

Abstract
Certain indolyl-pyridinyl-propenone analogues kill glioblastoma cells that have become resistant to conventional therapeutic drugs. Some of these analogues induce a novel form of non-apoptotic cell death called methuosis, while others primarily cause microtubule disruption. Ready access to 5-indole substitution has allowed characterization of this position to be important for both types of mechanisms when a simple methoxy group is present. We now report the syntheses and biological effects of isomeric methoxy substitutions on the indole ring. Additionally, analogues containing a trimethoxyphenyl group in place of the pyridinyl moiety were evaluated for anticancer activity. The results demonstrate that the location of the methoxy group can alter both the potency and the mechanism of cell death. Remarkably, changing the methoxy from the 5-position to the 6-position switched the biological activity from induction of methuosis to disruption of microtubules. The latter may therefore represent a prototype for a new class of mitotic inhibitors with potential therapeutic utility.
Keywords: Methuosis, indolyl-pyridinyl-propenones, glioblastoma, microtubule disruption, cell death
Graphical Abstract

1. Introduction
Glioblastoma multiforme (GM) remains a lethal cancer due to rapid progression and limited treatment options, namely surgical removal of the tumor followed by combined radiotherapy and chemotherapy with temozolomide [1,2]. Recurrence of disease is usually untreatable as a result of acquired drug-resistance and invasive dissemination of the tumor. Temozolomide relies on triggering programmed cell death via activation of apoptosis [3]. However, GM cells harbor specific mutations in genes that are required to promote an efficient apoptotic response [4,5]. Stimulation of nonconventional cell death pathways offers a possible solution for treating drug-resistant cancers that are able to circumvent apoptosis [6,7]. Methuosis is a recently identified caspase-independent form of cell death that displays characteristics distinct from other types of non-apoptotic cell death, such as necroptosis or autophagy [8,9]. In cultured glioblastoma cells, methuosis begins with defective macropinocytotic trafficking, causing the formation of large fluid-filled vacuoles. Accumulation of vacuoles ultimately displaces the cytoplasm and the cell membrane loses integrity and ruptures. While dysfunctional vesicular trafficking and accumulation of vacuoles appear to contribute to cell death, there is evidence that additional metabolic or cellular insults are required for execution of the methuosis cell death program [8,10,11].
The methuosis phenotype was initially observed by the ectopic expression of activated Ras and Rac GTPases in GM cells [12,13]. More recent studies have focused on the pursuit of small molecules with the potential to induce this form of cell death in a therapeutic context. An initial search for compounds reported to induce cellular vacuolization led us to an indolyl-pyridinyl-propenone (IPP, also referred to as indole-derived chalcone) as a potential lead [13]. Associated structure-activity relationship (SAR) studies revealed that the optimized scaffold for induction of methuosis consists of a 2,5-disubstituted indole and a pyridine in the para-configuration, bridged by an α,β-unsaturated ketone [11,14,15]. Our previously reported IPP compounds, and their modes of biological activity, are summarized in Fig. 1.
Figure 1.

Previously reported analogues illustrating the various biological activities of substituted IPP’s.
To date, compounds 1a–1e are the most potent inducers of methuosis, possessing activity between 2–3 μM when assayed against the human glioblastoma cell line, U251. Among these compounds 1a has been studied most thoroughly. Comparisons of structurally similar IPP’s have revealed intriguing and unexpected results suggesting that the morphological appearance of vacuoles in the treated cells is not always associated with cell death. For instance, analogues with larger aliphatic substitutions (2e–2g) on the 2-indolyl position caused vacuolization but had surprisingly less cytotoxicity than the vacuole-inducing compounds with Me (1a) or Et (1b) at this position (Fig. 1) [11]. Similarly, certain 5-substituted analogues (2a–2c), as well as the 2-des-methyl derivative 2d, also induced vacuole formation but were not cytotoxic [15]. While 5-methoxy (1a) and 5-propoxyindole (1e) analogues triggered cell death by methuosis, their structurally similar counterparts, namely 5-ethoxy (2a) and 5-isopropoxyindole (2b) caused cytoplasmic vacuolization without cell death. Studies are currently underway with this series of analogues to explore the mechanistic basis for their differential cytotoxicity.
Another novel insight into the biological effects of the IPP compounds was gathered from derivatives containing electron-withdrawing functionalities at the 2-indolyl position [15]. Derivatives containing trifluoromethyl (3a) or alkyl carboxylate (3b–3d) substitutions caused minimal to no vacuolization but remained highly cytotoxic. Morphologically, cells treated with the latter series of compounds did not resemble cells undergoing methuosis. Instead, the cells displayed features consistent with disruption of tubulin polymerization and microtubule architecture. Cell cycle analysis demonstrated an accumulation of cells in the G2/M phase, with eventual death by mitotic catastrophe. In this respect, 3a–3d were quite distinct from the methuosis-inducing compounds, which did not disrupt microtubules or cause mitotic arrest at the same concentrations. The redirection of cytotoxicity from methuosis to microtubule disruption for derivatives 3a–3d was associated with a significant increase in growth inhibitory potency.
While our previous synthetic work focused on substitution at either the 2- or 5-indole positions, a lack of information exists for substitutions at the 4-, 6-, or 7- positions (Scheme 1). We noted that the importance of a 5-methoxy group for either methuosis or microtubule disruption is further dependent upon the electron withdrawing properties of the 2-substituent. In the present study we have synthesized and evaluated methoxy isomers of 1a to sequentially survey the 4-, 6- and 7-positions while initially holding the 2-position constant. Upon finding significant anti-mitotic activity for the 6-position isomer 9b, we immediately prepared its 2-trifluoromethyl version (15) by analogy to 3a where this type of functionality also led to microtubule disruption [15]. Finally, drawing from several reports describing N-methyl-indole-based trimethoxyphenyl chalcones as compounds affecting tubulin polymerization, we examined replacing the para-pyridine in our structural template with a trimethoxyphenyl group and, likewise, separately examined the effect of adding a methyl group to the indole nitrogen. The results reveal that the position of the methoxy group on the indole ring and the para-pyridine are critical determinants of the biological activities of the IPP compounds.
Scheme 1.

Synthesis of either mono- or di-methoxyindole substituted IPP’s (9a–9d). Reagents and conditions: (i) TBAB, 50% NaOH, THF; then benzenesulfonyl chloride, rt; (ii) THF, t-butyllithium, −30 °C to 0 °C; then CH3-I, −30°C to rt; (iii) 3 M NaOH, EtOH, reflux; (iv) POCl3, DMF, 0 °C to rt; then 1 N NaOH; (v) 4-Acetylpyridine, piperidine, MeOH, reflux.
2. Results and discussion
2.1. Chemistry
Scheme 1 illustrates the synthesis of isomers of 1a at the 4-, 6- or 7- indole position (9a-9c). A disubstituted 5,6-dimethoxy derivative (9d) was also synthesized. From commercially available 4a–4d, the indole nitrogen was protected with benzenesulfonyl chloride (5a–5d). The benzenesulfonyl group ensured regioselective methylation at the 2-indolyl position (6a–6d), which was accomplished under conditions of tert-butyllithium and iodomethane [16]. Removal of benzenesulfonyl in a mixed solvent system of EtOH/aqueous NaOH provided 7a–7d. Formylation reactions utilizing Vilsmeier conditions (8a–8d) followed by Claissen-Schmidt condensation reactions produced target compounds 9a–9d. This approach generally provided compounds in reasonable yields; however, intermediate 7b was not stable under these conditions and resulted in low yields for 9b. When 9b was synthesized by the alternative method shown in Scheme 2, intermediates were stable and produced high yields. Compound 15 was also prepared according to Scheme 2. Targets 9b and 15 were synthesized from aniline derivative 10, which was protected with BOC. Regioselective acylation of 11 with Weinreb amide [17] 12a or 12b controlled by sec-butyllithium provided a ketone intermediate, which subsequently cyclized to indole. The BOC protecting group was cleaved with TFA to produce 7b or 13 [18]. Typical conditions of formylation (8b or 14) and condensation afforded final targets 9b and 15.
Scheme 2.

Synthesis of trifluoromethyl compound 15 and a more efficient synthesis of 9b. Reagents and conditions: (i) Boc2O, THF, reflux; (ii) THF, sec-butyllithium, −40 °C to 0 °C; then Weinreb amide (12a or 12b)/THF, −40 °C to rt; (iii) DCM/TFA; (iv) POCl3, DMF, 0 °C to rt; then 1N NaOH; (v) 4-Acetylpyridine, MeOH, reflux.
The compounds synthesized in Scheme 3 probe the effects of a trimethoxyphenyl functionality in place of para-pyridine. Aldehyde 16 was condensed with either 2,4,6-trimethoxyacetophenone or 3,4,5-trimethoxyacetophenone to yield 17 or 18, respectively. Piperidine is typically employed as the base to form the enolate from aryl acetates. This was appropriate during the synthesis of 18, as well as target compounds 9a–9d and 15, but yielded no reaction for 17. Presumably, the steric hindrance of methoxy groups present at the adjacent 2- and 6- positions of acetophenone prevented the formation of enolate by piperidine. Compound 17 was synthesized using 50% aqueous KOH/MeOH, albeit in relatively low yields as compared to other analogues in this series. Additionally, derivative 19 was synthesized from 1a in one step using sodium hydride as the base and methyl iodide as the alkylating reagent (Scheme 4).
Scheme 3.

Synthesis of indole-trimethoxyphenyl-propenones 17 and 18. Reagents and conditions: (i) 2′,4′,6′-Trimethoxyacetophenone, KOH, MeOH/H2O, reflux; (ii) 3′,4′,5′-Trimethoxyacetophenone, piperidine, MeOH, reflux.
Scheme 4.

Synthesis of N-Methyl analogue 19. Reagents and conditions: (i) 1. DMF, NaH; then CH3-I.
