clinical trials.
These preclinical multicentre studies should focus on efficacy, and, although some pharmacokinetics and toxicology data can be collected, this should not be the primary goal. These studies should use the best, most clinically relevant model for the targeted epilepsy syndrome. Assessment of the efficacy of the potential new therapy should be against endpoints where a treatment gap in clinical practice exists (drug-resistant seizures, epileptogenesis, or comorbidities). The studies should use rigorous blinding to minimise observer bias. Studies should be multicentred, ideally involving four to ten laboratories, which would minimise biases associated with a specific laboratory, and data and sites should be carefully monitored as in clinical trials. A central coordinating site should be set up, which should be independent from the data collection sites. The studies should compare the new therapy with an inactive control (ie, vehicle), and ideally also with at least one appropriate, established antiepileptic drug in clinical use to generate rigorous evidence for incremental efficacy of the test therapy. Ideally, at least two different models should be used to minimise the chances that results are model-specific. When possible, these models should be true epilepsy models that show spontaneous recurrent seizures. The primary study endpoints and statistical analysis should be predetermined by expert biomedical statisticians with experience in clinical trial design, and the animal numbers should be based on power calculations from phase 1 preclinical studies.
Phase 2 multicentre preclinical studies will be more expensive and resource-intensive than would traditional preclinical studies; however, they will still need far fewer resources than would failed phase 2 or 3 clinical trials. Such an approach would probably be most useful in a situation in which mounting a clinical trial would be high-risk, lengthy, and expensive—eg, a trial of antiepileptogenesis. For successful implementation, a combination of government and industry funding will be needed. The government funding could establish the basic structures, protocols, laboratory credentialing, and databases. Industry or venture capital (ie, the sponsor) would fund the primary costs of the study, potentially supplemented with government and philanthropic grants. Such a radical new model would need validation, which could be done by testing of drugs that have been successful in clinical trials and practice in a phase 2 multicentre preclinical study, ideally compared with a drug that was promising in traditional phase 1 preclinical studies but showed poor efficacy in subsequent clinical trials or in clinical practice.
Publication issues
To fully and objectively assess the therapeutic potential of tested antiepilepsy therapies and the validity of biomarkers, the publication of good-quality preclinical studies, even if negative or not novel, will be essential. A negative study testing the treatment on a different seizure model might provide crucial information about its therapeutic indications. A replication study for the same therapeutic indication would strengthen the initial findings. Negative and replication studies are crucial to de-risk the process of selection of the most promising antiepilepsy therapies and the best study design for clinical testing. For the same reasons, results of studies that could not be completed should be made available through publication or logging in depositories that could be used for meta-analyses.
It is also important to recognise that a single preclinical study might not be able to address all of the issues that need to be answered before transitioning to a first-in-human study. To know the efficacy and tolerability of a drug in both sexes or in several age groups, species, or models would be important (table 4). Inclusion of all these variables in a single study would be beyond the capabilities of a single laboratory. Therefore, our recommendations in table 4 should not be taken as reasons to reject good-quality, rigorous proof-of-principle preclinical studies that appropriately address their study goals, simply because these goals might relate to a specific focal area of therapy development and validation.
Table 4.
Issues to be considered before selection of antiepileptic therapies for clinical testing
| Recommendations | |
|---|---|
| Species | Insufficient information is available to recommend efficacy testing in more than one species. Toxicity testing in at least two species is, however, a regulatory requirement |
| Sex | One sex might be sufficient for a particular study, but both male and female animals should be tested before Investigational New Drug (IND)-enabling studies |
| Age | Antiepileptic therapies intended for use in newborn babies, infants, or children should be tested in age-specific models |
| Animal model | Testing in at least two different animal models (if more than one validated model is available) might inform on the validity of the findings and the broadness of the therapeutic indications of the antiepilepsy therapy. Cautionary interpretation of disparate results is advised if the predictive validity of a model is unknown |
| Tolerability in a disease model | Tolerability testing in disease models might identify relevant toxicities that do not occur in healthy animals |
| Intent of intervention | Testing for antiepileptogenesis, disease modification, or antiseizure effects in animal models should mirror intended future trials in human beings |
| Therapeutic window | Obtaining information on the optimum therapeutic timepoint of intervention or dose range is advised. Care should be taken to avoid studies that intervene at a timepoint that cannot be duplicated in human trials (eg, before insult) |
| Target validation in humans | Validation of the target mechanism in human beings might help to de-risk selection of promising therapies for clinical testing |
Conclusions
Despite its important achievements, epilepsy therapy development still needs to address the major clinical gaps in provision of more effective and better-tolerated antiseizure treatments, including for drug-resistant seizures, and therapies for disease-modification and comorbidities. We advocate four policies: adoption of better practice standards in the design, analysis, and reporting of preclinical studies, to assist transparency and translation to clinical practice; validation and selection of animal models for drug-resistant seizures and epileptogenesis, including epilepsies affecting specific sub-populations; development and validation of epilepsy biomarkers and surrogate endpoints that could substantially de-risk therapy development; and creation of platforms to report sound but negative or fragmentary results. We further propose that phase 2 multicentre preclinical studies have the potential to de-risk phase 2 and 3 clinical trials (panel).
