

Summary
Our findings demonstrated that DNA lesions that induce major helix distortion only form in euchromatin, and SIRT1 plays a critical role in restricting the formation of this kind of DNA lesions in cells.
Abstract
Discretely orchestrated chromatin condensation is important for chromosome protection from DNA damage. However, it is still unclear how different chromatin states affect the formation and repair of nucleotide excision repair (NER) substrates, e.g. ultraviolet (UV)-induced cyclobutane pyrimidine dimers (CPD) and the pyrimidine (6-4) pyrimidone photoproducts (6-4PP), as well as cisplatin-induced intrastrand crosslinks (Pt-GG). Here, by using immunofluorescence and chromatin immunoprecipitation assays, we have demonstrated that CPD, which cause minor distortion of DNA double helix, can be detected in both euchromatic and heterochromatic regions, while 6-4PP and Pt-GG, which cause major distortion of DNA helix, can exclusively be detected in euchromatin, indicating that the condensed chromatin environment specifically interferes with the formation of these DNA lesions. Mechanistic investigation revealed that the class III histone deacetylase SIRT1 is responsible for restricting the formation of 6-4PP and Pt-GG in cells, probably by facilitating the maintenance of highly condensed heterochromatin. In addition, we also showed that the repair of CPD in heterochromatin is slower than that in euchromatin, and DNA damage binding protein 2 (DDB2) can promote the removal of CPD from heterochromatic region. In summary, our data provide evidence for differential formation and repair of DNA lesions that are substrates of NER. Both the sensitivity of DNA to damage and the kinetics of repair can be affected by the underlying level of chromatin compaction.
Introduction
DNA damage occurs in the context of native chromatin state. The basic unit of chromatin is the nucleosome consisting of 147bp of DNA wrapped around a histone octamer composed of two H2A–H2B dimers and a H3–H4 tetramer. Linear arrays of nucleosomes are then folded into more compact structures and stabilized by linker histones such as histone H1. Structurally, chromatin in mammalian cells is organized as the loosely euchromatin and highly condensed heterochromatin. Euchromatin, which is characterized by hyperacetylation of the histone tails, represents transcriptionally active, relatively decondensed chromatin, whereas heterochromatin, which is characterized by hypoacetylation of the histone tails and the presence of trimethylation on lysine 9 and 27 of histone H3 (H3K9me3, H3K27me3), represents chromatin that is predominantly transcriptionally inactive and highly compacted. Protein factors, like heterochromatin protein 1 (HP1), bind to H3K9me3 and help in higher order chromatin packing (1,2). Depending on the cell type, ~10–25% of mammalian DNA is transcriptionally inactive or silenced and is highly compacted by heterochromatinization (3–5).
Histone posttranslational modifications play a pivotal role in the assembly of heterochromatin. Histone deacetylase (HDAC) mediated histone hypoacetylation correlates with heterochromatin assembly. HDACs are divided into four classes depending on sequence homology to the yeast original enzymes and domain organization (6), class I (HDAC1, 2, 3, 8), class II (HDAC4, 5, 6, 7, 9, 10), class III (Sirtuins in mammals, including SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6 and SIRT7) and class IV (HDAC11). Among them, SIRT1 is believed to be involved in the formation of repressive chromatin through the coordination of several events, including deacetylating histone H4 at lysine 16, recruiting histone H1 and modulating the activity of SUV39H1, a histone methyltransferase responsible for the maintenance of H3K9me3 (7–9).
It is believed that both the sensitivity of DNA to damage and the kinetics of repair can be affected by the underlying level of chromatin compaction, which presents a demanding obstacle to the function of various DNA templated processes including DNA repair (10,11). For example, ionizing radiation (IR)-induced double-strand breaks (DSBs) within heterochromatin are repaired more slowly compared with that in euchromatin, and ATM signaling is specifically required for DSB repair within heterochromatin (5). However, the formation and repair of nucleotide excision repair (NER) substrates, e.g. ultraviolet (UV) light-induced DNA crosslinks, in different entities of chromatin is still unclear.
UV light irradiation produces several types of mutagenic DNA photoproducts. The two most frequent types of DNA lesions induced by UV irradiation are the cis-syn cyclobutane pyrimidine dimers (CPD) and the pyrimidine (6-4) pyrimidone photoproducts (6-4PP). Another common substrate of NER is cisplatin-induced DNA intrastrand crosslinks. This chemotherapeutic agent forms primarily 1, 2-intrastrand crosslinks between adjacent purines in DNA, e.g. cis-Pt(NH3)2d(GpG) (Pt-GG), with Pt bound to two adjacent guanines, and cis-Pt(NH3)2d(ApG) (Pt-AG), in which the Pt is bound to adenine and an adjacent guanine. It is still unclear whether these NER substrates are formed differentially in different entities of chromatin, and whether their repair can be affected by different chromatin structure.
In this study, we have provided evidence showing that CPD are in fact detectable in both euchromatic and heterochromatic regions, while 6-4PP and Pt-GG are exclusively seen only in euchromatin. Disrupting heterochromatin by targeting SIRT1 was able to increase the formation of both 6-4PP and Pt-GG, but not CPD, indicating that the condensed chromatin state specifically interferes with the formation of those lesions that are processed through NER. Furthermore, we also demonstrated that repair of CPD is impeded in heterochromatin, and DNA damage binding protein 2 (DDB2) could facilitate the removal of CPD in this condensed chromatin region.