2.2. Biological activity
Previous studies with U251 glioblastoma cells have established that the sulforhodamine B (SRB) colorimetric assay is useful for evaluating the loss of viable cells and for ranking the relative potency [11,15]. This assay is sensitive to both methuosis and microtubule disruption. Growth inhibitory activities for all compounds at a 48 h end-point are listed in Table 1 as the dose able to achieve 50% growth inhibition (GI50) compared to growth within control cultures treated with only vehicle (DMSO).
Table 1.
Summary of growth inhibition results and phenotypical analysis for final targets.
| Compound | GI50 (in μM)* | Vacuoles |
|---|---|---|
| 1a | 2.32 ± 0.04 | Abundant, Persistent |
| 9a | 8.67 ± 1.23 | Few, Transient |
| 9b | 0.09 ± 0.01 | Few, Transient |
| 9c | 4.42 ± 0.39 | No |
| 9d | >10 | No |
| 15 | >10 | No |
| 17 | 3.74 ± 0.52 | No |
| 18 | >10 | No |
| 19 | >10 | Few, Transient |
Results are the mean ± S.D. derived from three separate SRB assays. >10 indicates that growth inhibition relative to control did not reach 50% at the highest concentration tested (10 μM).
In the early stages of methuosis, cells become filled with a large number of phase-lucent macropinosome-derived vacuoles, which can be readily observed by phase contrast microscopy. As the cells begin to die (usually between 24–48 h), they detach from the culture surface and lose membrane integrity. Phase-contrast microscopy pictures of cells treated with the compounds at 2.5 μM for 4 h or 48 h are compiled in Fig. 2. Consistent with our previous studies, the 5-methoxy compound 1a induced methuosis with a GI50 of 2.30 μM. By 48 h, tumor cells treated with the compound at concentrations ≥ 2.5 μM began to detach from the dish and lose membrane integrity. By comparison, the 4-methoxy compound 9a elicited much fewer vacuoles and possessed weak growth inhibitory activity. The 7-methoxy compound 9c produced no vacuoles, but was moderately growth inhibitory. A strikingly different pattern of activity was observed for the 6-methoxy compound 9b. Growth inhibitory activity increased more than 25 fold, with a GI50 of 0.09 μM. At the same concentration that 1a induced methuosis (2.5 μM), 9b caused early formation of extensive membrane blebs on the cell surface and general rounding of the cell body, with only a few vacuoles detected (Fig. 2). By 24 h the majority of the cells had rounded up and detached from the dish, and by 48 h most of the cells had disintegrated. The few remaining attached cells were either rounded or enlarged with multiple micronuclei. The higher potency and distinct morphological characteristics observed with 9b were similar to the effects we previously observed when testing compounds 3a–3d in Fig. 1 [15]. Examination of cells treated with 9b by immunofluorescence microscopy with an antibody against tubulin (Fig. 3) confirmed disruption of microtubules. Cells treated with 9b at 100 nM exhibited a dense network of microtubules, similar to the DMSO control. However, at higher concentrations, 9b caused a complete loss of normal microtubule architecture in the few cells that remained attached to the culture dish. In contrast, the microtubule network in cells treated with the methuosis inducer, 1a, was generally intact, except for distortions created by the presence of the large cytoplasmic vacuoles.
Figure 2.

Morphological effects of the listed compounds on U251 glioblastoma cells. The control cells received an equivalent volume of DMSO vehicle. Phase-contrast images were obtained at the indicated time intervals after addition of compounds at 2.5 μM.
Figure 3.

Immunofluorescence imaging of tubulin (red) in cells treated for 24 h with 9b and 1a. The nuclei are visualized with DAPI (blue).
DNA histograms obtained by flow cytometry of cells treated with 9b for 24 h (attached and detached cells combined) were indicative of mitotic arrest at concentrations > 500 nM, with a large increase in the G2/M phase population (Fig. 4). In contrast, cells treated with 1a were predominantly in the G1/G0 phase of the cell cycle (Fig. 4). It is interesting to note that cells treated with 100 nM 9b exhibited an intact microtubule network (Fig. 3) and a near-normal cell cycle distribution (Fig. 4), despite the growth inhibition observed at this concentration in the SRB assay (Table 1). This raises the possibility that at low concentrations, near the GI50, 9b may inhibit cell proliferation via an unidentified mechanism separate from microtubule disruption. Based on the distinct phenotypes elicited by 1a (methuosis) and 9b (microtubule disruption), we were curious to determine what type of activity might occur with a 5,6-dimethoxyindole derivative, 9d. Surprisingly, the dimethoxy compound lost all activity in the growth assay and neither mode of action was observed in the morphology assays (Table 1, Fig. 2).
Figure 4.
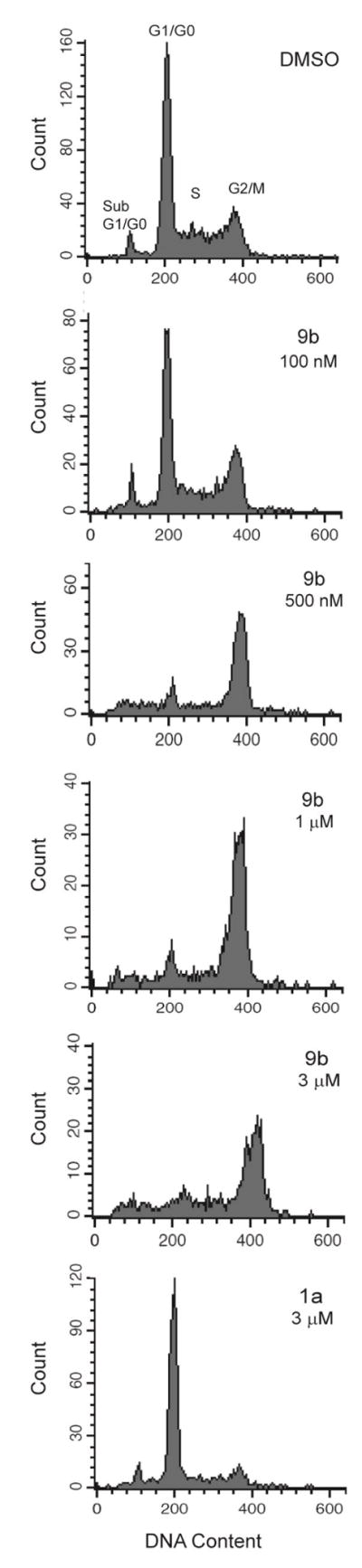
DNA histograms of cells treated with the indicated compounds for 24 h were generated by flow cytometry.
We previously reported that the methuosis-inducing activity of 1a could be switched to microtubule-disrupting activity when trifluoromethyl was substituted for methyl at the 2-position on the indole ring (3a, Fig. 1) [15]. Therefore, we postulated that a 2-trifluoromethyl substitution might increase the activity of 9b. Interestingly, the resulting compound 15 lost all detectable growth inhibitory activity (Table 1, Fig. 2).
The trimethoxyphenyl moiety and the indole scaffold are common motifs in many compounds that possess anticancer activity mediated by microtubule disruption, including several indole-based chalcones [19–23]. Features that distinguish the latter compounds from our IPP series include methylation of the indolyl nitrogen and presence of either a 3,4,5-trimethoxyphenyl [24,25] or 2,4,6-trimethoxyphenyl [26,27] group in place of the pyridine ring. Based on these observations, we evaluated whether substitution of the pyridinyl moiety in 1a with a trimethoxyphenyl moiety would switch activity from methuosis to microtubule disruption. The results indicate that neither the 2,4,6-trimethoxyphenyl (17) nor the 3,4,5-trimethoxyphenyl (18) derivative had any morphological effects on U251 cells that would be consistent with methuosis or microtubule disruption (Fig. 2). Compound 18 had no detectable effect on cell proliferation, whereas 17 was moderately growth inhibitory (Table 1). Similarly, we asked what effect methylation of the indole nitrogen might have on the activity of potent methuosis inducer 1a. The resulting N-methyl derivative 19 (Scheme 4) showed substantially diminished growth inhibition (GI50 >10 μM) and only transient vacuole-inducing activity at 2.5 μM (Fig. 2).
3. Conclusions
The present SAR studies demonstrate that the position of methoxy substitutions on the indoly-pyridinyl-propenone scaffold have a significant influence on anti-cancer activity. The 5-methoxy substitution is optimal for the induction of methuosis (1a). Changing the methoxy from the 5-position (1a) to the 4-position (9a) or the 7-position (9c) of the indole ring attenuates or eliminates methuosis activity. Unexpectedly, moving the methoxy group to the 6-position (9b) provided a striking enhancement of growth inhibitory potency by conferring a different type of anti-cancer activity to the compound; i.e., disruption of microtubules leading to mitotic arrest and cell death. Rather than enhancing either methuosis or microtubule-disrupting activity, combining the 5- and 6-methoxy indole modifications so as to produce dimethoxy compound 9d essentially abolished both activities. Thus, two mutually distinct substitution patterns emerge from our parent template, the 5-methoxy (1a) which induces methuosis and the 6-methoxy (9b) which can disrupt microtubules without additionally having an electron withdrawing substituent at the 2-position. Both types of arrangements are promising leads for the development of new therapeutic agents that can remain effective when cancer cells become resistant to apoptotic cell death induced by traditional anti-cancer drugs.
In addition to providing new insights regarding isomeric methoxy substitutions on the indole, the present studies also reinforce the importance of the pyridinyl moiety for the continued development of IPPs as potential anti-cancer therapeutics. Our previous studies demonstrated that changing the configuration of the pyridinyl nitrogen (e.g., from para to meta) in the context of various IPP’s with either methuosis or microtubule-disrupting activity eliminated or markedly reduced activity [11,14,15]. Here we show that replacing the pyridine ring with trimethoxyphenyl substituents (17 and 18) markedly reduced or eliminated the morphological effects and growth inhibitory activity. These trimethoxyphenyl substitutions previously were shown to impart potent microtubule-disrupting activity on various indole-based chalcones containing N-methyl indole [24,25]. It thus appears that the trimethoxyphenyl substituents are not compatible with the 5-methoxy-2-methylindole moiety for generating either microtubule-targeted compounds or methuosis-inducing compounds.