In conclusion, epilepsy research has been successful in translation in the past and is entering a new era with new challenges and opportunities. Our optimistic view is that a wise, careful, and highly determined and internationally coordinated effort, if adequately supported, will lead to those truly innovative new therapies that the tens of millions of people with epilepsy worldwide are waiting for.
Panel: Key issues in anti-epileptic therapy development in animal models.
Animal models
Develop and validate models of paediatric epilepsy useful for therapy development
Pay attention to the stage of the disease: markers and therapies might be stage-specific
Align diagnostic methods used in preclinical research with those used in clinical epileptology
Increase the use of chronic models
Validate treatment mechanisms in treatment-responsive and treatment-refractory populations
Therapy discovery
Improve standards of preclinical research
Consider manifestations other than seizures (comorbidities) when assessing therapeutic effect
In antiepileptogenesis, consider that effects could be specifically time-linked to the epileptogenic insult, which is not always identifiable in human beings
Better understand the networks underlying ictogenesis and epileptogenesis
Develop predictive biomarkers
General issues
Overcome the resistance of pharmaceutical industries to invest in epilepsy therapy by focusing on existing therapeutic gaps (epileptogenesis, drug-resistance, comorbidities)
Pursue changes in regulation (increased patent life, orphan disease status for certain epilepsies, etc)
Increase the rigour and statistical power of preclinical studies by organisation of multicentre preclinical trials
Create platforms to report incomplete or fragmentary findings in a way that might permit future rigorous analyses
Search strategy and selection criteria.
We searched PubMed and Embase for papers published up to the end of March, 2014, using the MeSH term “epilepsy”, subheading “therapy”, and crossing it with the MeSH terms “Drug Evaluation, Preclinical” or “Translational Medical Research”. Moreover, we did a free search using the terms “epilepsy”, “epileptogenesis”, “preclinical”, and “development”. We also searched references of relevant publications. We included only papers published in English. We generated the final reference list on the basis of relevance to the topic of this Personal View.
Acknowledgments
The contents of this article are based on proposals made at a workshop of the Joint International League Against Epilepsy (ILAE) and American Epilepsy Society (AES) Translational Task Force to optimise and accelerate preclinical epilepsy research (London, UK, Sept 28–29, 2012). MS, ASG, TJO’B, and JAF organised the workshop. The work of this Task Force has received co-sponsorship from the ILAE, AES, Citizens United for Research in Epilepsy (CURE), Epilepsy Therapy Project, and Autism Speaks. The opinions expressed in this manuscript are ours and do not necessarily reflect the individual opinions of the workshop participants. We thank contributors to the Task Force for the wonderful discussions at the London workshop. We also thank Andrea Pizzirani for preparation of the figures before submission. MS has received funding from the European Community (FP7-PEOPLE-2011-IAPP project 285827 [EPIXCHANGE] and FP7-HEALTH project 602102 [EPITARGET]) and the Italian Ministry for Education, University and Research (PRIN 2010–11 project 2010N8PBAA [INBDNF]). ARB-K has received funding from the US National Institute of Neurological Disorders and Stroke (NINDS; R01NS38595; 1R21NS083057-02) and the US Department of Defense (W81XWH-11-1-0501). JE has received funding from the US National Institutes of Health (NIH; P01 NS02808, R01 NS33310, U01 NS42372, and P20 NS80181), AES, CURE, and the Epilepsy Therapy Project. ASG has received funding from NINDS (NS078333), CURE, Autism Speaks, the US Department of Defense (W81XWH-13-1-0180), and the Heffer Family and Segal Family Foundations. FEJ has received grants from NINDS (NS031718, NS 080268, NS-080565). SLM has received grants from NINDS (NS20253, NS43209, NS45911, NS78333), the US Department of Defense (W81XWH-13-1-0180), CURE, and the Heffer Family and Segal Family Foundations. TJO’B has received funding from the National Health and Medical Research Council, Australia (grants 1059858, 1059860, 1075117, 1017063, 1006077, and 1001206), and the Royal Melbourne Hospital Neuroscience Foundation. AP has received funding from the European Community (FP7-HEALTH project 602102 [EPITARGET]), the Academy of Finland (grant numbers 272249, 273909, 267199), the Sigrid Juselius Foundation, the European Science Foundation (EpiGENet), and ERA-NET Neuron II. KSW has received funding from NINDS (contract 271201100029C; grants NS078331 and NS065434). JAF has received funding from The Milken Foundation, the Epilepsy Research Foundation, and NINDS (2U01NS038455-11A1).