Materials and methods
Cell culture and treatment
Normal human skin fibroblasts OSU-2 cells were established and maintained in culture in our laboratory, DDB2-deficient Li-Fraumeni Syndrome fibroblasts strain, designated 041 cells, were kindly provided by Dr. Michael Tainsky (MD Anderson Cancer Center, Houston, TX), DDB2-expressing 041 cell line (041-N22) was established in our laboratory by stably transfecting pcDNA3.1-His-DDB2 into 041 cells (12). NIH3T3 fibroblasts were purchased from American Type Culture Collection (ATCC, Manassas, VA). These cells were grown in DMEM supplemented with 10% fetal calf serum and antibiotics. Ovarian cancer cell line 2008C13 and SKOV3 were kindly provided by Dr Francois Claret (MD Anderson Cancer Center) and Dr Thomas Hamilton (Fox Chase Cancer Center, Philadelphia, PA), respectively, and cultured using RPMI 1640 medium supplemented with 10% fetal calf serum and antibiotics. Their corresponding cancer stem cells (CSCs) were isolated and cultured as described previously (13). All cell lines were authenticated by DNA (STR) profiling, and were grown at 37°C in humidified atmosphere of 5% CO2 in air.
For UV irradiation, cells were washed with PBS, irradiated with UV with a germicidal lamp at a dose rate of 0.8 J/m2/s as measured by a Kettering model 65 radiometer (Cole Palmer Instrument Co., Vernon Hill, IL) and then harvested immediately or incubated in suitable medium for the desired time periods. For cisplatin treatment, cells were maintained in medium with the desired dose of cisplatin (Sigma, St. Louis, MO), which was prepared freshly with PBS, for 1h and then harvested for analysis of DNA lesion formation.
For HDAC inhibitor treatment, cells were treated with sodium butyrate (NaBu, Millipore, Billerica, MA), AR-42 (Ohio State University) or Sirtinol (Cayman, Ann Arbor, MI) for 24h, then harvested or further treated with UV or cisplatin.
RNA interference
Small hairpin RNA (shRNA) targeting human SIRT1 and non-target control shRNA were purchased from Sigma. shRNA transfection was performed with FuGene 6 (Promega, Madison, WI) according to manufacturer’s instruction.
Immunofluorescence
NIH3T3 cells growing on glass coverslips were UV irradiated at 10 J/m2. Cells were fixed and permeabilized with 2% paraformaldehyde and 0.5% Triton X-100. For double staining of histone H3K9me3 and DNA lesions, the coverslips were first incubated with rabbit anti-H3K9me3 antibody (Active Motif, Carlsbad, CA), and stained with goat antirabbit antibody conjugated with Alexa Fluor 488 (Life Technologies, Grand Island, NY). The cells were then denatured with 2M HCl at 37°C for 10min, incubated with mouse anti-CPD (TDM2, 1:1000, Cosmo Bio USA, Carlsbad, CA), mouse anti-6-4PP (1:1000, 64M-2, Cosmo Bio USA) or rat anti-Pt-GG (1: 200, ab103261, Abcam, Cambridge, MA), and stained with goat antimouse antibody or goat-antirat antibody conjugated with Alexa Fluor 594 (Life Technologies). The cells were mounted in an antifade containing medium with 0.25 μg/ml of 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA) as a DNA counter stain. Fluorescence images were obtained with a Nikon Fluorescence Microscope E80i (Nikon, Tokyo, Japan) fitted with appropriate filters for FITC and Texas Red. The digital images were then captured with a 100× oil lens and a cooled CCD camera and processed with the help of its SPOT software (Diagnostic Instruments, Sterling Heights, MI).
Chromatin immunoprecipitation
After desired UV or cisplatin treatments, cells were fixed with 1% formaldehyde in PBS, sonicated to shear the chromatin, and the chromatin immunoprecipitation (ChIP) assay was performed as previously described (14) with rabbit anti-H3K9me3 antibody (Active Motif), rabbit anti-H3K9ac antibody (Cell Signaling, Danvers, MA) or normal rabbit IgG (Santa Cruz, Dallas, TX), in combination with ChIP-Grade Protein G Magnetic Beads (Cell Signaling). The beads-bound proteins/DNA were recovered by incubating in elution buffer (1% SDS and 0.1M NaHCO3) at room temperature followed by incubating in 0.2M NaCl for 5h at 65°C to reverse formaldehyde cross-linking. DNA was finally isolated from immunoprecipitates with phenol/chloroform and subjected to immuno-slot blotting (ISB) for analyses of the amount of DNA lesions.
Immuno-slot blotting analysis
The ISB analysis was used to quantitate DNA lesions as described previously (15). In short, DNA was isolated from UV or cisplatin-treated cells or from immunoprecipitated chromatin. Equal amounts of DNA were loaded onto nitrocellulose membranes, and the amounts of CPD, 6-4PP or Pt-GG were determined using monoclonal anti-CPD (Cosmo Bio USA), anti-6-4PP (Cosmo Bio USA) or anti-Pt-GG antibody (Abcam). Membranes were stripped and re-blotted with mouse anti-single strand DNA (ssDNA) antibody (Millipore) to serve as a loading control. The intensity of each band was quantified with ImageJ software.
Immunoblotting
Whole cell lysates were prepared by direct boiling cells in SDS lysis buffer [2% SDS, 10% glycerol, 62 mmol/l Tris–HCl pH 6.8 and a complete miniprotease inhibitor cocktail (Roche Applied Science)]. After protein quantification with Bio-Rad Dc Protein Assay (Bio-Rad Laboratories, Hercules, CA), equal amounts of total protein were subjected to 8–16% SDS-PAGE and the specific proteins were detected by immunoblotting with desired antibodies, e.g. rabbit anti-H3K9ac (Cell Signaling), rabbit anti-H3K9me3 (Active Motif), rabbit anti-H4K16ac (Active Motif), rabbit anti-H4 (Millipore), mouse anti-Tubulin (Santa Cruz) and rabbit anti-SIRT1 (Millipore).