Compounds that alter microtubule stability are widely used in cancer therapy [28–30]. However, these agents are not without drawbacks, such as dose-limiting toxicity, development of drug resistance, and restricted penetrance of the blood-brain barrier [31,32]. Because of these issues, there continues to be significant interest in discovery of new microtubule-targeted compounds with distinct properties. In this regard, our findings suggest that further exploration of the anti-neoplastic potential and pharmacological properties of the 6-methoxy derivative, 9b, could be productive.
4. Experimental section
4.1. Chemistry - General description
All reactions were performed in oven-dried 2-neck round-bottom flasks under an atmosphere of either Ar or N2 and stirred with teflon-coated magnetic bars. TLC (silica gel F254 plates, Baker-flex) was used to monitor progress of all reactions with visualization performed under 254 nm UV light. Reagent grade and anhydrous solvents were purchased from Sigma-Aldrich and used without further purification unless otherwise noted. Compounds 4a–4d were purchased from Alfa-Aesar, while compound 10 was purchased from Sigma-Aldrich. Compounds 12a, 12b and 16 were reported previously [11,14,15]. Chromatography was conducted on normal phase silica gel sorbent (Fisher Scientific, 230–400 mesh) by flash column methods as described previously [15,33] utilizing a gradient of increasing polar eluent specifically indicated for each compound. Isocratic separations are denoted on an individual basis. Samples to be purified were prepared by adsorption onto silica gel before performing chromatography (previously described as “dry loading”) [15]. TLC was used to monitor product elution during flash column chromatography. Appropriate fractions were combined, solvents distilled in vacuo (rotary evaporator under water aspirator vacuum) and then further dried by a vacuum pump (0.5 mm Hg) for 24 h unless described otherwise. Samples that were heated in a vacuum desiccator were equipped to a vacuum pump (0.5 mm Hg) and dried for a specified time and temperature denoted in the individual procedure. Solvent solutions dried with Na2SO4 were stored in a sealed flask and allowed to sit for at least 4 h. Upon completion, the drying agent was removed by filtration and filtrate was evaporated in vacuo and then further dried by a vacuum pump (0.5 mm Hg) for 24 h. Melting points were performed in triplicate on an Electrothermal digital melting point apparatus and are uncorrected. Proton (1H) and carbon (13C) NMR experiments were recorded on either a 600 MHz Bruker Avance, Inova 600 MHz or an Inova 400 MHz instrument. Samples were referenced to TMS when present, or the solvent residual peak for 1H and 13C, respectively: (CDCl3; 7.27, 77.13; d6-DMSO; 2.50, 39.51; d6-acetone; 2.05, 29.92). 1H NMR chemical shifts were given in ppm and coupling constants (J values) were expressed in hertz (Hz) using the following designations: s (singlet), d (doublet), t (triplet), dd (doublet of doublets), m (multiplet). The 13C chemical shifts were reported in ppm for each compound in the experimental section and in all cases confirm structure. In a few cases 13C shifts were found to double-up in their peak locations. Fluorine (19F) NMR was recorded on an Inova 400 MHz instrument at 376 MHz. Samples were referenced externally to CFCl3. Purity for tested compounds was determined by combustion analysis (Atlantic Microlabs, Norcross GA) and HPLC. All tested compounds possess ≥95% as determined by both purity methods. Observed values for combustion analysis were considered acceptable within ±0.4% of calculated values. Synthetic derivatives reported as solvates are denoted in the text. HPLC was performed on an Alliance® instrument (#2659) equipped with a quaternary pump, an inline membrane degasser, autosampler and Photodiode Array (PDA) Detector (#2996) from Waters Corporation (Milford, MA). The column was a Nova-Pak®C18 column, 4 μm particle size (150 mm × 3.9 mm). Samples were dissolved in 60% eluent A (H2O) and 40% eluent B (CH3CN) for injection. The following procedure, termed “Gradient 1”, was employed for final targets (9a–9d, 15, 17–19) and intermediate 8c: Time 0.01–2.00 min (isocratic, 20% eluent B); Time 2.01–15.00 min (linear gradient, 45% eluent B to 80% eluent B); Time 15.01–20.00 min (isocratic, 20% eluent B). Details for HPLC analysis are denoted in the individual procedures. Chromatograms are illustrated in Figure S1 (supplementary data) and were recorded at the UVmax for each compound.
4.2. Preparation of individual compounds
4.2.1. 1-Benzenesulfonyl-4-methoxyindole (5a)
A suspension of 4-methoxyindole (500 mg, 3.4 mmol), TBAB (10 mol%, 109 mg, 0.34 mmol) and 50% sodium hydroxide (5 mL) in THF (10 mL) and H2O (3 mL) was stirred vigorously for 20 min at rt. Benzenesulfonyl chloride (2 eq, 6.8 mmol, 1.2 g) in THF (15 mL) was added dropwise to the reaction mixture. The reaction mixture was stirred overnight then extracted with EtOAc (20 mL X 3). The combined organic layers were dried with anhydrous Na2SO4 and then concentrated in vacuo to yield a yellow oil. The product was purified using column chromatography (DCM) to yield white solid (910 mg, 92%): mp 86–88 °C. TLC Rf 0.71 (DCM). 1H NMR (600 MHz, CDCl3) δ 7.87-7.86 (d, 2H, J = 7.68 Hz), 7.61-7.59 (d, 1H, J = 8.34 Hz), 7.5155 (t, 1H, J = 7.38 Hz), 7.47 (d, 1H, J= 3.66 Hz), 7.43-7.41 (t, 2H, J = 7.8 Hz), 7.25-7.22 (t, 1H, J = 8.16 Hz), 6.78-6.77 (d, 1H, J = 3.66 Hz), 6.65-6.64 (d, 1H, J = 7.98 Hz), 3.88 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 153.3, 138.4, 136.3, 134.0, 129.4, 127.0, 125.89, 124.97, 121.3, 106.68, 106.49, 103.7, 55.6. Elemental analysis calcd for C15H13NO3S: C, 62.70; H, 4.56; N, 4.87. Found: C, 62.71; H, 4.60; N, 4.97.
4.2.2. 1-Benzenesulfonyl-6-methoxyindole (5b)
A suspension of 6-methoxyindole (1.0 g, 6.79 mmol), TBAB (218 mg, 0.679 mmol) and 50% sodium hydroxide (10 mL) in THF (10 mL) and H2O (3 mL) was stirred vigorously for 20 min at rt. Benzenesulfonyl chloride (13.6 mmol, 2.4 g) in THF (8 mL) was added dropwise to the reaction mixture. The reaction mixture was stirred overnight then extracted with EtOAc (50 mL X 3). The combined organic layer was dried over Na2SO4 then concentrated in vacuo. The product was purified using column chromatography (5% to 25% EtOAc/hexanes) to yield white solid (1.80 g, 92%): mp 144–146 °C (140–142 °C [34]). TLC Rf 0.38 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.87-7.85 (m, 2H), 7.55-7.52 (m, 2H), 7.45-7.43 (m, 3H), 7.39-7.38 (d, 1H, J = 8.58 Hz), 6.87-6.85 (dd, 1H, J1 = 8.58 Hz, J2 = 2.34 Hz), 6.59-6.58 (dd, 1H, J1 = 3.66 Hz, J2 = 0.72 Hz), 3.88 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 157.9, 138.2, 135.9, 133.8, 129.2, 126.7, 125.06, 124.45, 121.8, 112.6, 109.2, 97.9, 55.8.
4.2.3. 1-Benzenesulfonyl-7-methoxyindole (5c)
A suspension of 7-methoxyindole (1 g, 6.8 mmol), TBAB (219 mg, 0.68 mmol) and 50% sodium hydroxide (10 mL) in THF (20 mL) and water (6 mL) was reacted and purified in a manner similar to that for 5a to yield a white solid (1.5 g, 73%): mp 88–90 °C (89–90 °C [35]). TLC Rf 0.75 (DCM). 1H NMR (600 MHz, CDCl3) δ 7.85-7.84 (m, 3H), 7.57-7.54 (m, 1H), 7.49-7.46 (m, 2H), 7.17-7.16 (m, 1H), 7.13–7.11 (m, 1H), 6.68-6.66 (d, 1H, J = 7.86 Hz), 6.66-6.65 (d, 1H, J = 3.66 Hz), 3.64 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 147.3, 140.4, 133.67, 133.11, 128.72, 128.59, 127.1, 124.55, 124.11, 114.0, 107.12, 106.76, 55.3.
4.2.4. 1-Benzenesulfonyl-5,6-dimethoxyindole (5d)
A suspension of 6-methoxyindole (1.01 g, 5.70 mmol), TBAB (184 mg, 0.570 mmol) and 50% sodium hydroxide (5 mL) in THF (10 mL) and H2O (10 mL) was reacted and purified in a similar manner to that for 5b to yield a white solid (1.71 g, 92%): mp 140–143 °C (130–133 °C [36]). TLC Rf 0.18 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.87-7.85 (m, 2H), 7.55-7.52 (m, 2H), 7.45-7.43 (m, 3H), 7.39-7.38 (d, 1H, J = 8.58 Hz), 6.87-6.85 (dd, 1H, J1 = 8.58 Hz, J2 = 2.34 Hz), 6.59-6.58 (dd, 1H, J1 = 3.66 Hz, J2 = 0.72 Hz), 3.88 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 157.9, 138.2, 135.9, 133.8, 129.2, 126.7, 125.06, 124.45, 121.8, 112.6, 109.2, 97.9, 55.8. 1H NMR (600 MHz, d6-acetone) δ 7.99-7.97 (m, 2H), 7.67-7.65 (m, 1H), 7.59-7.56 (m, 3H), 7.52 (d, 1H, J = 3.66 Hz), 7.08 (s, 1H), 6.67-6.66 (dd, 1H, J1 = 3.60 Hz, J2 = 0.66 Hz), 3.90 (s, 3H), 3.79 (s, 3H). 13C NMR (150 MHz, d6-acetone) δ 149.61, 148.71, 138.9, 135.1, 130.47, 130.06, 127.7, 126.0, 124.8, 110.7, 104.3, 98.4, 56.53, 56.30. Elemental analysis calcd for C16H15NO4S: C, 60.55; H, 4.76; N, 4.41. Found: C, 60.40; H, 4.84; N, 4.52.