Footnotes
For more on MultiPART see http://www.dcn.ed.ac.uk/multipart
Contributors
MS, ARB-K, JE, ASG, TJO’B, AP, KSW, and JAF contributed equally to the conception, design, literature search, and writing of this Personal View. FEJ and SLM contributed to the conception and design, and the editing of the second and third versions.
Declaration of interests
JE has a patent pending for a magnetonanoparticle for functional MRI; JE has received no fees to date. (WO2009/123734A1 and WO2009/123735A1). ASG has received royalties from Morgan & Claypool Publishers and John Libbey Eurotext, and consultancy honorarium from Viropharma. FEJ has received investigator-initiated grant support from Eisai Pharma and Lundbeck Pharma. SLM has received consultancy honorarium from Lundbeck and UCB Pharma. TO’B has received research funding from Sanofi-Aventis, UCB, SciGen, GlaxoSmithKline, Novartis, and Janssen-Cilag, and speakers’ honoraria from Sanofi-Aventis, UCB, SciGen, GlaxoSmithKline, and Janssen-Cilag. JAF is the president of The Epilepsy Study Consortium, a non-profit organisation; New York University (NYU) receives a fixed amount from the Epilepsy Study Consortium towards her salary. The money is for work by JAF on behalf of The Epilepsy Study Consortium, for consulting and clinical trial-related activities. She receives no personal income for these activities. Within the past year, The Epilepsy Study Consortium received payments for research services from: Acorda, Eisai Medical Research, GlaxoSmithKline, Impax, Johnson & Johnson, Mapp Pharmaceuticals, Marinus, Novartis, Lundbeck, Pfizer, Sepracor, Sunovion, SK Life Science, Supernus Pharmaceuticals, UCB/Schwarz Pharma, Upsher Smith, Vertex. JAF is an investigator at NYU on studies for Eisai Medical Research, LCGH, Impax, Mapp Pharmaceuticals, Novartis, UCB/Schwarz Pharma, Upsher Smith, and Vertex. MS, ARB-K, SLM, AP, and KSW declare no competing interests.
References
- 1.Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. 2011;52:657–78. doi: 10.1111/j.1528-1167.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 2.Galanopoulou AS, Buckmaster PS, Staley KJ, et al. the American Epilepsy Society Basic Science Committee And The International League Against Epilepsy Working Group On Recommendations For Preclinical Epilepsy Drug Discovery Identification of new epilepsy treatments: issues in preclinical methodology. Epilepsia. 2012;53:571–82. doi: 10.1111/j.1528-1167.2011.03391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher M, Feuerstein G, Howells DW, et al. the STAIR Group Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–50. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shineman DW, Basi GS, Bizon JL, et al. Accelerating drug discovery for Alzheimer’s disease: best practices for preclinical animal studies. Alzheimers Res Ther. 2011;3:28. doi: 10.1186/alzrt90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steward O, Popovich PG, Dietrich WD, Kleitman N. Replication and reproducibility in spinal cord injury research. Exp Neurol. 2012;233:597–605. doi: 10.1016/j.expneurol.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 6.Ludolph AC, Bendotti C, Blaugrund E, et al. Guidelines for preclinical animal research in ALS/MND: A consensus meeting. Amyotroph Lateral Scler. 2010;11:38–45. doi: 10.3109/17482960903545334. [DOI] [PubMed] [Google Scholar]
- 7.Brooks-Kayal AR, Bath KG, Berg AT, et al. Issues related to symptomatic and disease-modifying treatments affecting cognitive and neuropsychiatric comorbidities of epilepsy. Epilepsia. 2013;54(suppl 4):44–60. doi: 10.1111/epi.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galanopoulou AS, Kokaia M, Loeb JA, et al. Epilepsy therapy development: technical and methodologic issues in studies with animal models. Epilepsia. 2013;54(suppl 4):13–23. doi: 10.1111/epi.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hooijmans CR, Leenaars M, Ritskes-Hoitinga M. A gold standard publication checklist to improve the quality of animal studies, to fully integrate the Three Rs, and to make systematic reviews more feasible. Altern Lab Anim. 2010;38:167–82. doi: 10.1177/026119291003800208. [DOI] [PubMed] [Google Scholar]