Statistical analysis
Two-sample t-tests were used for the studies. P values < 0.05 were considered as significant for single tests.
Results
Differential formation of DNA lesions in two structural entities of chromatin
Both UV-induced CPD and 6-4PP form characteristic deformations of the DNA double helix (16,17). However, the DNA distortions caused by these DNA lesions are different. Thus, we reasoned that these DNA lesions may form within distinct locations in chromatin. To test the preferable location of these DNA lesions in chromatin, we used NIH3T3 mouse fibroblasts, which have large pericentromeric heterochromatin domains that are easily visible after DAPI staining. We first marked heterochromatin by using DAPI staining and its characteristic histone modification, H3K9me3 and visualized the existence of UV-induced CPD and 6-4PP in differentially marked regions. As shown in Figure 1A, CPD can be detected throughout the nucleus, especially accumulating in intense DAPI staining regions and H3K9me3-enriched regions. However, these heterochromatic regions are devoid of 6-4PP. This result indicates that the formation of CPD is not affected by the chromatin structure, while 6-4PP only form in euchromatic regions. We further confirmed this finding by analyzing the amounts of CPD and 6-4PP in different chromatin entities by using the modified ChIP assay. Given that heterochromatin and euchromatin are characterized by histone H3K9me3 and H3K9ac, respectively, we enriched H3K9me3-associated and H3K9ac-associated chromatin from human fibroblast OSU-2 cells after UV irradiation, and determined the amounts of CPD and 6-4PP via DNA damage specific antibody binding. As shown in Figure 1B and E, CPD can be detected in both entities, with a dominant amount in H3K9me3-enriched heterochromatin due to high density of DNA, while 6-4PP are exclusively detected in H3K9ac-marked euchromatin, further confirming the differential formation of CPD and 6-4PP in chromatin.
Figure 1.
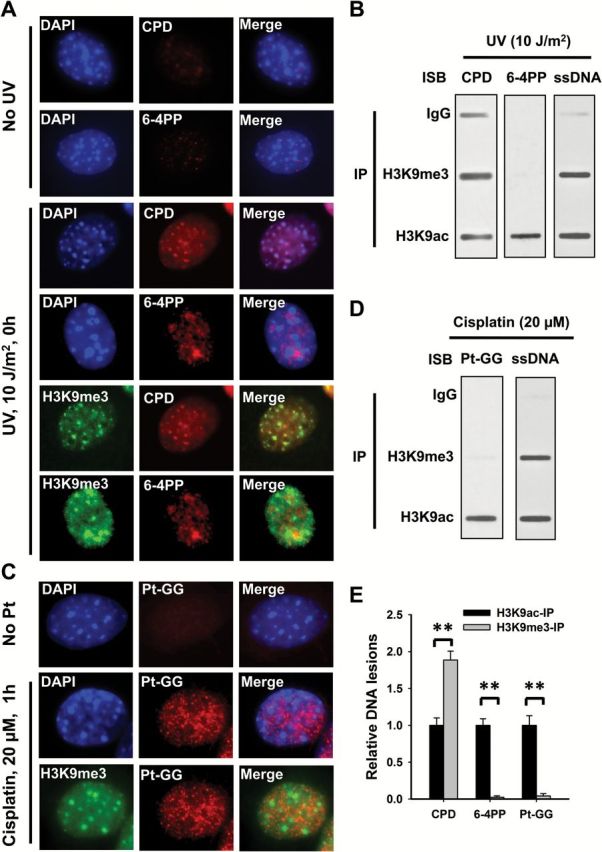
The differential formation of UV and cisplatin induced DNA lesions in chromatin. (A, C) NIH3T3 cells were UV irradiated at 10 J/m2 (A) or treated with cisplatin at 20 μM for 1h (C), or mock-treated, fixed and analyzed with mouse anti-CPD, mouse anti-6-4PP or rat anti-Pt-GG, along with rabbit anti-H3K9me3 antibodies. Detection was with cognate secondary antibodies conjugated with either AlexaFluor 594 or AlexaFluor 488. The slides were counterstained with DAPI. (B, D) Human skin fibroblast OSU-2 cells were UV irradiated at 10 J/m2 (B) or treated with 20 μM cisplatin for 1h (D), the cross-linked and sonicated DNA was subjected to the ChIP assay with either normal IgG, anti-H3K9me3 or anti-H3K9ac antibodies. The recovered DNA was subjected to the immuno-slot blot (ISB) analysis to quantitate CPD, 6-4PP or Pt-GG. ssDNA detection was used as the loading control. (E) The bands in B and D were scanned for intensity, and relative amounts of DNA lesions were calculated by first normalizing to ssDNA, then to the corresponding H3K9ac-IP samples. N = 3; Bar: SD. **P < 0.01.
Cisplatin-induced 1, 2-intrastrand crosslinks also unwind the DNA duplex in the vicinity of the site of platination, induce DNA structure distortion (18,19), and are removed through the NER pathway. We treated NIH3T3 and OSU-2 cells with cisplatin for 1h, and determined the location of Pt-GG, the primary DNA lesion induced by cisplatin, in chromatin by using immunofluorescence and the ChIP assay, respectively. Similar to UV-induced 6-4PP, Pt-GG are also absent in intense DAPI staining and H3K9me3-marked heterochromatin (Figure 1C–E). Given that 6-4PP and Pt-GG cause major helix distortion (17–19), these results indicate that the condensed chromatin environment specifically interferes with the formation of these lesions that are processed through NER.
