

Ataxia telangiectasia-mutated kinase (ATM) deficiency attenuates cardiac dysfunction early postmyocardial infarction (post-MI). Here, we provide evidence that ATM deficiency is associated with worse late post-MI outcomes in terms of function, fibrosis, apoptosis, and hypertrophy. Further identification of molecular targets involved in cell survival and growth may help explain the role of ATM in myocardial remodeling post-MI.
Keywords: ataxia telangiectasia-mutated kinase, cardiac remodeling, fibrosis, apoptosis, hypertrophy, myocardial infarction
Abstract
Ataxia telangiectasia-mutated kinase (ATM), a cell cycle checkpoint protein, is activated in response to DNA damage and oxidative stress. We have previously shown that ATM deficiency is associated with increased apoptosis and fibrosis and attenuation of cardiac dysfunction early (1–7 days) following myocardial infarction (MI). Here, we tested the hypothesis that enhanced fibrosis and apoptosis, as observed early post-MI during ATM deficiency, exacerbate cardiac dysfunction and remodeling in ATM-deficient mice late post-MI. MIs were induced in wild-type (WT) and ATM heterozygous knockout (hKO) mice by ligation of the left anterior descending artery. Left ventricular (LV) structural and functional parameters were assessed by echocardiography 14 and 28 days post-MI, whereas biochemical parameters were measured 28 days post-MI. hKO-MI mice exhibited exacerbated LV dysfunction as observed by increased LV end-systolic volume and decreased percent fractional shortening and ejection fraction. Infarct size and thickness were not different between the two genotypes. Myocyte cross-sectional area was greater in hKO-MI group. The hKO-MI group exhibited increased fibrosis in the noninfarct and higher expression of α-smooth muscle actin (myofibroblast marker) in the infarct region. Apoptosis and activation of GSK-3β (proapoptotic kinase) were significantly lower in the infarct region of hKO-MI group. Matrix metalloproteinase 2 (MMP-2) expression was not different between the two genotypes. However, MMP-9 expression was significantly lower in the noninfarct region of hKO-MI group. Thus ATM deficiency exacerbates cardiac remodeling late post-MI with effects on cardiac function, fibrosis, apoptosis, and myocyte hypertrophy.
NEW & NOTEWORTHY
Ataxia telangiectasia-mutated kinase (ATM) deficiency attenuates cardiac dysfunction early postmyocardial infarction (post-MI). Here, we provide evidence that ATM deficiency is associated with worse late post-MI outcomes in terms of function, fibrosis, apoptosis, and hypertrophy. Further identification of molecular targets involved in cell survival and growth may help explain the role of ATM in myocardial remodeling post-MI.
following myocardial infarction (MI), the heart undergoes a remodeling process whereby the architecture and composition of the left ventricle (LV) changes. Factors affecting these changes include myocyte hypertrophy, myocyte apoptosis, myofibroblast proliferation, and interstitial fibrosis (13). During the early stages of LV repair, there is differentiation of fibroblasts into myofibroblasts. The myofibroblast is a proliferative cell type that secretes a majority of the collagen that ultimately forms the post-MI scar (27). The early changes in LV architecture include formation of a scar and thinning of the infarcted region (13). During the later stages of cardiac remodeling, cross linking of the newly formed scar occurs, resulting in a subsequent reduction of myofibroblasts in the infarct region (24, 29). The later stages of structural remodeling are predominated by myocyte elongation in the noninfarcted zone, increased septal wall mass, LV chamber enlargement, and a shift from an elliptical to a spherical chamber configuration. The changes in myocyte size as well as increase in interstitial fibrosis result in a progressive decline in ventricular performance ultimately leading to heart failure (13).
Ataxia telangiectasia-mutated kinase (ATM) is a serine/threonine kinase that is activated in response to DNA damage. Once activated, ATM phosphorylates several proteins involved in cell cycle arrest, DNA repair, and apoptosis (20). Mutations in the ATM gene cause an autosomal recessive disorder known as ataxia-telangiectasia (AT). Individuals with AT exhibit neuronal degeneration and are at increased risk for developing cancer (26). Patients with one mutated allele make up ∼1.4–2.0% of the general population. These patients are spared from most of the symptoms of AT; however, they are predisposed to cancer and ischemic heart disease (23). Previously, we examined the role of ATM in cardiac remodeling during the inflammatory and proliferative phases of the post-MI healing process. It was observed that ATM deficiency results in attenuation of cardiac dysfunction early (1 and 7 days) post-MI. In addition, ATM deficiency has been associated with increased cardiac cell apoptosis and fibrosis post-MI (7, 9). An early increase in fibrosis post-MI may help maintain heart function but can ultimately result in cardiac dysfunction (22). Apoptosis has also been associated with a negative post-MI outcome, and inhibition of cardiac cell apoptosis has been shown to reduce infarct size and preserve ventricular function (12, 21, 32). Here, we tested the hypothesis that enhanced fibrosis and apoptosis, as observed early post-MI during ATM deficiency, would exacerbate cardiac dysfunction and remodeling in ATM-deficient hearts late (14 and 28 days) post-MI. The data presented here demonstrate that ATM deficiency worsens cardiac function and remodeling late post-MI with effects on fibrosis, apoptosis, and myocyte hypertrophy.