- 10.Kahle MP, Bix GJ. Successfully Climbing the “STAIRs”: Surmounting Failed Translation of Experimental Ischemic Stroke Treatments. Stroke Res Treat. 2012;2012:374098. doi: 10.1155/2012/374098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landis SC, Amara SG, Asadullah K, et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187–91. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Editorial. Making methods clearer. Nat Neurosci. 2013;16:1. [Google Scholar]
- 14.Rigor in Science Working Group. Improving the Quality of NINDS-Supported Preclinical and Clinical Research through Rigorous Study Design and Transparent Reporting. 2011 http://www.ninds.nih.gov/funding/transparency_in_reporting_guidance.pdf.
- 15.Simonato M, Löscher W, Cole AJ, et al. Finding a better drug for epilepsy: preclinical screening strategies and experimental trial design. Epilepsia. 2012;53:1860–67. doi: 10.1111/j.1528-1167.2012.03541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simonato M, French JA, Galanopoulou AS, O’Brien TJ. Issues for new antiepilepsy drug development. Curr Opin Neurol. 2013;26:195–200. doi: 10.1097/WCO.0b013e32835efe29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Löscher W, Klitgaard H, Twyman RE, Schmidt D. New avenues for anti-epileptic drug discovery and development. Nat Rev Drug Discov. 2013;12:757–76. doi: 10.1038/nrd4126. [DOI] [PubMed] [Google Scholar]
- 18.Fisher RS, van Emde Boas W, Blume W, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) Epilepsia. 2005;46:470–72. doi: 10.1111/j.0013-9580.2005.66104.x. [DOI] [PubMed] [Google Scholar]
- 19.Fisher RS, Acevedo C, Arzimanoglou A, et al. A practical clinical definition of epilepsy. Epilepsia. 2014;55:475–82. doi: 10.1111/epi.12550. [DOI] [PubMed] [Google Scholar]
- 20.Pitkänen A, Nehlig A, Brooks-Kayal AR, et al. Issues related to development of antiepileptogenic therapies. Epilepsia. 2013;54(suppl 4):35–43. doi: 10.1111/epi.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1069–77. doi: 10.1111/j.1528-1167.2009.02397.x. [DOI] [PubMed] [Google Scholar]
- 22.Wilcox KS, Dixon-Salazar T, Sills GJ, et al. Issues related to development of new antiseizure treatments. Epilepsia. 2013;54(suppl 4):24–34. doi: 10.1111/epi.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engel J, Jr, Pitkänen A, Loeb JA, et al. Epilepsy biomarkers. Epilepsia. 2013;54(suppl 4):61–69. doi: 10.1111/epi.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katz R. Biomarkers and surrogate markers: an FDA perspective. NeuroRx. 2004;1:189–95. doi: 10.1602/neurorx.1.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.National Institute of Neurological Disorders and Stroke. http://www.ninds.nih.gov/research/asp/asp_working_group_report_022712.htm (accessed Aug 4, 2014)
- 26.French JA, White HS, Klitgaard H, et al. Development of new treatment approaches for epilepsy: unmet needs and opportunities. Epilepsia. 2013;54(suppl 4):3–12. doi: 10.1111/epi.12294. [DOI] [PubMed] [Google Scholar]
- 27.Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–53. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74:679–87. doi: 10.1002/ana.23960. [DOI] [PubMed] [Google Scholar]
- 29.Rakhade SN, Jensen FE. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 2009;5:380–91. doi: 10.1038/nrneurol.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meador KJ, French J, Loring DW, Pennell PB. Disparities in NIH funding for epilepsy research. Neurology. 2011;77:1305–07. doi: 10.1212/WNL.0b013e318230a18f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pitkänen A, Lukasiuk K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011;10:173–86. doi: 10.1016/S1474-4422(10)70310-0. [DOI] [PubMed] [Google Scholar]