To further confirm that heterochromatin blocks the formation of DNA lesions leading to major helix distortion, we analyzed the amount of UV-induced CPD and 6-4PP in CSCs and their corresponding bulk cancer cells. Stem cells have been classically described as having a typical open chromatin conformation that is mostly devoid of heterochromatin (20). We isolated CSCs from ovarian cancer cell line 2008C13 and SKOV3 as described previously (13), and determined the histone modification markers as well as amounts of CPD and 6-4PP in these cells upon UV irradiation. As shown in Figure 2A and D, CSCs exhibit elevated levels of euchromatin markers, e.g. H3K9ac, and H4K16ac, while display a lowered level of heterochromatin marker, e.g. H3K9me3, indicating that these CSCs are characterized with a reduced heterochromatin structure. As expected, UV-induced CPD formed equally in both CSCs and their counterpart bulk cancer cells, while significantly increased amount of 6-4PP was detected in CSCs compared to their corresponding bulk cancer cells (Figure 2B, C, E and F). These results further indicate that the formation of 6-4PP, but not CPD, is affected by the chromatin structure, and 6-4PP is favorably formed in relaxed euchromatin.
Figure 2.

Cancer stem cells characterized with relaxed chromatin structure exhibited increased formation of UV-induced 6-4PP, but not CPD. CSCs were isolated from ovarian cancer cell lines 2008C13 (A–C) and SKOV3 (D–F). Immunoblotting was conducted to determine histone modifications in these CSCs and their corresponding bulk cancer cells (A, D). Cells were UV irradiated at 10 J/m2, genomic DNA was isolated and subjected to ISB to detect the formation of UV-induced 6-4PP and CPD. ssDNA was detected as a loading control (B, E). The intensity of each band was quantitated, and relative amounts of varying DNA lesions were calculated by normalizing to ssDNA first, and then to their corresponding bulk cancer cells (C, F). N = 3; Bar: SD. **P < 0.01 compared with bulk cancer cells.
SIRT1 is responsible for blocking the formation of 6-4PP and Pt-GG in cells
The heterochromatin is generally characterized by hypoacetylation of the histone tails, which is attributed to the function of HDACs. Thus, treatment with HDAC inhibitors can be expected to reduce the amount of the heterochromatin in cells and increase the formation of UV-induced 6-4PP and cisplatin-induced Pt-GG. To test this hypothesis, we first treated OSU-2 cells with sodium butyrate (NaBu), an inhibitor of most HDACs except class III HDAC and class II HDAC6 and -10, and then UV irradiated or treated with cisplatin to determine the formation of CPD, 6-4PP and Pt-GG. As expected, although NaBu had no effect on H3K9me3, it did induce dramatic increase in acetylation of histone H3K9 and H4K16 (Figure 3A). However, the formation of 6-4PP and Pt-GG did not exhibit a corresponding increase (Figure 3B–D). Similar results were obtained by using another HDAC inhibitor AR-42, a class I and II HDAC inhibitor (21) (Figure 3E–H).
Figure 3.
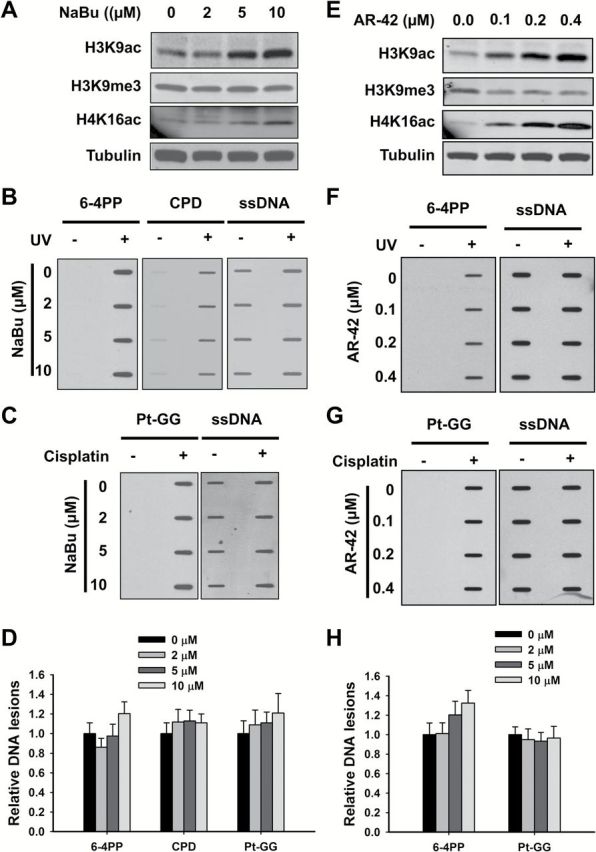
Classes I and II HDAC inhibitors do not affect the formation of 6-4PP and Pt-GG. (A–D) Effect of NaBu on the formation of DNA lesions. OSU-2 cells were treated with NaBu for 48h, whole cell lysates were prepared and subjected to immunoblotting to determine histone modifications (A). OSU-2 cells pre-treated with NaBu were further UV irradiated at 10 J/m2 (B), or treated with cisplatin at 20 μM for 1h (C). Genomic DNA was isolated and subjected to ISB to detect the formation of UV-induced 6-4PP and CPD (B), as well as cisplatin-induced Pt-GG (C) using their corresponding antibodies. ssDNA was detected as a loading control. Representative ISB images from three independent experiments were shown (B, C). The intensity of each band was quantitated using ImageJ. Relative amounts of varying DNA lesions were calculated by normalizing to ssDNA first, and then to non-NaBu-treated samples (D). N = 3; Bar: SD. (E–H) Effect of AR-42 on the formation of 6-4PP and Pt-GG. OSU-2 cells were treated with AR-42 for 48h, whole cell lysates were prepared and subjected to immunoblotting to determine histone modifications (E). OSU-2 cells pre-treated with AR-42 were further UV irradiated at 10 J/m2 (F), or treated with cisplatin at 20 μM for 1h (G). Genomic DNA was isolated and subjected to ISB to detect the formation of UV-induced 6-4PP (F), as well as cisplatin-induced Pt-GG (G) using their corresponding antibodies. ssDNA was detected as a loading control. Representative ISB images from three independent experiments were shown (F, G). The relative amounts of varying DNA lesions were calculated as described above (H). N = 3; Bar: SD.