4.2.5. 1-Benzenesulfonyl-6-methoxy-2-methylindole (6b)
Compound 5b (1.50 g, 5.22 mmol) was dissolved in THF (25 mL). The solution was cooled to −30 °C and treated with tert-butyllithium (1.7 M in pentane, 6.79 mmol, 4.0 mL) slowly to maintain an internal temperature of −30 °C. After completion, the reaction mixture stirred for an additional 30 min at −30 °C, then allowed to warm to 0 °C and stirred for another 20 min. The reaction mixture was cooled to −30 °C and iodomethane (15.7 mmol, 0.97 mL) was added drop-wise. The reaction mixture was warmed to rt and stirred for 16 h. The reaction mixture was concentrated in vacuo and redissolved in DCM (100 mL) then washed with saturated NaHCO3 (75 mL X 3). The organic layer was dried over Na2SO4, the filtrate collected and concentrated in vacuo to yield crude material which was purified by chromatography (0% to 20% EtOAc/hexanes) to yield a brown solid (984 mg, 63%): mp 68–71 °C. TLC Rf 0.29 (10% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.76-7.74 (m, 3H), 7.54-7.51 (m, 1H), 7.43-7.41 (m, 2H), 7.27-7.26 (d, 1H, J = 8.58 Hz), 6.85-6.84 (dd, 1H, J1 = 8.52 Hz, J2 = 2.34 Hz), 6.26 (t, 1H, J = 0.84 Hz), 3.87 (s, 3H), 2.55 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 157.3, 139.2, 138.0, 136.0, 133.6, 129.3, 126.2, 123.4, 120.3, 112.3, 109.4, 99.4, 55.8, 15.8. Elemental analysis calcd for C16H15NO3S: C, 63.77; H, 5.02; N, 4.65. Found: C, 63.88; H, 5.17; N, 4.57.
4.2.6. 1-Benzenesulfonyl-5,6-dimethoxy-2-methylindole (6d)
Compound 5d (1.70 g, 5.36 mmol) was reacted and purified in a manner similar to that for 6b to yield a cream-colored solid (742 mg, 42%): mp 129–131 °C. TLC Rf 0.23 (20% EtOAc/hexanes). 1H NMR (600 MHz, d6-acetone) δ 7.88-7.86 (m, 2H), 7.73 (s, 1H), 7.68-7.65 (m, 1H), 7.58-7.55 (m, 2H), 6.96 (s, 1H), 6.38-6.37 (t, 1H, J = 0.84 Hz), 3.88 (s, 3H), 3.79 (s, 3H), 2.56 (s, 3H). 13C NMR (150 MHz, d6-acetone) δ 148.7, 139.8, 136.6, 135.0, 131.8, 130.5, 127.2, 123.9, 111.1, 103.4, 100.1, 56.60, 56.27, 15.9. Elemental analysis calcd for C17H17NO4S: C, 61.62; H, 5.17; N, 4.23. Found: C, 61.34; H, 5.17; N, 4.19.
4.2.7. 4-Methoxy-2-methylindole (7a)
Compound 5a (700 mg, 2.44 mmol) was dissolved in THF (25 mL) under an Ar (g) atmosphere. The solution was cooled to −30 °C and then treated with tert-butyllithium (1.7 M in pentane, 3.16 mmol, 1.86 mL) slowly to maintain an internal temperature of −30 °C. After completion, the reaction mixture was allowed to stir for an additional 30 min at −30 °C then was allowed to warm up to 0 °C and stirred for another 20 min. The reaction mixture was cooled to −30 °C and iodomethane (0.46 mL, 7.3 mmol) was added dropwise to the stirring mixture. After the addition was complete, the reaction mixture was stirred and warmed to rt overnight. The mixture was concentrated in vacuo and redissolved in DCM (75 mL) and washed with saturated NaHCO3 (50 mL x 3). The organic layer was separated and then dried over anhydrous sodium sulfate. The filtrate was collected and concentrated in vacuo to yield black oil which was immediately taken to the next step. Crude 6a (1.1 g) was dissolved in a solution of 3 N NaOH/EtOH (50 mL, 1:1) and heated at 90 °C for 24 h. The reaction mixture was concentrated in vacuo and extracted with DCM (50 mL X 3). The combined organic layers were dried over Na2SO4. The filtrate was collected and concentrated in vacuo to yield a brown oil. The product was purified using column chromatography (0% to 10% EtOAc/hexanes) to yield a white solid (250 mg, 64%): mp 88 °C. TLC Rf 0.34 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.85 (s, 1H), 7.05-7.02 (t, 1H, J = 8.10 Hz), 6.94-6.93 (d, 1H, J = 7.68 Hz), 6.51-6.50 (d, 1H, J = 7.56 Hz), 6.31 (s, 1H), 3.94 (s, 3H), 2.44 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 152.5, 137.3, 133.4, 121.6, 119.3, 103.8, 99.7, 97.5, 55.3, 14.1. Elemental analysis calcd for C10H11NO • 0.15 EtOH: C, 73.64; H, 7.14; N, 8.34. Found: C, 73.83; H, 7.23; N, 7.97.
4.2.8. 6-Methoxy-2-methylindole (7b, Scheme 2)
Compound 11 (1.20 g, 5.06 mmol) was dissolved in THF (15 mL) under an atmosphere of Argon. The solution was cooled to −40 °C over 10 min and sec-butyllithium (1.4 M, 7.93 mL) was added slowly as to maintain an internal temperature of < −25 °C. After reaching 1 equivalent of sec-butyllithium (~3.96 mL) the reaction mixture turned a bright yellow signifying de-protonation of the amide nitrogen. The reaction mixture was then cooled to −50 °C and a solution of N-methoxy-N-methylacetamide 12a (575 mg, 5.57 mmol) in THF (3 mL) was added over 5 min. The reaction mixture was warmed to −10 °C over 30 min. The mixture was partitioned between Et2O (75 mL) and 0.5 N HCl (75 mL). The aqueous layer was separated and extracted an additional two times with Et2O (50 mL). The Et2O phases were combined and washed with brine (75 mL) and then dried over Na2SO4 to yield a dark oil. The crude intermediate was dissolved in DCM (20 mL). TFA (3 mL) was added to the mixture which was then stirred at rt for 24 h. Upon completion, the reaction mixture was added to a separatory funnel and washed with saturated NaHCO3 (50mL) followed by brine (50 mL). The organic layer was separated, dried with Na2SO4 and concentrated in vacuo to provide a crude oil which was purified by chromatography (0% to 20% EtOAc/hexanes) to yield a white solid (146 mg, 18%): mp 106–108 °C. TLC Rf 0.31 (20% EtOAc/hexanes). 1H NMR (600 MHz, d6-acetone) δ 9.77 (s, 1H), 7.27-7.26 (d, 1H, J = 8.46 Hz), 6.84 (d, 1H, J = 2.22 Hz), 6.62-6.61 (dd, 1H, J1 = 8.52 Hz, J2 = 2.28 Hz), 6.04 (m, 1H) 3.77 (s, 3H), 2.36 (s, 3H). 13C NMR (150 MHz, d6-acetone) δ 156.5, 138.2, 134.8, 124.4, 120.4, 109.4, 100.2, 95.1, 55.7, 13.6. Elemental analysis calcd for C10H11NO: C, 74.51; H, 6.88; N, 8.69. Found: C, 74.67; H, 6.90; N, 8.66.
4.2.9. 7-Methoxy-2-methylindole (7c)
Compound 5c (890 mg, 3.10 mmol) was reacted and purified in a manner similar to that for 7a to yield a white solid (120 mg, 24%): mp 83 °C (83–83.5 °C [37]). TLC Rf 0.74 (DCM). 1H NMR (600 MHz, CDCl3) δ 8.09 (s, 1H), 7.13-7.12 (d, 1H, J = 7.92 Hz), 6.99-6.96 (t, 1H, J = 7.86 Hz), 6.58-6.57 (d, 1H, J = 7.44 Hz), 6.18 (s, 1H) 3.93 (s, 3H), 2.41 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 145.4, 134.6, 130.3, 126.2, 119.9, 112.5, 101.11, 100.65, 55.3, 13.6.
4.2.10. 5,6-Dimethoxy-2-methylindole (7d)
Compound 6d (0.732 g, 2.21 mmol) was dissolved in a solution of 3 N NaOH/EtOH (75 mL, 1:1) and refluxed at 90 °C for 60 h. The reaction mixture was then concentrated in vacuo and extracted using DCM (50 mL X 3). The combined organic layers were dried over Na2SO4, collected and then concentrated to provide a brown oil. The product was purified using column chromatography (0% to 20% EtOAc/hexanes) to yield a yellow solid (352 mg, 83%): mp 94–95 °C (90–91 °C [38]). TLC Rf 0.15 (20% EtOAc/hexanes). 1H NMR (600 MHz, d6-acetone) δ 9.66 (s, 1H), 6.95 (s, 1H), 6.88 (s, 1H), 6.00-5.99 (m, 1H), 3.77 (s, 6H), 2.35 (s, 3H). 13C NMR (150 MHz, d6-acetone) δ 147.4, 146.1, 134.5, 132.0, 123.2, 103.8, 100.3, 96.3, 56.85, 56.63, 13.7.