MATERIALS AND METHODS
Vertebrate animals.
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All the experiments were performed in accordance with the protocols approved by the East Tennessee State University Committee on Animal Care. ATM-deficient mice (129xblack Swiss hybrid background), generated as described (2), were purchased from Jackson Laboratory. Genotyping was performed by PCR using primers suggested by the Jackson Laboratory. Age-matched (∼4 mo old) male and female mice were used for the study. Because homozygous knockout mice die at ∼2 mo of age because of thymic lymphomas (2), the study used ATM heterozygous knockout (hKO; deficient) mice. ATM-deficient mice did not exhibit visible signs of thymic lymphomas during the course of the investigation. All mice undergoing surgery (sham and MI) received buprenorphine injections before surgery and 24 h after surgery. Mice were monitored on a daily basis to evaluate survival rate.
MI.
MI was performed as previously described (7). Briefly, mice were anesthetized with a mixture of isoflurane (2%) and oxygen (0.5 l/min) inhalation and ventilated using a rodent ventilator (Harvard Apparatus). Body temperature was maintained at ∼37°C using a heating pad. The heart was exposed by a left thoracotomy followed by ligation of the left anterior descending artery (LAD) with a 7-0 polypropylene suture. Mice in the sham group underwent the same procedure without ligation of the LAD. At the end of the study period (28 days post-MI), isolated hearts were used for either histology or molecular analyses.
Echocardiography.
M-mode echocardiography images were obtained using transthoracic short-axis imaging at midpapillary level 14 and 28 days post-MI using a Toshiba Aplio 80 Imaging Systemequipped with a 12-MHz linear transducer as previously described (7). Measurements were performed on the M-mode images using the leading edge to leading edge method (10). An individual blinded to the experimental groups recorded the images, while a second individual assessed the images and calculated the structural and functional parameters of the heart.
Morphometric analyses.
Twenty-eight days following MI, hearts were excised and aortically perfused with Krebs buffer and then stopped in diastole using KCl (30 mmol/l). Hearts were then fixed using 10% buffered formalin for 24–48 h. The heart was divided into three transverse sections (base, mid, and apex) and embedded in paraffin. Tissue sections (4 μm thick) were stained using Masson's trichrome stain to determine infarct size. Percent infarct size was determined by adding the midline infarct lengths of three histological sections and dividing them by the sum of the midline LV circumference and multiplying by 100 (19). Masson's trichrome-stained sections were also used to measure percent fibrosis. For this, at least five images were analyzed from the noninfarct and infarct regions. Percent fibrosis was calculated by dividing total fibrosis by total tissue area and multiplying by 100.
TUNEL assay.
TUNEL staining was performed according to manufacturer's instruction (cell death detection kit; Roche). Heart tissue sections (4 μm thick) were stained with rhodamine-conjugated wheat germ agglutinin (WGA) to identify myocytes and Hoechst 33258 (Sigma) to identify nuclei. Index of apoptosis was calculated as the total number of apoptotic cells divided by the total number of nuclei and multiplied by 100.
Immunohistochemistry.
Cross sections of the heart (4 μm thick) were deparaffinized and rehydrated with xylene and ethanol rinses. Immunohistochemical staining for myofibroblasts was performed using anti-α-smooth muscle actin (α-SMA) antibodies (Sigma). All images (at least 5 each from infarct and noninfarct regions) were acquired using a Nikon TE-2000 microscope equipped with an Andor Zyla sCMOS camera. Quantitative analysis was carried out using Nikon NIS-Elements software.
Myocyte cross-sectional area.
Cross sections of the heart (4 μm thick) were stained with rhodamine-conjugated WGA to measure myocyte cross-sectional area. Images from the noninfarct region were acquired using a fluorescent microscope equipped with an Andor Zyla sCMOS camera. Suitable areas of the section were defined as myocytes with nearly circular profiles. Myocyte cross-sectional areas were measured using Nikon NIS-Elements software as described (7).
Western blot analysis.
LV lysates were prepared in RIPA buffer, separated by SDS-PAGE, and transferred to a PVDF membrane. The membrane was blocked using either 5% nonfat milk or 5% BSA. The membranes were then incubated overnight with antibodies against p-GSK-3β (Ser-9), matrix metalloproteinase 2 (MMP-2), or MMP-9 (Santa Cruz Biotechnology). There were no significant differences in GAPDH levels between genotypes or groups. Therefore, GAPDH (Santa Cruz Biotechnology) immunostaining was used as a protein-loading control. The immune complexes were detected using chemiluminescence reagents (Pierce Biotechnology) in conjunction with X-ray film. Band intensities were quantified using Kodak photo documentation system.