Given that both NaBu and AR-42 are unable to inhibit HDAC class III activity, it appears that HDAC class III might be the critical HDAC responsible for the maintenance of heterochromatin structure and restriction of the formation of 6-4PP and Pt-GG. Therefore, we pre-treated OSU-2 cells with HDAC class III inhibitor Sirtinol, and analyzed the formation of various DNA lesions. As shown in Figure 4A, Sirtinol dramatically increased histone H4K16ac, but had no influence on H3K9ac and H3K9me3, in OSU-2 cells. The formation of UV-induced 6-4PP (Figure 4B and D) and cisplatin-induced Pt-GG (Figure 4C and D) was enhanced by Sirtinol in a dose–response manner, but the formation of CPD was not affected (Figure 4B and D). Sirtinol can inhibit the activity of both SIRT1 and SIRT2. SIRT1 plays a critical role in promoting the formation and maintenance of heterochromatin (8,22,23). To further determine the requirement of SIRT1 in confining the formation of 6-4PP and Pt-GG, we knocked down the expression of SIRT1 in OSU-2 cells and analyzed the amount of various DNA lesions upon UV or cisplatin treatments. We first confirmed the downregulation of SIRT1 and the increase of H4K16ac in cells upon SIRT1 shRNA transfection (Figure 4E). Similar to Sirtinol treatment, knockdown of SIRT1 enhanced the formation of 6-4PP and Pt-GG, but not of CPD (Figure 4F–H). Taken together, these data indicate that HDAC class III, particularly SIRT1, is central to restricting the formation of UV-induced 6-4PP and cisplatin-induced Pt-GG, possibly by promoting the assembly and maintenance of heterochromatin in cells.
Figure 4.
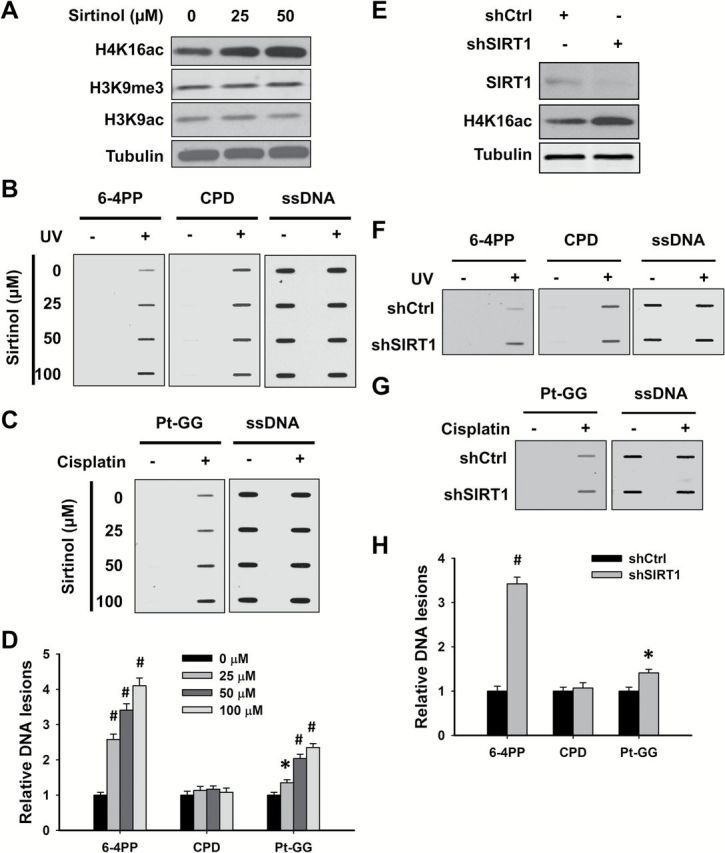
SIRT1 is responsible for restricting the formation of 6-4PP and Pt-GG. (A–D) Sirtuin inhibitor Sirtinol promotes the formation of 6-4PP and Pt-GG, but not CPD in human cells. OSU-2 cells were treated with Sirtinol for 48h, whole cell lysates were prepared and subjected to immunoblotting to determine histone modifications (A). OSU-2 cells pre-treated with Sirtinol were further UV irradiated at 10 J/m2 (B), or treated with cisplatin at 20 μM for 1h (C). Genomic DNA was isolated and subjected to ISB to detect the formation of UV-induced 6-4PP and CPD (B), as well as cisplatin-induced Pt-GG (C) using their corresponding antibodies. ssDNA was detected as a loading control. Representative ISB images from three independent experiments were shown (B, C). The relative amounts of varying DNA lesions were calculated as described in Figure 3 (D). N = 3; Bar: SD; Compared with non-Sirtinol-treated sample, *P < 0.05; # P < 0.01. (E–H) Knockdown of SIRT1 promotes the formation of 6-4PP and Pt-GG, but not CPD in human cells. OSU-2 cells were transfected with shRNA specific for SIRT1 (shSIRT1) or control shRNA (shCtrl) for 48h, whole cell lysates were prepared and subjected to immunoblotting to detect histone modifications (E). Cells were further treated with UV radiation at 10 J/m2 (F), or cisplatin at 20 μM for 1h (G). The formation of DNA lesions were determined as described in Fig. 3. Representative ISB images from three independent experiments were shown (F, G). Relative amounts of varying DNA lesions were calculated as described in Figure 3H. N = 3; Bar: SD; Compared with shSIRT1, *P < 0.05; # P < 0.01.