4.2.11. 4-Methoxy-2-methylindole-3-carboxaldehyde (8a)
DMF (2 mL) was cooled to 0 °C. POCl3 (0.5 mL) was added and the reaction mixture was stirred for 10 min at 0 °C. A solution of 7a (220 mg, 1.37 mmol) in DMF (2 mL) was added to the reaction mixture drop-wise over 10 min. The solution was stirred for an additional 40 min while warming to rt, then slowly poured into ice-cold 1 N NaOH (50 mL) and stirred for 10 min. The precipitate was collected, washed with ice-cold H2O and dried at 40 °C for 24 h in a vacuum desiccator yielding a tan solid (177 mg, 68%): mp 190–192 °C. TLC Rf 0.71 (80% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 12.08 (s, 1H), 10.44 (s, 1H), 7.10-7.08 (t, 1H, J = 7.92 Hz), 7.02-7.01 (d, 1H, J = 7.98 Hz), 6.73-6.72 (d, 1H, J = 7.8 Hz), 3.92 (s, 3H), 2.65 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 187.3, 153.2, 142.7, 136.0, 122.7, 116.4, 113.6, 104.8, 102.2, 55.1, 13.8. Elemental analysis calcd for C11H11NO2: C, 69.83; H, 5.86; N, 7.40. Found: C, 69.59; H, 6.00; N, 7.27.
4.2.12. 6-Methoxy-2-methylindole-3-carboxaldehyde (8b)
Compound 6b (0.718 g, 2.38 mmol) was dissolved in a solution of 3 N NaOH/EtOH (75 mL, 1:1) and refluxed at 90 °C for 36 h. The reaction mixture was then concentrated in vacuo and extracted using DCM (50 mL X 3). The combined organic layers were dried over Na2SO4, collected and concentrated in vacuo to provide brown oil 7b (205 mg, 53%). TLC and NMR of 7b suggested the sample was rapidly decomposing. The product was immediately taken to the next step. DMF (2 mL) was cooled to 0 °C. POCl3 (0.4 mL) was added and the reaction mixture was stirred for 10 min. A solution of crude 7b (205 mg, 1.27 mmol) in DMF (1 mL) was added to the reaction mixture dropwise over 10 min. The solution was stirred for an additional 2 h. The reaction mixture was added to ice-cold 1 N NaOH (40 mL) and stirred for 10 min. The precipitate was collected, washed with ice-cold H2O and dried overnight in a vacuum desiccator set at 40 °C yielding a tan solid (50 mg, 21%): mp 219–222 °C. TLC Rf 0.47 (75% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 11.79 (s, 1H), 9.99 (s, 1H), 7.89-7.88 (d, 1H, J = 8.58 Hz), 6.87 (d, 1H, J = 2.22 Hz), 6.79-6.78 (dd, 1H, J1 = 8.52 Hz, J2 = 2.28 Hz), 3.77 (s, 3H), 2.64 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 184.0, 156.2, 147.8, 136.3, 120.7, 119.4, 113.7, 110.9, 95.1, 55.2, 11.4. Elemental analysis calcd for C11H11NO2: C, 69.83; H, 5.86; N, 7.40. Found: C, 69.74; H, 5.98; N, 7.23.
4.2.13. 6-Methoxy-2-methylindole-3-carboxaldehyde (8b, Scheme 2)
This compound was prepared from 7b-Scheme 2 (280 mg, 1.74 mmol) in a similar manner to that for 8a except the reaction was stirred for 2 h while warming to rt to yield a tan solid (297 mg, 90%): mp 223–225 °C. TLC Rf 0.57 (EtOAc). 1H NMR (600 MHz, d6-DMSO) δ 11.79 (s, 1H), 9.99 (s, 1H), 7.89-7.88 (d, 1H, J = 8.58 Hz), 6.87 (d, 1H, J = 2.22 Hz), 6.79-6.78 (dd, 1H, J1 = 8.58 Hz, J2 = 2.28 Hz), 3.77 (s, 3H), 2.64 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 184.0, 156.2, 147.8, 136.3, 120.7, 119.4, 113.7, 110.9, 95.1, 55.2, 11.4. Elemental analysis calcd for C11H11NO2 • 0.15 H2O: C, 68.84; H, 5.93; N, 7.30. Found: C, 68.48; H, 5.90; N, 7.24.
4.2.14. 7-Methoxy-2-methylindole-3-carboxaldehyde (8c)
This compound was prepared from 7c (100 mg, 0.62 mmol) in a manner similar to that for 8a to yield a light brown solid (97 mg, 83%): mp 209 °C. TLC Rf 0.53 (80% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 12.10 (s, 1H), 10.03 (s, 1H), 7.62-7.61 (d, 1H, J = 7.80 Hz), 7.09-7.06 (t, 1H, J = 7.86 Hz), 6.78-6.77 (d, 1H, J = 7.74 Hz), 3.93 (s, 3H), 2.65 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 184.2, 147.8, 145.5, 126.9, 124.9, 122.6, 114.0, 112.5, 103.6, 55.1, 11.2. HPLC analysis: retention time = 3.338 min; peak area, 99.45%; eluent A, H2O; eluent B, CH3CN; Gradient 1 over 20 min with a flow rate of 1 mL min−1 and detection at 215 nm; injection of 10 μL of 20 μM 8c.
4.2.15. 5,6-Dimethoxy-2-methylindole-3-carboxaldehyde (8d)
DMF (2 mL) was cooled to 0 °C. POCl3 (0.6 mL) was added and the reaction mixture was stirred for 10 min at 0 °C. A solution of 7d (344 mg, 1.80 mmol) in DMF (2 mL) was added to the reaction mixture drop-wise over 10 min. The solution was stirred for an additional 3 h at rt, then slowly poured into ice-cold 1 N NaOH (50 mL) and stirred for 10 min. The solution was transferred to a separatory funnel and extracted with EtOAc (50 mL X 4). The combined organic layers were washed with brine (100 mL) and then dried over Na2SO4. The filtrate was collected and concentrated in vacuo. The material was purified by chromatography (20% to 100% EtOAc/hexanes) to yield a beige solid (368 mg, 93%): mp 204–208 °C. TLC Rf 0.25 (75% EtOAc/hexanes). 1H NMR (600 MHz, d6-acetone) δ 10.64 (s, 1H), 10.10 (s, 1H), 7.69 (s, 1H), 6.98 (s, 1H), 3.83 (s, 3H), 3.80 (s, 3H), 2.69 (s, 3H). 13C NMR (150 MHz, d6-acetone) δ 184.4, 148.64, 148.00, 146.6, 130.8, 120.0, 115.6, 104.2, 96.3, 56.51, 56.44, 11.6. Elemental analysis calcd for C12H13NO3: C, 65.74; H, 5.98; N, 6.39. Found: C, 65.47; H, 5.95; N, 6.28.
4.2.16. trans-3-(4-Methoxy-2-methyl-1H-indole-3-yl)-1-(4-pyridinyl)-2-propen-1-one (9a)
Compound 8a (100 mg, 0.53 mmol) was dissolved in anhydrous methanol (15 mL). 4-Acetylpyridine (96 mg, 0.79 mmol) and piperidine (67 mg, 0.79 mmol) were added and the solution was heated to reflux for 24 h. A precipitate slowly formed which was collected, washed with ice-cold MeOH (50 mL) and dried at 40 °C in a vacuum desiccator for 24 h to yield a bright orange powder (143 mg, 92%): mp 234 °C. TLC Rf 0.27 (80% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 11.98, (s, 1H), 8.82-8.81 (m, 2H), 8.46-8.44 (d, 1H, J = 15.54 Hz), 7.84-7.83 (m, 2H), 7.44-7.41 (d, 1H, J = 15.54 Hz), 7.10-7.08 (t, 1H, J = 7.92 Hz), 7.00-6.99 (d, 1H, J = 7.98 Hz), 6.70-6.68 (d, 1H, J = 7.74 Hz), 3.93 (s, 3H), 2.64 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 188.1, 153.2, 150.5, 145.3, 141.68, 141.46, 136.9, 123.1, 121.1, 116.13, 115.46, 109.3, 104.8, 102.1, 55.2, 14.0. Elemental analysis calcd for C18H16N2O2 • 0.25 MeOH: C, 72.98; H, 5.70; N, 9.33. Found: C, 72.62; H, 5.51; N, 9.14. HPLC analysis: retention time = 5.020 min; peak area, 98.99%; eluent A, H2O; eluent B, CH3CN; Gradient 1 over 20 min with a flow rate of 1 mL min−1 and detection at 224 nm; injection of 10 μL of 20 μM 9a.
4.2.17. trans-3-(6-Methoxy-2-methyl-1H-indole-3-yl)-1-(4-pyridinyl)-2-propen-1-one (9b)
This compound was prepared from 8b (50 mg, 0.26 mmol) in a manner similar to that for 9a to yield a bright orange powder (33 mg, 43%): mp 250–252 °C. TLC Rf 0.39 (80% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 11.83, (s, 1H), 8.81-8.80 (m, 2H), 8.07-8.04 (d, 1H, J = 15.18 Hz), 7.96-7.94 (m, 3H), 7.43-7.40 (d, 1H, J = 15.18 Hz), 6.91-6.90 (d, 1H, J = 2.28 Hz), 6.84-6.82 (dd, 1H, J1 = 8.64 Hz, J2 = 2.34 Hz), 3.80 (s, 3H), 2.56 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 187.9, 156.0, 150.6, 144.96, 144.92, 139.4, 137.3, 121.43, 121.10, 119.6, 112.6, 110.44, 109.55, 95.3, 55.2, 11.8. Elemental analysis calcd for C18H16N2O2 • 0.7 H2O: C, 70.90; H, 5.75; N, 9.19. Found: C, 70.51; H, 5.32; N, 8.93. HPLC analysis: retention time = 5.779 min; peak area, 98.44%; eluent A, H2O; eluent B, CH3CN; Gradient 1 over 20 min with a flow rate of 1 mL min−1 and detection at 230 nm; injection of 10 μL of 20 μM 9b.