Statistical analyses.
Data are presented as means ± SE. Data were tested for normality using a Shapiro-Wilk test. Multiple comparisons were analyzed using either one-way ANOVA or a Kruskal-Wallis test. All pairwise comparisons were carried out using either a Student's t-test or Mann-Whitney U-test. Survival analysis was performed using a Kaplan-Meier survival curve and a log-rank test. Probability (P) values <0.05 were considered significant.
RESULTS
Morphological analyses.
There was no significant difference in the body weights (BW) between the two sham groups. BWs remained unchanged between the two genotypes post-MI. MI increased heart weight (HW) and HW/BW ratios to a similar extent in both genotypes (Table 1). Infarct size (% infarct size: WT-MI, 38.8 ± 3.5 vs. hKO-MI, 42.7 ± 4.4; P = 0.25; n = 6–7) and infarct width (WT-MI, 48.9 ± 11.1 μm vs. hKO 39.1 ± 4.3 μm; P = 0.22; n = 6–7) were not significantly different between the two genotypes 28 days post-MI.
Table 1.
Body weight and heart weight for all four groups
| Body Weight | Heart Weight | HW/BW | |
|---|---|---|---|
| WT-sham (n = 7) | 26.20 ± 0.59 | 109.59 ± 2.97 | 4.19 ± 0.10 |
| hKO-sham (n = 7) | 28.15 ± 1.52 | 122.31 ± 8.59 | 4.34 ± 0.18 |
| WT-MI (n = 22) | 25.29 ± 0.70 | 164.69 ± 7.28* | 6.66 ± 0.44* |
| hKO-MI (n = 22) | 26.12 ± 0.69 | 158.94 ± 6.28* | 6.11 ± 0.21* |
Values are means ± SE.
P < 0.05 vs. respective sham. HW, heart weight; BW, body weight; WT, wild-type; hKO, heterozygous knockout; MI, myocardial infarction.
Survival rate was 56.3% (36/64), 54.7% (35/64), and 53.1% (34/64) at 7, 14, and 28 days post-MI, respectively, in the WT-MI group, whereas it was 52.2% (36/69), 43.5% (30/69), and 39.1% (27/69) at 7, 14, and 28 days post-MI, respectively, in the hKO-MI group. Although the survival rate was not found to be statistically significant (P = 0.2; Fig. 1), the survival curve showed a separation between two groups ∼7 days post-MI.
Fig. 1.
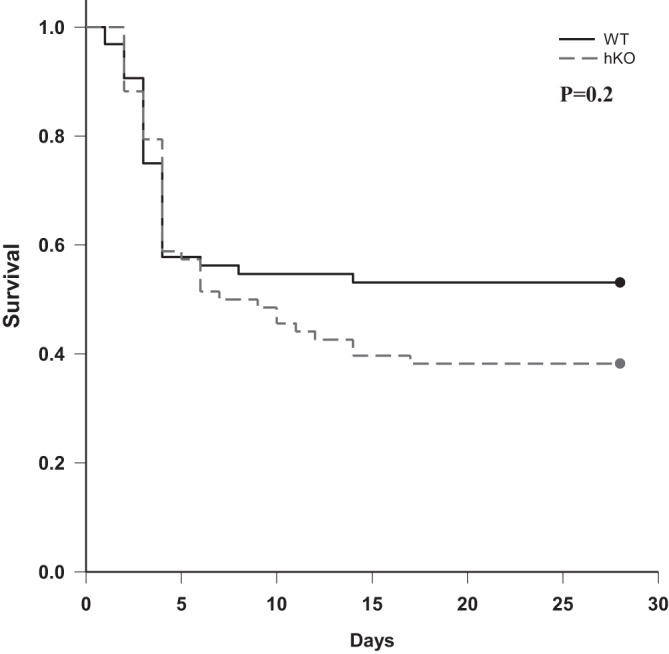
Kaplan-Meier survival curve and log-rank test. Survival was monitored over 28 days post-myocardial infarction (MI). There was a trend toward decreased survival in the heterozygous knockout (hKO)-MI group compared with the wild-type (WT)-MI group (P = 0.2 using log-rank test; WT, n = 64; hKO, n = 69).
Echocardiographic studies.
M-mode echocardiography revealed no differences in percent fractional shortening (%FS), ejection fraction (%EF), or LV end-systolic volume (LVESV) between the two sham groups. Echocardiographic parameters were not significantly different between 14-day vs. 28-day sham groups. Therefore, the sham data were pooled to increase the sample size. MI significantly decreased %FS and %EF and increased LVESV in both genotypes compared with their respective sham groups 14 and 28 days post-MI. However, the decrease in %FS and %EF and increase in LVESV were significantly greater in the hKO-MI group compared with the WT-MI group at both time points (Fig. 2).