Heterochromatin impedes the repair of CPD
Chromatin creates a natural barrier against access to DNA during damage repair. Therefore, it is logical to surmise that DNA lesions formed in the highly condensed heterochromatin are repaired with a slow dynamics. Amongst three NER substrates analyzed in this study, only CPD is able to form in both euchromatin and heterochromatin. Thus, we enriched the euchromatin and heterochromatin via their corresponding marks H3K9ac and H3K9me3, respectively, in our ChIP assay, and analyzed the removal rate of UV-induced CPD. As shown in Figure 5A and B, CPD within euchromatin were repaired faster than those formed within heterochromatin. Given the different chromatin condensation status in euchromatin and heterochromatin, we reason that these two chromatin structures utilize distinct remodeling complexes and pathways to process and repair UV-induced CPD.
Figure 5.

CPD is repaired slower in heterochromatin than that in euchromatin. OSU-2 cells were UV irradiated at 10 J/m2 and cultured for 0, 8 and 24h. The cells were fixed and subjected to the ChIP assay with either anti-H3K9ac or anti-H3K9me3 antibody. The recovered DNA was subjected to the ISB analysis to quantitate CPD. ssDNA detection was used as the loading control (A). The intensity of each band was quantitated, and relative amounts of CPD were calculated by normalizing to ssDNA first, and then to 0h (B). N = 3; Bar: SD. **P < 0.01 compared to H3K9ac-IP samples at the same time point.
DDB2 facilitates the removal of CPD in heterochromatin, but not in euchromatin
DDB2 is a 48-kDa protein originally identified as a component of the damage-specific DNA-binding heterodimeric complex DDB (24). DDB2 is able to bind UV-damaged DNA and serves as the initial damage recognition factor during repair (25). We have previously shown that DDB2 redistributed to nuclease-resistant heterochromatin compartment upon UV irradiation (26), suggesting that DDB2 may be involved in the repair of DNA lesions located in heterochromatin. To test this hypothesis, we irradiated DDB2-deficient human Li–Fraumeni Syndrome fibroblasts 041 and DDB2-restored 041-N22 cells with UV (12), purified H3K9me3-enriched and H3K9ac-enriched chromatin from these cells immediately after UV irradiation or after further culturing for 8 and 24h, and determined the amount of CPD in these chromatin entities. As shown in Figure 6A–C, equivalent removal rates of CPD in H3K9ac-enriched chromatin were found in DDB2-deficient and -proficient cells, indicating that DDB2 is not required for CPD repair in the euchromatic region. In contrast, CPD in H3K9me3-enriched chromatin region was repaired faster in DDB2-proficient cells than in DDB2-deficient cells (Figure 6A, B and D), indicating that DDB2 facilitates the repair of CPD located in heterochromatin.
Figure 6.
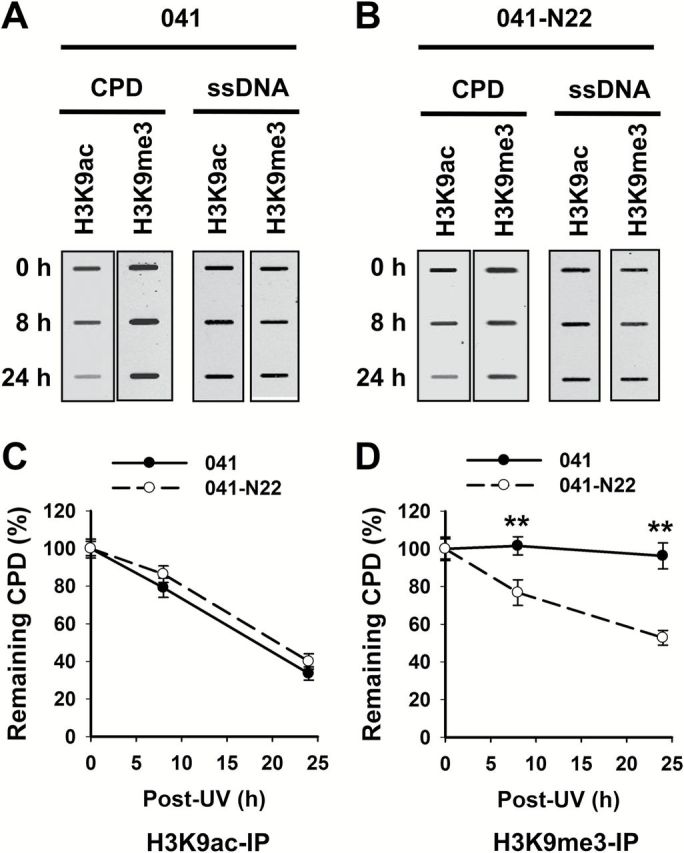
DDB2 facilitates the removal of heterochromatic CPD. DDB2-deficient (041) and -proficient (041-N22) cells were UV irradiated at 10 J/m2 and further cultured for 0, 8 and 24h. The cells were fixed and subjected to the ChIP assay with either anti-H3K9ac or anti-H3K9me3 antibody. The recovered DNA was subjected to the ISB analysis to quantitate CPD. ssDNA detection was used as the loading control (A). The intensity of each band was quantitated, and relative amounts of CPD were calculated by normalizing to ssDNA first, and then to corresponding samples at 0h (B, C). N = 3; Bar: SD. **P < 0.01 compared to 041 cells at the same time point.