4.2.18. trans-3-(6-Methoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (9b,Scheme 2)
This compound was prepared from 8b-Scheme 2 (252 mg, 1.33 mmol) in a manner similar to that for 9a to yield an orange solid (338 mg, 87%): mp 248–250 °C. TLC Rf 0.32 (80% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 11.83, (s, 1H), 8.81-8.80 (m, 2H), 8.07-8.04 (d, 1H, J = 15.18 Hz), 7.96-7.94 (m, 3H), 7.43-7.40 (d, 1H, J = 15.18 Hz), 6.91-6.90 (d, 1H, J = 2.28 Hz), 6.84-6.82 (dd, 1H, J1 = 8.64 Hz, J2 = 2.34 Hz), 3.80 (s, 3H), 2.56 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 187.9, 156.0, 150.6, 144.96, 144.92, 139.4, 137.3, 121.43, 121.10, 119.6, 112.6, 110.44, 109.55, 95.3, 55.3, 11.8. Elemental analysis calcd for C18H16N2O2 • 0.025 H2O: C, 73.84; H, 5.53; N, 9.57. Found: C, 73.45; H, 5.52; N, 9.32. HPLC analysis: retention time = 5.932 min; peak area, 97.97%; eluent A, H2O; eluent B, CH3CN; Gradient 1 over 20 min with a flow rate of 1 mL min−1 and detection at 229 nm; injection of 10 μL of 20 μM 9b.
4.2.19. trans-3-(7-Methoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (9c)
This compound was prepared from 8c (76 mg, 0.40 mmol) in a manner similar to that for 9a to yield a bright orange powder (104 mg, 89%): mp 265–266 °C. TLC Rf 0.43 (80% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 12.10 (s, 1H), 8.81-8.80 (m, 2H), 8.09-8.06 (d, 1H, J = 15.24 Hz), 7.95-7.94 (m, 2H), 7.63-7.62 (d, 1H, J = 7.92 Hz), 7.44-7.41 (d, 1H, J = 15.18 Hz), 7.16-7.13 (t, 1H, J = 7.92 Hz), 6.82-6.81 (d, 1H, J = 7.74 Hz), 3.95 (s, 3H), 2.57 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 188.0, 150.6, 145.80, 144.98, 144.91, 139.6, 127.2, 125.9, 122.26, 121.41, 113.09, 112.94, 109.9, 103.6, 55.3, 11.8. Elemental analysis calcd for C18H16N2O2 • 0.875 H2O: C, 70.17; H, 5.81; N, 9.09. Found: C, 69.78; H, 5.88; N, 8.79. HPLC analysis: retention time = 5.342 min; peak area, 98.91%; eluent A, H2O; eluent B, CH3CN; Gradient 1 over 20 min with a flow rate of 1 mL min−1 and detection at 225 nm; injection of 10 μL of 20 μM 9c.
4.2.20. trans-3-(5,6-Dimethoxy-2-methylindol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (9d)
This compound was prepared from 8d (355 mg, 1.62 mmol) in a manner similar to that for 9a to yield a dark orange gum (438 mg, 83%): mp 222–223 °C. TLC Rf 0.14 (80% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 11.75 (s, 1H), 8.81-8.80 (m, 2H), 8.07-8.05 (d, 1H, J = 15.24 Hz), 7.94-7.93 (m, 2H), 7.43 (s, 1H), 7.36-7.33 (d, 1H, J = 15.24 Hz), 6.94 (s, 1H), 3.88 (s, 3H), 3.80 (s, 3H), 2.55 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 188.1, 150.6, 146.8, 145.76, 145.17, 143.8, 139.7, 130.5, 121.4, 118.4, 112.5, 109.6, 103.7, 95.6, 56.40, 55.71, 12.0. Elemental analysis calcd for C19H18N2O3 • 0.075 H2O: C, 70.50; H, 5.65; N, 8.65. Found: C, 70.07; H, 5.64; N, 8.39. HPLC analysis: retention time = 5.311 min; peak area, 96.50%; eluent A, H2O; eluent B, CH3CN; Gradient 1 over 20 min with a flow rate of 1 mL min−1 and detection at 222 nm; injection of 10 μL of 20 μM 9d.
4.2.21. N-Boc-5-methoxy-2-methylaniline (11)
Compound 10 (2.0 g, 14.6 mmol) and di-tert-butyl-dicarbonate (3.51 g, 16.1 mmol) in THF (50 mL) were heated to reflux for 20 h. The reaction mixture was concentrated in vacuo and redissolved in DCM (75 mL). This mixture was washed with saturated NaHCO3 (75 mL), and then brine (50 mL). The organic layer was separated, dried over Na2SO4 and concentrated in vacuo to produce an oil which was purified by chromatography (0% to 20% EtOAc/hexanes) to yield a white solid (2.80 g, 81%): mp 79–80 °C (76–80 °C [39]). TLC Rf 0.55 (20% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.56 (s, 1H), 7.03-7.02 (d, 1H, J = 8.34 Hz), 6.56-6.54 (dd, 1H, J1 = 8.34 Hz, J2 = 2.64 Hz), 6.28 (s, 1H), 3.80 (s, 3H), 2.18 (s, 3H), 1.54 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 158.8, 153.0, 137.4, 130.9, 118.3, 109.6, 105.6, 80.7, 55.6, 28.6, 17.1.
4.2.22. 6-Methoxy-2-trifluoromethylindole (13)
This compound was prepared from 11 (1.18 g, 4.97 mmol) in a manner similar to that for compound 7b-Scheme 2 except Weinreb amide 12b (859 mg, 5.47 mmol) was employed to obtain a white solid (446 mg, 42%): mp 91–95 ° C. TLC Rf 0.32 (15% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.29 (s, 1H), 7.56-7.55 (d, 1H, J = 8.46 Hz), 6.89-6.87 (m, 3H), 3.86 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 158.4, 137.3, 124.6 (q, 2JFC = 39 Hz), 123.0, 121.52 (q, 1JFC = 266 Hz), 120.97, 112.1, 104.6 (q, 3JFC = 3 Hz), 94.4, 55.8. 19F NMR (376 MHz, CDCl3) δ −60.6 (s, 3F). Elemental analysis calcd for C10H8F3NO • 0.11 CH2Cl2: C, 54.09; H, 3.69; N, 6.24. Found: C, 54.54; H, 3.34; N, 5.78.
4.2.23. 6-Methoxy-2-trifluoromethylindole-3-carboxaldehyde (14)
DMF (2 mL) was cooled to 0 °C. POCl3 (0.6 mL) was added and the reaction mixture was stirred for 10 min at 0 °C. A solution of 13 (286 mg, 1.33 mmol) in DMF (2 mL) was added to the reaction mixture drop-wise over 10 min. The solution was stirred for 30 min while warming to rt and then heated to 80 °C for 3 h. The mixture was slowly poured into ice-cold 1 N NaOH (50 mL) and stirred for 10 min. The solution was transferred to a separatory funnel and extracted with EtOAc (50 mL X 4). The combined organic layers were washed with brine (100 mL) and then dried over Na2SO4. The filtrate was collected and concentrated in vacuo. The material was purified by chromatography (10% to 50% EtOAc/hexanes) to yield a white solid (105 mg, 32%): mp 243–245 ° C. TLC Rf 0.19 (20% EtOAc/hexanes). 1H NMR (600 MHz, d6-acetone) δ 11.91 (s, 1H), 10.29 (s, 1H), 8.21-8.20 (d, 1H, J = 8.82 Hz), 7.07 (d, 1H, J = 2.22 Hz), 7.03-7.01 (dd, 1H, J1 = 8.82 Hz, J2 = 2.28 Hz), 3.85 (s, 3H). 13C NMR (150 MHz, d6-acetone) δ 184.7, 160.1, 137.7, 130.9 (q, 2JFC = 39 Hz), 124.3, 122.1 (q, 1JFC = 268 Hz), 119.6, 117.6, 115.5, 95.6, 55.9. 19F NMR (376 MHz, d6-acetone) δ −51.9 (s, 3F). Elemental analysis calcd for C11H8F3NO2: C, 54.33; H, 3.32; N, 5.76. Found: C, 54.49; H, 3.32; N, 5.71.
4.2.24. trans-3-(6-Methoxy-2-trifluoromethyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (15)
Compound 14 (53 mg, 0.22 mmol) was dissolved in anhydrous MeOH (10 mL). 4-Acetylpyridine (40 mg, 0.33 mmol) and piperdine (28 mg, 0.33 mmol) were added and the mixture heated to reflux for 20 h. Upon completion, volatiles were evaporated in vacuo and the sample was purified by column chromatography (30% to 70% EtOAc/hexanes). The resulting solid was recrystallized from MeOH and dried at 40 °C in a vacuum desiccator for 36 h to yield a yellow solid (40 mg, 53%): mp 305–308 °C. TLC Rf 0.37 (75% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 12.35, (s, 1H), 8.85-8.84 (m, 2H), 8.38 (s, 1H), 8.21-8.18 (d, 1H, J = 15.72 Hz), 7.98-7.97 (m, 2H), 7.87-7.85 (d, 1H, J = 15.72 Hz), 7.01-6.98 (m, 2H), 3.95 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 189.0, 156.8, 150.8, 144.1, 141.7, 139.7, 125.0 (q, 2JFC = 38 Hz), 123.2, 121.55, 121.36 (q, 1JFC = 266 Hz), 120.19, 119.46, 118.40, 104.2 (q, 3JFC = 3 Hz), 93.8, 55.9. 19F NMR (376 MHz, d6-DMSO) δ −54.5 (s, 3F). Elemental analysis calcd for C18H13F3N2O2: C, 62.43; H, 3.78; N, 8.09. Found: C, 62.22; H, 3.84; N, 8.00. HPLC analysis: retention time = 8.239 min; peak area, 98.78%; eluent A, H2O; eluent B, CH3CN; Gradient 1 over 20 min with a flow rate of 1 mL min−1 and detection at 236 nm; injection of 10 μL of 20 μM 15.