- 32.Blumenfeld H, Klein JP, Schridde U, et al. Early treatment suppresses the development of spike-wave epilepsy in a rat model. Epilepsia. 2008;49:400–09. doi: 10.1111/j.1528-1167.2007.01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dezsi G, Ozturk E, Stanic D, et al. Ethosuximide reduces epileptogenesis and behavioral comorbidity in the GAERS model of genetic generalized epilepsy. Epilepsia. 2013;54:635–43. doi: 10.1111/epi.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cook MJ, O’Brien TJ, Berkovic SF, et al. Prediction of seizure likelihood with a long-term, implanted seizure advisory system in patients with drug-resistant epilepsy: a first-in-man study. Lancet Neurol. 2013;12:563–71. doi: 10.1016/S1474-4422(13)70075-9. [DOI] [PubMed] [Google Scholar]
- 35.Fisher RS, Blum DE, DiVentura B, et al. Seizure diaries for clinical research and practice: limitations and future prospects. Epilepsy Behav. 2012;24:304–10. doi: 10.1016/j.yebeh.2012.04.128. [DOI] [PubMed] [Google Scholar]
- 36.Engel J., Jr Biomarkers in epilepsy: introduction. Biomarkers Med. 2011;5:537–44. doi: 10.2217/bmm.11.62. [DOI] [PubMed] [Google Scholar]
- 37.Engel J, Jr, da Silva FL. High-frequency oscillations - where we are and where we need to go. Prog Neurobiol. 2012;98:316–18. doi: 10.1016/j.pneurobio.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Worrell G, Gotman J. High-frequency oscillations and other electrophysiological biomarkers of epilepsy: clinical studies. Biomarkers Med. 2011;5:557–66. doi: 10.2217/bmm.11.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomes WA, Shinnar S. Prospects for imaging-related biomarkers of human epileptogenesis: a critical review. Biomarkers Med. 2011;5:599–606. doi: 10.2217/bmm.11.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar A, Asano E, Chugani HT. α-[¹¹C]-methyl-L-tryptophan PET for tracer localization of epileptogenic brain regions: clinical studies. Biomarkers Med. 2011;5:577–84. doi: 10.2217/bmm.11.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pitkänen A, Lukasiuk K. Molecular biomarkers of epileptogenesis. Biomarkers Med. 2011;5:629–33. doi: 10.2217/bmm.11.67. [DOI] [PubMed] [Google Scholar]
- 42.Vezzani A, Friedman A. Brain inflammation as a biomarker in epilepsy. Biomarkers Med. 2011;5:607–14. doi: 10.2217/bmm.11.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Brien TJ, Ben-Menachem E, Bertram EH, 3rd, et al. Proposal for a “phase II” multicenter trial model for preclinical new antiepilepsy therapy development. Epilepsia. 2013;54(suppl 4):70–74. doi: 10.1111/epi.12300. [DOI] [PubMed] [Google Scholar]
- 44.Mullard A. Reliability of ‘new drug target’ claims called into question. Nat Rev Drug Discov. 2011;10:643–44. doi: 10.1038/nrd3545. [DOI] [PubMed] [Google Scholar]
- 45.Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011;10:712. doi: 10.1038/nrd3439-c1. [DOI] [PubMed] [Google Scholar]
- 46.Kimmelman J, Anderson JA. Should preclinical studies be registered? Nat Biotechnol. 2012;30:488–89. doi: 10.1038/nbt.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Button KS, Ioannidis JP, Mokrysz C, et al. Power failure: why small sample size undermines the reliability of neuroscience. Nat Rev Neurosci. 2013;14:365–76. doi: 10.1038/nrn3475. [DOI] [PubMed] [Google Scholar]
- 48.Brodie MJ, Barry SJ, Bamagous GA, Norrie JD, Kwan P. Patterns of treatment response in newly diagnosed epilepsy. Neurology. 2012;78:1548–54. doi: 10.1212/WNL.0b013e3182563b19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baillie TA, Rettie AE. Role of biotransformation in drug-induced toxicity: influence of intra- and inter-species differences in drug metabolism. Drug Metab Pharmacokinet. 2011;26:15–29. doi: 10.2133/dmpk.dmpk-10-rv-089. [DOI] [PMC free article] [PubMed] [Google Scholar]