Fig. 2.

Ataxia telangiectasia-mutated kinase (ATM) deficiency worsens left ventricular (LV) function post-MI. MI was performed in WT and hKO mice. Indices of cardiac function; percent fractional shortening (%FS), percent ejection fraction (%EF), and LV end-systolic volume (LVESV) were calculated using echocardiographic images 14 and 28 days post-MI. A: M-mode echocardiographic images. B: LVESV. C: %FS; D: %EF. *P < 0.05 vs. respective sham, #P < 0.05 vs. WT-MI; n = 8–14. PW, posterior wall; SW, septal wall; LVESD, LV end-systolic diameter; LVEDD, LV end-diastolic diameter.
Fibrosis, apoptosis, and myocyte cross-sectional area.
Quantitative analysis of fibrosis using Masson's trichrome-stained sections indicated increased fibrosis in the LV of hKO-sham compared with the WT-sham (% fibrosis: WT-sham, 0.07 ± 0.01; hKO-sham: 0.14 ± 0.02*; *P < 0.05 vs. WT-sham; n = 5–7). In hKO-sham, the increase in fibrosis was mainly observed in the perivascular space. Late post-MI, there was a significant increase in fibrosis in the infarct and noninfarct regions (mainly in the interstitial space) of both genotypes. However, the increase in fibrosis was significantly greater in the noninfarct region of hKO-MI vs. the WT-MI (% fibrosis: WT-MI, 1.26 ± 0.7*; hKO-MI, 3.21 ± 0.7*$; *P < 0.05 vs. sham; $P < 0.05 vs. WT-MI; n = 4–8; Fig. 3). Fibrosis in the infarct region was not significantly different between the two genotypes (% fibrosis: WT-MI, 61.4 ± 4.3; hKO-MI, 56.3 ± 4.7; P = 0.23; n = 4–8).
Fig. 3.

ATM deficiency is associated with increased fibrosis in the noninfarct region. Masson's trichrome-stained sections of the heart were used for quantitative measurement of fibrosis. Blue stain indicates fibrosis, while red stain indicates living tissue. Top: Masson's trichrome-stained sections demonstrating fibrosis in the WT-sham, hKO-sham, and noninfarct regions from WT and hKO hearts 28 days post-MI. Bottom: quantitative analysis of fibrosis (*P < 0.05 vs. WT-sham, #P < 0.05 vs. hKO-sham, $P < 0.05 vs. WT-MI; n = 4–8).
Cardiac cell apoptosis was not different between the two sham groups. MI increased apoptosis in the noninfarct and infarct regions of both genotypes late post-MI vs. their respective sham groups. There was no significant difference in apoptosis in the noninfarct region between the two genotypes. However, apoptosis was significantly lower in the infarct region of the hKO-MI vs. WT-MI (Fig. 4).
Fig. 4.

ATM deficiency decreases apoptosis in the infarct region 28 days post-MI. Top: TUNEL-stained and Hoechst-stained images obtained from the infarct regions of WT and hKO hearts post-MI. Green fluorescent staining indicates TUNEL-positive (apoptotic) nuclei, while blue fluorescent staining indicates total number of nuclei. Bottom: quantitative analysis of apoptosis in the infarct (Inf) and noninfarct (Non) regions. *P < 0.05 vs. respective sham, #P < 0.05 vs. respective Non, $P < 0.05 vs. WT-Inf; n = 5–8.
Myocyte cross-sectional area, measured in the septal wall, was greater in the hKO-sham vs. WT-sham group. Following MI, there was a significant increase in myocyte cross-sectional area in the noninfarct (septal wall) region of both genotypes compared with their respective sham groups. However, the increase in myocyte cross-sectional area was significantly larger in the hKO-MI vs. WT-MI (Fig. 5).
Fig. 5.

ATM deficiency increases myocyte cross-sectional area. Cross sections of the heart were stained using rhodamine-conjugated wheat germ agglutinin (WGA). Top: WGA-stained images obtained from the WT-sham, hKO-sham, and noninfarct regions of WT and hKO hearts post-MI. Bottom: quantitative analysis of myocyte cross-sectional areas in the sham and noninfarct regions 28 days post-MI (*P < 0.05 vs. respective sham, #P < 0.05 vs. WT-sham, $P < 0.05 vs. WT-MI; n = 7–9).
Expression of α-SMA.
Myofibroblasts are the main producers of collagen post-MI. Expression of α-SMA is commonly used to identify myofibroblasts (8). There was no significant difference in α-SMA expression between the two sham groups. Late post-MI, α-SMA expression was significantly greater in the infarct region of both genotypes compared with their respective sham groups or noninfarct regions. However, α-SMA expression was significantly higher in the infarct region of the hKO-MI vs. WT-MI (Fig. 6).