4.2.25. trans-3-(5-Methoxy-2-methyl-1H-indol-3-yl)-1-(2,4,6-trimethoxyphenyl)-2-propen-1-one (17)
Compound 16 (189 mg, 1 mmol) and 2′,4′,6′-trimethoxyphenylacetophenone (210 mg, 1 mmol) were dissolved in MeOH (10 mL). KOH (50%, 10 mL) was added and the solution was heated to reflux for 7 d. Upon completion, MeOH was distilled in vacuo and the aqueous layer was extracted with DCM (50 mL X 3). The combined organic layers were washed with brine (75 mL) and dried over Na2SO4. The filtrate was collected and evaporated in vacuo. The resulting material was purified by chromatography (50% to 90% EtOAc/hexanes) to yield a yellow solid (65 mg, 17%): mp 197–200 °C. TLC Rf 0.34 (75% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 11.66 (s, 1H), 7.44-7.42 (d, 1H, J = 15.90 Hz), 7.27-7.25 (d, 1H, J = 8.70 Hz), 7.14 (d, 1H, J = 2.28 Hz), 6.79-6.77 (dd, 1H, J1 = 8.70 Hz, J2 = 2.34 Hz), 6.65-6.63 (d, 1H, J = 15.90 Hz), 6.31 (s, 2H), 3.83 (s, 3H), 3.80 (s, 3H), 3.72 (s, 6H), 2.36 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 192.5, 161.5, 157.9, 154.8, 142.9, 137.6, 130.9, 126.3, 122.0, 112.19, 111.82, 110.98, 108.1, 102.1, 91.0, 55.72, 55.41, 55.39, 11.8. Elemental analysis calcd for C22H23NO5 • 0.075 H2O: C, 69.03; H, 6.10; N, 3.66. Found: C, 68.63; H, 6.17; N, 3.59. HPLC analysis: retention time = 5.345 min; peak area, 96.94%; eluent A, H2O; eluent B, CH3CN; Gradient 1 over 20 min with a flow rate of 1 mL min−1 and detection at 204 nm; injection of 10 μL of 20 μM 17.
4.2.26. trans-3-(5-Methoxy-2-methyl-1H-indol-3-yl)-1-(3,4,5-trimethoxyphenyl)-2-propen-1-one (18)
This compound was prepared from 16 (100 mg, 0.53 mmol) in a manner similar to that for 9a to yield a yellow solid (131 mg, 65%): mp 250–253 °C. TLC Rf 0.45 (75% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 11.75 (s, 1H), 8.03-8.00 (d, 1H, J = 15.24 Hz), 7.50-7.48 (d, 1H, J = 15.24 Hz), 7.44 (d, 1H, J = 2.34 Hz), 7.37 (s, 2H), 7.29-7.28 (d, 1H, J = 8.70 Hz), 6.81-6.79 (dd, 1H, J1 = 8.70 Hz, J2 = 2.34 Hz), 3.90 (s, 6H), 3.86 (s, 3H), 3.75 (s, 3H), 2.57 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 187.5, 154.9, 152.8, 144.2, 141.0, 137.6, 134.3, 130.8, 126.5, 113.8, 112.23, 111.45, 109.2, 105.4, 102.4, 60.2, 55.89, 55.10, 12.1. Elemental analysis calcd for C22H23NO5 • 0.2 MeOH: C, 68.75; H, 6.19; N, 3.61. Found: C, 68.35; H, 5.94; N, 3.66. HPLC analysis: retention time = 6.131 min; peak area, 97.89%; eluent A, H2O; eluent B, CH3CN; Gradient 1 over 20 min with a flow rate of 1 mL min−1 and detection at 204 nm; injection of 10 μL of 20 μM 18.
4.2.27. trans-3-(5-Methoxy-1,2-dimethyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (19)
Compound 1a ‘MOMIPP’ (100 mg, 0.34 mmol) was dissolved in DMF (2.5 mL). NaH (16.4 mg, 0.68 mmol, 60% dispersion in mineral oil, unwashed) was added and stirred for 10 min. Methyl iodide (32 μL, 0.51 mmol) was then added and the reaction mixture was stirred at rt for 5 h. Upon completion, saturated NH4Cl solution (30 mL) was added to the reaction mixture and transferred to a separatory funnel. The aqueous later was extracted with EtOAc (25 mL X 3). The combined organic layer was dried over Na2SO4, the filtrate was collected and volatiles were distilled. The resulting material was purified by chromatography (2% to 5% MeOH/DCM). The resulting solid was recrystallized from MeOH to yield a yellow-orange solid (13 mg, 12%): mp 160–161 °C. TLC Rf 0.33 (75% EtOAc/hexanes). 1H NMR (600 MHz, d6-DMSO) δ 8.81-8.80 (m, 2H), 8.13-10 (d, 1H, J = 15.18 Hz), 7.95-7.94 (m, 2H), 7.50-7.48 (d, 1H, J = 8.88 Hz), 7.48 (d, 1H, J = 2.40 Hz), 7.41-7.38 (d, 1H, J = 15.18 Hz), 6.93-6.91 (dd, 1H, J1 = 8.82 Hz, J2 = 2.40 Hz), 3.88 (s, 3H), 3.74 (s, 3H), 2.58 (s, 3H). 13C NMR (150 MHz, d6-DMSO) δ 188.0, 155.5, 150.6, 146.7, 145.1, 139.4, 132.7, 125.7, 121.4, 112.8, 111.16, 110.82, 108.9, 103.7, 55.6, 30.3, 10.7. Elemental analysis calcd for C19H18N2O2 • 0.055 MeOH: C, 74.28; H, 5.96; N, 9.09. Found: C, 73.84; H, 5.90; N, 9.06. HPLC analysis: retention time = 5.788 min; peak area, 96.78%; eluent A, H2O; eluent B, CH3CN; Gradient 1 over 20 min with a flow rate of 1 mL min−1 and detection at 429 nm; injection of 10 μL of 20 μM 19.
4.3. Biological evaluation
4.3.1. Cell culture
U251 human glioblastoma cells were obtained from the DCT Tumor Repository (National Cancer Institute) and were maintained in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10% (v/v) fetal bovine serum (FBS) (JR Scientific, Woodland, CA) at 37°C with 5% CO2/95% air. For live cell imaging, the cells were plated in 35mm dishes at 100,000 cells per dish. On the following day compounds, dissolved in DMSO, were added at a final concentration of 2.5 μM. Controls received an equivalent volume of DMSO. Phase-contrast images were captured at 4 h and 48 h after addition of the compounds using an Olympus IX70 inverted microscope equipped with a DP-80 digital camera and Cellsense imaging software.
4.3.2. SRB assays
The effects of compounds on cell growth were assessed using the sulphorhodamine B (SRB) colorimetric assay, as described previously [11,15]. The concentration of each compound producing 50% growth inhibition (GI50) relative to the control without drug was calculated as described in the NCI-60 human cell line screening protocol (http://dtp.nci.nih.gov/branches/btb/ivclsp.html).
4.3.3. Imaging of microtubules
U251 cells were seeded on glass coverslips in 60 mm dishes at 350,000 cells dish. One day after plating, fresh medium was added with compounds at the indicated concentration. Cells were fixed and stained with a monoclonal antibody against α-tubulin, followed by Alexa Fluor 568-labeled goat anti-mouse IgG as described previously [15]. Nuclear DNA was stained with 4′,6-diamidino-2-phenyl-indole (DAPI).
4.3.4. Cell cycle analysis
U251 cells were seeded at 3.5 × 105 cells in 60 mm dishes. On the next day the cells were treated with 9b or 1a at the indicated concentrations. After 24 h the cells were harvested by trypsinization, fixed in ice-cold 70% ethanol, washed twice by centrifugation/resuspension in PBS, and then suspended in 900 μl PBS containing 6.25 mM MgSO4 and 1 mM CaCl2. After incubation at rt for 15 min, 20 μl of a 10 mg/ml solution of RNAse A was added and cells were incubated at 37° C for 15 min. Finally, 100 μl of a 500 mg/ml aqueous solution of propidium iodide was added and the cells were analyzed with a Becton-Dickinson FACS-Calibur flow cytometer. DNA histograms were generated with CellQuest Pro software.
Supplementary Material
Indole-derived chalcones were synthesized and then evaluated in glioblastoma cells.
The position of indole methoxy induces either methuosis or microtubule disruption.
9b disrupts microtubules (GI50 90 nM) and 1a induces methuosis (GI50 2.32 μM).
Both activities were reduced when trimethoxyphenyl was substituted for pyridinyl.
The 6-methoxy analogue may be a prototype for a new class of mitotic inhibitors.
Acknowledgments
This work was supported by the NIH (R01CA115495) and by the Harold and Helen McMaster Endowment for Biochemistry and Molecular Biology. We thank Dr. Jeff Sarver for his intellectual input. We also thank Dr. Yong Wah Kim for maintenance of the UT NMR facility.
ABBREVIATIONS
- EtOAc
ethyl acetate
- GM
glioblastoma multiforme
- h
hour
- IPP
indolyl-pyridinyl-propenone
- min
minute
- NaH
sodium hydride
- SAR
structure-activity relationship
- SRB
Sulforhodamine B
- PBS
phosphate-buffered saline
- rt
room temperature
- TBAB
tetrabutylammonium bromide
- TFA
trifluoroacetic acid
Appendix A. Supplementary Data
Supplementary data related to this article, including 1H NMR spectra for all synthesized compounds, 13C NMR for final targets and HPLC chromatograms for final targets can be found at http://dx.doi.org/.