Discussion
Because unwinding and bending of DNA are important determinants of UV damage induction (27), the chromatin structure is supposed to affect the formation of these DNA lesions. Here, we show that highly condensed heterochromatin allows the formation of UV-induced CPD, but not UV-induced 6-4PP and cisplatin-induced Pt-GG. CPD are characterized by small deformations of the DNA double helix, the DNA is unwound and kinked by approximately 7–9° relative to B-form DNA (16,17), while 6-4PP produce even more significant structural distortions in the DNA double helix than CPD because the pyrimidine planes within the 6-4PP are almost perpendicular, and a bending angle of 44° was observed (17). Similar to UV damage, cisplatin-induced 1, 2-intrastrand crosslinks also unwind the DNA duplex in the vicinity of the site of platination, bending it 50–78° toward the major groove (18,19). Therefore, highly condensed heterochromatin impedes the unwinding and bending of DNA, restricting the formation of DNA lesions that may cause major helix distortion in this chromatin region. This is also supported by the findings that 6-4PP and Pt-GG preferentially form in internucleosomal (linker) region (28–31), and transcription factor binding can significantly increase the formation of 6-4PP in the promoter regions, probably due to the structural alterations induced by factor binding (27).
Heterochromatin assembly and maintenance require histone hypoacetylation, which can be achieved by HDAC. Thus, HDAC inhibitors are supposed to increase histone acetylation and relax chromatin structure, leading to an enhancement of 6-4PP and Pt-GG formation. However, we found that only the HDAC class III inhibitor, but not classes I and class II inhibitors, was able to increase the formation of 6-4PP and Pt-GG in mammalian cells. Although the most prominent histone feature in heterochromatin is global hypoacetylation, acetylation of the H4 tail seems to be most important in histone deposition to newly replicated DNA and in chromatin structure (32). H4K16 acetylation neutralizes the positive charge on the H4 tail, which then poses a structural constraint on the formation of higher order chromatin and forces the chromatin to stay in a more open configuration, thus determining the ability of chromatin to fold into a higher order structure. Although all these HDAC inhibitors are able to increase H4K16ac, inhibiting zinc-containing HDACs (classes I and II) only stabilized the acetylation state of histone H4 in euchromatin regions, whereas has no effect on heterochromatin regions (33). Given that the formation of 6-4PP and Pt-GG is excluded from heterochromatin, it is reasonable that only the Class III HDAC inhibitor is able to increase the formation of 6-4PP and Pt-GG in cells upon UV or cisplatin treatments.
Chromatin presents a formidable natural barrier against access to DNA during processing of diverse DNA damage. It is logical to surmise that DNA lesions forming in the highly condensed heterochromatin are repaired at a slower kinetics. It has already been reported that IR-induced DSB within heterochromatin are repaired more slowly compared with that in euchromatin, and ATM signaling as well as p53 are specifically required for DSB repair within heterochromatin (5,34). In this study, we have shown that CPD within euchromatin were repaired at a faster rate than those forming within heterochromatin. Given the different chromatin condensation states in euchromatin and heterochromatin, we reason that these two chromatin compartments utilize distinct remodeling complexes and pathways to repair UV-induced photolesions and other bulky genomic adducts.
In this study, we also revealed that DDB2 is able to facilitate the repair of CPD in H3K9me3-enriched heterochromatin. DDB2 has specific affinity for UV-damaged DNA and serves as the initial damage recognition factor to assemble NER machinery (25). Interestingly, DDB2 is not at all needed for CPD excision from naked DNA, thus pointing to an unidentified function related to modulation of chromatin structure. Although DDB2 binds to both 6-4PP and CPD in vitro (35–38), it is only required for the efficient repair of CPD in vivo (39–41). Given our finding that 6-4PP are only seen in euchromatin, it seems plausible that DDB2 is not required for the repair of DNA lesions in this loosely packed chromatin. In contrast, DDB2 is required for the repair of CPD, which form in both euchromatin and heterochromatin. In addition, DDB2 can detect lesions with small structural and thermodynamic perturbation and embedded by nucleosomes (42). Therefore, we believe that DDB2 is a critical factor in repairing DNA lesions within highly condensed heterochromatic regions. This conclusion is also supported by a report showing that DDB2 is able to unfold heterochromatin through recruiting ATP-dependent chromatin remodeling factors to decondense local heterochromatin (43).
In conclusion, our study has shown that the DNA lesions are differentially induced in euchromatic and heterochromatic regions, and the repair of DNA lesions residing in heterochromatic regions require additional complementary processing machinery to ensure genomic integrity. Impaired DNA repair within heterochromatin will de-repress the normally silenced tandem repetitive satellite DNA, which contributes to the evolution of the cancer cell through the induction of genomic instability by increased abnormal mitotic figures (44). Thus, it will be interesting to investigate mechanisms through which chromatin structure affects the formation of DNA lesions, and how DNA lesions in heterochromatin are completely repaired. Understanding the interplay between DNA damage formation/repair and chromatin structure would provide novel insights into DNA damage-induced cancer initiation, and DNA damaging agent-mediated cancer therapy.