Fig. 6.

ATM deficiency increases α-smooth muscle actin (α-SMA) expression 28 days post-MI. Cross sections of the hearts were immunostained using anti-α-SMA antibodies. Top: α-SMA-stained images from the infarct regions of WT and hKO hearts 28 days post-MI. Bottom: quantitative immunohistochemical analysis of α-SMA expression (*P < 0.05 vs. respective sham; #P < 0.05 vs. respective Non; $P < 0.05 vs. WT-MI Inf; n = 5–7).
Activation of GSK-3β.
GSK-3β is a proapoptotic kinase that is inactivated by phosphorylation at serine-9 (25). GSK-3β phosphorylation (inactivation) remained unchanged between the sham groups and noninfarct regions of the two groups late post-MI. However, GSK-3β phosphorylation was significantly higher in the infarct region of the hKO-MI vs. the infarct region of the WT-MI group, implying decreased activity of GSK-3β in the hKO-MI group (Fig. 7).
Fig. 7.
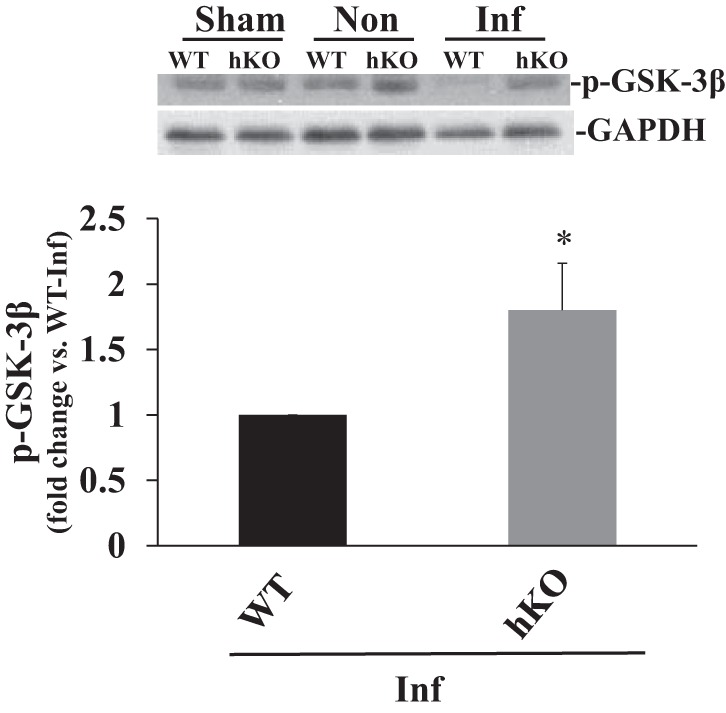
ATM deficiency decreases GSK-3β activation 28 days post-MI. Total LV lysates, prepared from sham, noninfarct (Non), and infarct (Inf) regions post-MI, were analyzed by Western blot using phospho-specific GSK-3β (Ser-9) antibodies. Top: autoradiograms indicating immunostaining for p-GSK-3β and GAPDH. Bottom: quantitative analyses of p-GSK-3β normalized to GAPDH (*P < 0.05 vs. WT-Inf; n = 7).
Expression of MMP-2 and MMP-9.
MMP-2 and MMP-9 play an important role in modifying the composition of the extracellular matrix (ECM) post-MI (15). MMP-2 protein levels were significantly lower in the infarct region of both MI groups compared with their respective sham and noninfarct groups with no significant differences between the two genotypes (Fig. 8A). MMP-9 protein levels were also lower in the infarct region vs. their respective sham groups. However, there was no significant difference between the two genotypes. Interestingly, MMP-9 protein levels were significantly lower in the noninfarct region of the hKO-MI vs. the WT-MI group and its respective sham (Fig. 8B).
Fig. 8.

ATM deficiency decreases expression of matrix metalloproteinase 9 (MMP-9) in the noninfarct region 28 days post-MI. Total LV lysates, prepared from sham, noninfarct (Non), and infarct (Inf) regions 28 days post-MI, were analyzed by Western blot using anti-MMP-2 (A) and anti-MMP-9 (B) antibodies. Top: autoradiograms indicating immunostaining for MMP-2, MMP-9, and GAPDH. Bottom: quantitative analyses of MMP-2 and MMP-9 normalized to GAPDH (*P < 0.05 vs. respective sham, #P < 0.05 vs. respective Non, $P < 0.05 vs. WT-Non; n = 5–6).