Footnotes
Author Contributions
All authors have given approval to the final version of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Eng J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, Fisher J Consortium NC. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res. 2010;16:2443–2449. doi: 10.1158/1078-0432.CCR-09-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hurley LH. DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer. 2002;2:188–200. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- 4.Delbridge AR, Valente LJ, Strasser A. The role of the apoptotic machinery in tumor suppression. Cold Spring Harbor Perspect Biol. 2012;4:a008789. doi: 10.1101/cshperspect.a008789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 6.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nunez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kreuzaler P, Watson CJ. Killing a cancer: what are the alternatives? Nat Rev Cancer. 2012;12:411–424. doi: 10.1038/nrc3264. [DOI] [PubMed] [Google Scholar]
- 8.Maltese WA, Overmeyer JH. Methuosis: nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments. Am J Pathol. 2014;184:1630–1642. doi: 10.1016/j.ajpath.2014.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitambi SS, Toledo EM, Usoskin D, Wee S, Harisankar A, Svensson R, Sigmundsson K, Kalderen C, Niklasson M, Kundu S, Aranda S, Westermark B, Uhrbom L, Andang M, Damberg P, Nelander S, Arenas E, Artursson P, Walfridsson J, Forsberg Nilsson K, Hammarstrom LG, Ernfors P. Vulnerability of glioblastoma cells to catastrophic vacuolization and death induced by a small molecule. Cell. 2014;157:313–328. doi: 10.1016/j.cell.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 10.Maltese WA, Overmeyer JH. Non-apoptotic cell death associated with perturbations of macropinocytosis. Frontiers in Physiology. 2015;6:38. doi: 10.3389/fphys.2015.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trabbic CJ, Dietsch HM, Alexander EM, Nagy PI, Robinson MW, Overmeyer JH, Maltese WA, Erhardt PW. Differential induction of cytoplasmic vacuolization and methuosis by novel 2-indolyl-substituted pyridinylpropenones. ACS Med Chem Lett. 2014;5:73–77. doi: 10.1021/ml4003925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Overmeyer JH, Kaul A, Johnson EE, Maltese WA. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol Cancer Res. 2008;6:965–977. doi: 10.1158/1541-7786.MCR-07-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Overmeyer JH, Young AM, Bhanot H, Maltese WA. A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells. Mol Cancer. 2011;10:69–85. doi: 10.1186/1476-4598-10-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson MW, Overmeyer JH, Young AM, Erhardt PW, Maltese WA. Synthesis and evaluation of indole-based chalcones as inducers of methuosis, a novel type of nonapoptotic cell death. J Med Chem. 2012;55:1940–1956. doi: 10.1021/jm201006x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trabbic CJ, Overmeyer JH, Alexander EM, Crissman EJ, Kvale HM, Smith MA, Erhardt PW, Maltese WA. Synthesis and biological evaluation of indolyl-pyridinyl-propenones having either methuosis or microtubule disruption activity. J Med Chem. 2015;58:2489–2512. doi: 10.1021/jm501997q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liou JP, Chang YL, Kuo FM, Chang CW, Tseng HY, Wang CC, Yang YN, Chang JY, Lee SJ, Hsieh HP. Concise synthesis and structure-activity relationships of combretastatin A-4 analogues, 1-aroylindoles and 3-aroylindoles, as novel classes of potent antitubulin agents. J Med Chem. 2004;47:4247–4257. doi: 10.1021/jm049802l. [DOI] [PubMed] [Google Scholar]
- 17.Nahm S, Weinreb SM. N-Methoxy-N-Methylamides as Effective Acylating Agents. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]
- 18.Clark RD, Muchowski JM, Fisher LE, Flippin LA, Repke DB, Souchet M. Preparation of indoles and oxindoles from N-(tert-butoxycarbonyl)-2-alkylanilines. Synthesis. 1991;10:871–878. [Google Scholar]
- 19.Valdameri G, Gauthier C, Terreux R, Kachadourian R, Day BJ, Winnischofer SM, Rocha ME, Frachet V, Ronot X, Di Pietro A, Boumendjel A. Investigation of chalcones as selective inhibitors of the breast cancer resistance protein: critical role of methoxylation in both inhibition potency and cytotoxicity. J Med Chem. 2012;55:3193–3200. doi: 10.1021/jm2016528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang DJ, Wang J, Li W, Miller DD. Structural optimization of indole derivatives acting at colchicine binding site as potential anticancer agents. ACS Med Chem Lett. 2015;6:993–997. doi: 10.1021/acsmedchemlett.5b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.La Regina G, Bai R, Coluccia A, Famiglini V, Pelliccia S, Passacantilli S, Mazzoccoli C, Ruggieri V, Verrico A, Miele A, Monti L, Nalli M, Alfonsi R, Di Marcotullio L, Gulino A, Ricci B, Soriani A, Santoni A, Caraglia M, Porto S, Da Pozzo E, Martini C, Brancale A, Marinelli L, Novellino E, Vultaggio S, Varasi M, Mercurio C, Bigogno C, Dondio G, Hamel E, Lavia P, Silvestri R. New indole tubulin assembly inhibitors cause stable arrest of mitotic progression, enhanced stimulation of natural killer cell cytotoxic activity, and repression of hedgehog-dependent cancer. J Med Chem. 2015;58:5789–5807. doi: 10.1021/acs.jmedchem.5b00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HY, Chang CY, Lai MJ, Chuang HY, Kuo CC, Chang CY, Chang JY, Liou JP. Antimitotic and antivascular activity of heteroaroyl-2-hydroxy-3,4,5-trimethoxybenzenes. Bioorg Med Chem. 2015;23:4230–4236. doi: 10.1016/j.bmc.2015.06.043. [DOI] [PubMed] [Google Scholar]
- 23.Sherer C, Snape TJ. Heterocyclic scaffolds as promising anticancer agents against tumours of the central nervous system: Exploring the scope of indole and carbazole derivatives. Eur J Med Chem. 2015;97:552–560. doi: 10.1016/j.ejmech.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Dupeyre G, Chabot GG, Thoret S, Cachet X, Seguin J, Guenard D, Tillequin F, Scherman D, Koch M, Michel S. Synthesis and biological evaluation of (3,4,5-trimethoxyphenyl)indol-3-ylmethane derivatives as potential antivascular agents. Bioorg Med Chem. 2006;14:4410–4426. doi: 10.1016/j.bmc.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 25.Kumar D, Kumar NM, Akamatsu K, Kusaka E, Harada H, Ito T. Synthesis and biological evaluation of indolyl chalcones as antitumor agents. Bioorg Med Chem Lett. 2010;20:3916–3919. doi: 10.1016/j.bmcl.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 26.Boumendjel A, McLeer-Florin A, Champelovier P, Allegro D, Muhammad D, Souard F, Derouazi M, Peyrot V, Toussaint B, Boutonnat J. A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP in in-vitro and in-vivo glioblastoma models. BMC Cancer. 2009;9:242–252. doi: 10.1186/1471-2407-9-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Champelovier P, Chauchet X, Hazane-Puch F, Vergnaud S, Garrel C, Laporte F, Boutonnat J, Boumendjel A. Cellular and molecular mechanisms activating the cell death processes by chalcones: Critical structural effects. Toxicol In Vitro. 2013;27:2305–2315. doi: 10.1016/j.tiv.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 28.Altmann KH. The cytoskeleton and its interactions with small molecules. Bioorg Med Chem. 2014;22:5038–5039. doi: 10.1016/j.bmc.2014.06.037. [DOI] [PubMed] [Google Scholar]
- 29.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nature Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 30.Mukhtar E, Adhami VM, Mukhtar H. Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther. 2014;13:275–284. doi: 10.1158/1535-7163.MCT-13-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyle FM, Eller SL, Grossman SA. Penetration of intra-arterially administered vincristine in experimental brain tumor. Neuro-oncology. 2004;6:300–305. doi: 10.1215/S1152851703000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fellner S, Bauer B, Miller DS, Schaffrik M, Fankhanel M, Spruss T, Bernhardt G, Graeff C, Farber L, Gschaidmeier H, Buschauer A, Fricker G. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest. 2002;110:1309–1318. doi: 10.1172/JCI15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Still WC, Kahn M, Mitra A. Rapid chromatographic technique for preparative separations with moderate resolution. J Org Chem. 1978;43:2923–2925. [Google Scholar]
- 34.Sundberg RJ, Parton RL. Lithiation of methoxyindoles. J Org Chem. 1976;41:163–165. [Google Scholar]
- 35.Gourdoupis CG, Stamos IK. The utility of the Pummerer rearrangement intermediates in the presence of lewis-acids - a short and practical synthesis of 4-(2-Di-N-propylaminoethyl)-7-methoxyindole. Synthetic Comm. 1994;24:1137–1144. [Google Scholar]
- 36.Ty N, Dupeyre G, Chabot GG, Seguin J, Tillequin F, Scherman D, Michel S, Cachet X. Synthesis and biological evaluation of new disubstituted analogues of 6-methoxy-3-(3′,4′,5′-trimethoxybenzoyl)-1H-indole (BPR0L075), as potential antivascular agents. Bioorg Med Chem. 2008;16:7494–7503. doi: 10.1016/j.bmc.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Cook JWL, JW, McCloskey P. The chemistry of the Mitragyna genus. Part III. Synthesis of the degradation product of Mitragynine. J ChemSoc. 1952:3904–3912. [Google Scholar]
- 38.Sinhababu AK, Borchardt RT. Silica-gel assisted reductive cyclization of alkoxy-2,Beta-dinitrostyrenes to alkoxyindoles. J Org Chem. 1983;48:3347–3349. [Google Scholar]
- 39.Dillard RD, Bach NJ, Draheim SE, Berry DR, Carlson DG, Chirgadze NY, Clawson DK, Hartley LW, Johnson LM, Jones ND, McKinney ER, Mihelich ED, Olkowski JL, Schevitz RW, Smith AC, Snyder DW, Sommers CD, Wery JP. Indole inhibitors of human nonpancreatic secretory phospholipase A2. 1. Indole-3-acetamides. J Med Chem. 1996;39:5119–5136. doi: 10.1021/jm960485v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.