DISCUSSION
A major finding of this study is that deficiency of ATM is associated with exacerbated heart dysfunction late post-MI. Survival 28 days post-MI was 14% lower in the ATM-deficient group. Myocardial fibrosis in the noninfarct region and α-SMA expression in the infarct region were higher in the ATM-deficient group, whereas apoptosis was higher in the infarct region of the WT group late post-MI. Myocyte cross-sectional area was greater in the ATM-deficient group. ATM deficiency was associated with decreased GSK-3β activation in the infarct region, whereas MMP-9 expression was higher in the noninfarct region. Thus ATM deficiency impairs heart function and remodeling late post-MI with effects on apoptosis, fibrosis, and hypertrophy.
Patients with AT (patients with mutations at both ATM allele) are reported to have increased plasma cholesterol and triglyceride levels (1). Ischemic heart disease (encompassing International Statistical Classification of Diseases codes 410–414) is the second major cause of death in patients carrying one mutated ATM allele. On average, carriers of one mutated ATM allele die ∼11 yr earlier than noncarriers because of ischemic heart disease (23). In apoE-deficient mice, ATM deficiency results in exaggerated hypercholesterolemia and atherosclerotic lesions (30), suggesting that ATM deficiency promotes atherosclerosis. Although not found to be significant, the survival curve showed a separation between two groups ∼7 days post-MI. It is possible that aberrant hypertrophic, apoptotic, and fibrotic responses during ATM deficiency may enhance adverse remodeling post-MI and susceptibility to death.
Late post-MI, ATM deficiency is associated with exacerbated systolic dysfunction as measured by decreased %FS and %EF. Increased LVESV is a predictor of mortality post-MI (28). Here we observed increased LVESV in the ATM-deficient group at both time points. The observations of exacerbated LV dysfunction and increased LVESV are in contrast to our early post-MI findings in which ATM deficiency was associated with reduced functional impairment (7, 9). There was no correlation between the exaggerated LV dysfunction late post-MI and infarct size and/or thickness. Late post-MI, infarct size and thickness were not different between the two genotypes. Other factors involved in the development of heart failure post-MI include cardiac hypertrophy, apoptosis, and fibrosis (33). Cardiac myocyte hypertrophy (pathological myocyte enlargement) occurs following MI to maintain cardiac output because of myocyte loss (11). However, increases in myocyte size often result in cardiac dysfunction late post-MI (13). Here, ATM deficiency is associated with increased myocyte cross-sectional area in sham animals. MI led to increased myocyte hypertrophy in both genotypes. However, the increase in myocyte hypertrophy was significantly greater in the ATM-deficient group. Previously, we have shown that ATM deficiency is associated with increased myocyte apoptosis and fibrosis in the border LV region early (7 days) post-MI (9). Here, apoptosis in the noninfarct regions was not different between the two genotypes late post-MI. ATM deficiency was associated with increased fibrosis in the sham group, which continued to be greater in the noninfarct region late post-MI. Therefore, increased myocyte hypertrophy, myocardial fibrosis, and myocyte apoptosis (in the region bordering the infarct early post-MI) may help explain exaggerated heart dysfunction during ATM deficiency late post-MI.
Myofibroblasts are generally absent in the healthy heart. However, they are present in the infarct region following MI and play an important role in wound healing. Myofibroblasts aid in wound closure and are the main source of collagen production during infarct healing (4). These cells may also aid in the contractile activity of the heart because of the expression of α-SMA (29). However, a persistent presence of myofibroblasts may cause adverse remodeling by turning the fibrous tissue into a living secretome (6). Here, we observed decreased apoptosis and increased α-SMA expression in the infarct region during ATM deficiency late post-MI. Furthermore, the decrease in apoptosis was associated with inactivation of a proapoptotic kinase, GSK-3β (17). Myofibroblasts have been identified as the major cell type undergoing apoptosis in the infarct region at this time point (31). Therefore, it is likely that the observed decrease in apoptosis in the infarct region during ATM deficiency is due to the decrease in myofibroblast apoptosis. Previously, we have shown that ATM deficiency is associated with increased α-SMA expression and decreased apoptosis in the infarct region of ATM-deficient hearts 7 days post-MI (9). Late post-MI, the amount of fibrosis in the infarct region was not different between the two genotypes. Therefore, it is plausible that an early increase in fibrosis and decreased apoptosis in the infarct region may also contribute to the observed LV dysfunction during ATM deficiency late post-MI. Taken together, these observations suggest that ATM deficiency affects heart function post-MI via the involvement of both cardiac fibroblasts and myocytes. Enhanced activation of fibroblasts in the infarct region early post-MI during ATM deficiency may enhance fibrosis in the infarct region, which in turn helps prevent dilative remodeling. However, increased myocyte hypertrophy and apoptosis (with increased fibrosis in the noninfarct region) may be accountable for late LV dysfunction and dilative remodeling during ATM deficiency.
Changes in MMP expression affect deposition of fibrosis (16). MMP-2 and MMP-9 have been suggested to play major roles in ECM remodeling via continual degradation of collagen, which leads to LV dilation and pump dysfunction (5). MMP-2 promoter activation peaks 7 days post-MI in the infarct region and 14 days post-MI in the noninfarct region. MMP-9 promoter activation peaks at 7 days post-MI in both infarct and noninfarct regions (18). Previously, we have shown that MI increases protein levels of MMP-2 in the infarct region 7 days post-MI. However, ATM deficiency had no effect on the expression and activity of MMP-2 (9). Here we observed decreased expression of MMP-2 in the infarct regions of both genotypes 28 days post-MI. ATM deficiency had no effect on MMP-2 expression, suggesting that MMP-2 may not play a major role in fibrosis during ATM deficiency post-MI. In contrast, ATM deficiency has been associated with increased expression and activation of MMP-9 in the infarct region 7 days post-MI (9). At 28 days, however, MI led to decreased expression of MMP-9 in the infarct regions of both genotypes with no differences between the genotypes. Interestingly, ATM-deficient hearts exhibited significantly lower expression of MMP-9 in the noninfarct regions. This decrease in MMP-9 expression was associated with increased fibrosis in the noninfarct regions of ATM-deficient hearts. These data point toward the possibility that ATM deficiency affects expression of MMP-9 and that MMP-9, not MMP-2, plays a role in fibrosis during ATM deficiency post-MI. MMP-9 degrades ECM; therefore, decrease in MMP-9 expression is expected to be associated with increased fibrosis. However, the relationship between MMP-9 and fibrosis may be more intricate than anticipated because deletion of MMP-9 has been shown to be associated with decreased fibrosis in aged mice (3) and improved LV function post-MI (14).
Conclusion.
The data presented here in conjunction with our previous findings (7, 9) provide evidence that ATM affects different phases of cardiac remodeling post-MI. Early post-MI, infarct healing may benefit from ATM deficiency in terms of cardiac function. However, it ultimately is associated with negative consequences on cardiac function, which can be attributed to increased hypertrophy, apoptosis, and fibrosis. Future investigations aimed at identifying the molecular signals in modulating cell survival and growth during ATM deficiency are needed to help explain the role of ATM in the healing process of the heart post-MI.
GRANTS
This work was supported by Merit Review awards (BX002332 and BX000640) from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development, National Institutes of Health (R15HL129140), and funds from Institutional Research and Improvement account (to K. Singh).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.L.D., C.R.F., M.S., and K.S. conception and design of research; L.L.D., P.T., C.R.D., and C.R.F. performed experiments; L.L.D., S.L.S., P.T., and S.D. analyzed data; L.L.D., S.L.S., P.T., and K.S. interpreted results of experiments; L.L.D., S.L.S., and S.D. prepared figures; L.L.D. drafted manuscript; L.L.D., S.L.S., S.D., C.R.F., M.S., and K.S. edited and revised manuscript; L.L.D., S.L.S., P.T., S.D., C.R.D., C.R.F., M.S., and K.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We would like to thank Barbara “Bobbie” Connelly for support and technical expertise.
REFERENCES
- 1.Badalian LO, Kalinina LV. [Lipid metabolism disorder in ataxia-telangiectasia]. Zh Nevropatol Psikhiatr Im S S Korsakova 76: 665–669, 1976. [PubMed] [Google Scholar]
- 2.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. ATM-deficient mice: A paradigm of ataxia telangiectasia. Cell 86: 159–171, 1996. [DOI] [PubMed] [Google Scholar]
- 3.Chiao YA, Ramirez TA, Zamilpa R, Okoronkwo SM, Dai Q, Zhang J, Jin YF, Lindsey ML. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc Res 96: 444–455, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol 147: 325–338, 1995. [PMC free article] [PubMed] [Google Scholar]
- 5.Creemers EE, Cleutjens JP, Smits JF, Daemen MJ. Matrix metalloproteinase inhibition after myocardial infarction: A new approach to prevent heart failure? Circ Res 89: 201–210, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Dadson K, Turdi S, Boo S, Hinz B, Sweeney G. Temporal and molecular analyses of cardiac extracellular matrix remodeling following pressure overload in adiponectin deficient mice. PLoS One 10: e0121049, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daniel LL, Daniels CR, Harirforoosh S, Foster CR, Singh M, Singh K. Deficiency of ataxia telangiectasia mutated kinase delays inflammatory response in the heart following myocardial infarction. J Am Heart Assoc 3: e001286, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eddy RJ, Petro JA, Tomasek JJ. Evidence for the nonmuscle nature of the “myofibroblast” of granulation tissue and hypertropic scar. An immunofluorescence study. Am J Pathol 130: 252–260, 1988. [PMC free article] [PubMed] [Google Scholar]
- 9.Foster CR, Daniel LL, Daniels CR, Dalal S, Singh M, Singh K. Deficiency of ataxia telangiectasia mutated kinase modulates cardiac remodeling following myocardial infarction: involvement in fibrosis and apoptosis. PLoS One 8: e83513, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gardin JM, Siri FM, Kitsis RN, Edwards JG, Leinwand LA. Echocardiographic assessment of left ventricular mass and systolic function in mice. Circ Res 76: 907–914, 1995. [DOI] [PubMed] [Google Scholar]
- 11.Gould KE, Taffet GE, Michael LH, Christie RM, Konkol DL, Pocius JS, Zachariah JP, Chaupin DF, Daniel SL, Sandusky GE, Hartley CJ, Entman ML. Heart failure and greater infarct expansion in middle-aged mice: A relevant model for postinfarction failure. Am J Physiol Heart Circ Physiol 282: H615–H621, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Holly TA, Drincic A, Byun Y, Nakamura S, Harris K, Klocke FJ, Cryns VL. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J Mol Cell Cardiol 31: 1709–1715, 1999. [DOI] [PubMed] [Google Scholar]
- 13.Konstam MA, Kramer DG, Patel AR, Maron MS, Udelson JE. Left ventricular remodeling in heart failure: Current concepts in clinical significance and assessment. JACC Cardiovasc Imaging 4: 98–108, 2011. [DOI] [PubMed] [Google Scholar]
- 14.Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, McClister DM, Su H, Gannon J, MacGillivray C, Lee RT, Sinusas AJ, Spinale FG. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol 290: H232–H239, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther 30: 31–41, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsui Y, Morimoto J, Uede T. Role of matricellular proteins in cardiac tissue remodeling after myocardial infarction. World J Biol Chem 1: 69–80, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menon B, Johnson JN, Ross RS, Singh M, Singh K. Glycogen synthase kinase-3beta plays a pro-apoptotic role in beta-adrenergic receptor-stimulated apoptosis in adult rat ventricular myocytes: Role of beta1 integrins. J Mol Cell Cardiol 42: 653–661, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukherjee R, Mingoia JT, Bruce JA, Austin JS, Stroud RE, Escobar GP, McClister DM, Allen CM, Alfonso-Jaume MA, Fini ME, Lovett DH, Spinale FG. Selective spatiotemporal induction of matrix metalloproteinase-2 and matrix metalloproteinase-9 transcription after myocardial infarction. Am J Physiol Heart Circ Physiol 291: H2216–H2228, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Nascimento DS, Valente M, Esteves T, de Pina Me F, Guedes JG, Freire A, Quelhas P, Pinto-do-Ó P. MIQuant—semi-automation of infarct size assessment in models of cardiac ischemic injury. PLoS One 6: e25045, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr 144: 505–511, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Rivard AL, Steer CJ, Kren BT, Rodrigues CM, Castro RE, Bianco RW, Low WC. Administration of tauroursodeoxycholic acid (TUDCA) reduces apoptosis following myocardial infarction in rat. Am J Chin Med 35: 279–295, 2007. [DOI] [PubMed] [Google Scholar]
- 22.See F, Watanabe M, Kompa AR, Wang BH, Boyle AJ, Kelly DJ, Gilbert RE, Krum H. Early and delayed t ranilast treatment reduces pathological fibrosis following myocardial infarction. Heart Lung Circ 22: 122–132, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Su Y, Swift M. Mortality rates among carriers of ataxia-telangiectasia mutant alleles. Ann Intern Med 133: 770–778, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Sun Y, Weber KT. Angiotensin converting enzyme and myofibroblasts during tissue repair in the rat heart. J Mol Cell Cardiol 28: 851–858, 1996. [DOI] [PubMed] [Google Scholar]
- 25.Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem J 296: 15–19, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor AM, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol 58: 1009–1015, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turner NA, Porter KE. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair 6: 5, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation 76: 44–51, 1987. [DOI] [PubMed] [Google Scholar]
- 29.Willems IE, Havenith MG, De Mey JG, Daemen MJ. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol 145: 868–875, 1994. [PMC free article] [PubMed] [Google Scholar]
- 30.Wu D, Yang H, Xiang W, Zhou L, Shi M, Julies G, Laplante JM, Ballard BR, Guo Z. Heterozygous mutation of ataxia-telangiectasia mutated gene aggravates hypercholesterolemia in apoE-deficient mice. J Lipid Res 46: 1380–1387, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Zhao W, Lu L, Chen SS, Sun Y. Temporal and spatial characteristics of apoptosis in the infarcted rat heart. Biochem Biophys Res Commun 325: 605–611, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Zhao ZQ, Morris CD, Budde JM, Wang NP, Muraki S, Sun HY, Guyton RA. Inhibition of myocardial apoptosis reduces infarct size and improves regional contractile dysfunction during reperfusion. Cardiovasc Res 59: 132–142, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Zhou S, Sun W, Zhang Z, Zheng Y. The role of Nrf2-mediated pathway in cardiac remodeling and heart failure. Oxid Med Cell Longev 2014: 260429, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]