Funding
National Institute of Health (CA151248 to Q.E.W., ES002388 and ES012991 to A.A.W.).
Acknowledgements
We are very grateful for Drs Michael Tainsky, Francois Claret and Thomas Hamilton for kindly providing various cell lines. We also thank members of Wani laboratory for their valuable input.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- ChIP
chromatin immunoprecipitation
- CPD
cyclobutane pyrimidine dimers
- CSC
cancer stem cell
- DAPI
4′-6′-diamidino-2-phenylindole
- DDB2
DNA damage binding protein 2
- DSB
double-strand breaks
- HDAC
histone deacetylase
- H3K9me3
trimethylation on lysine 9 of histone H3
- H3K9ac
acetylation on lysine 9 of histone H3
- H4K16ac
acetylation on lysine 16 of histone H4
- ISB
immune-slot blotting
- 6-4PP
pyrimidine (6-4) pyrimidone photoproducts
- NER
nucleotide excision repair
- Pt-GG
cis-Pt(NH3)2d(GpG)
- UV
ultraviolet
References
- 1. Campos E.I., et al. (2009) Histones: annotating chromatin. Annu. Rev. Genet., 43, 559–599. [DOI] [PubMed] [Google Scholar]
- 2. Dinant C., et al. (2009) The emerging role of HP1 in the DNA damage response. Mol. Cell. Biol., 29, 6335–6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yunis J.J., et al. (1971) Heterochromatin, satellite DNA, and cell function. Structural DNA of eucaryotes may support and protect genes and aid in speciation. Science, 174, 1200–1209. [DOI] [PubMed] [Google Scholar]
- 4. Miklos G.L., et al. (1979) Heterochromatin and satellite DNA in man: properties and prospects. Am. J. Hum. Genet., 31, 264–280. [PMC free article] [PubMed] [Google Scholar]
- 5. Goodarzi A.A., et al. (2008) ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell, 31, 167–177. [DOI] [PubMed] [Google Scholar]
- 6. Dokmanovic M., et al. (2007) Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res., 5, 981–989. [DOI] [PubMed] [Google Scholar]
- 7. Vaquero A., et al. (2004) Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell, 16, 93–105. [DOI] [PubMed] [Google Scholar]
- 8. Vaquero A., et al. (2007) SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature, 450, 440–444. [DOI] [PubMed] [Google Scholar]
- 9. Vaquero A., et al. (2006) SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev., 20, 1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Araki M., et al. (2000) Reconstitution of damage DNA excision reaction from SV40 minichromosomes with purified nucleotide excision repair proteins. Mutat. Res., 459, 147–160. [DOI] [PubMed] [Google Scholar]
- 11. Ura K., et al. (2001) ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. EMBO J., 20, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barakat B.M., et al. (2010) Overexpression of DDB2 enhances the sensitivity of human ovarian cancer cells to cisplatin by augmenting cellular apoptosis. Int. J. Cancer, 127, 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han C., et al. (2014) DDB2 suppresses tumorigenicity by limiting the cancer stem cell population in ovarian cancer. Mol. Cancer Res., 12, 784–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao Q., et al. (2009) Modulation of nucleotide excision repair by mammalian SWI/SNF chromatin-remodeling complex. J. Biol. Chem., 284, 30424–30432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han C., et al. (2015) Cdt2-mediated XPG degradation promotes gap-filling DNA synthesis in nucleotide excision repair. Cell Cycle, 14, 1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang C.I., et al. (1991) Site-specific effect of thymine dimer formation on dAn.dTn tract bending and its biological implications. Proc. Natl. Acad. Sci. USA, 88, 9072–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim J.K., et al. (1995) Contrasting structural impacts induced by cis-syn cyclobutane dimer and (6-4) adduct in DNA duplex decamers: implication in mutagenesis and repair activity. Photochem. Photobiol., 62, 44–50. [DOI] [PubMed] [Google Scholar]
- 18. Takahara P.M., et al. (1995) Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature, 377, 649–652. [DOI] [PubMed] [Google Scholar]
- 19. Gelasco A., et al. (1998) NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum(II) d(GpG) intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry, 37, 9230–9239. [DOI] [PubMed] [Google Scholar]
- 20. Gaspar-Maia A., et al. (2011) Open chromatin in pluripotency and reprogramming. Nat. Rev. Mol. Cell Biol., 12, 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lucas D.M., et al. (2010) The novel deacetylase inhibitor AR-42 demonstrates pre-clinical activity in B-cell malignancies in vitro and in vivo . PLoS One, 5, e10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katan-Khaykovich Y., et al. (2005) Heterochromatin formation involves changes in histone modifications over multiple cell generations. EMBO J., 24, 2138–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moazed D. (2001) Common themes in mechanisms of gene silencing. Mol. Cell, 8, 489–498. [DOI] [PubMed] [Google Scholar]
- 24. Dualan R., et al. (1995) Chromosomal localization and cDNA cloning of the genes (DDB1 and DDB2) for the p127 and p48 subunits of a human damage-specific DNA binding protein. Genomics, 29, 62–69. [DOI] [PubMed] [Google Scholar]
- 25. Tang J., et al. (2002) Xeroderma pigmentosum complementation group E and UmV-damaged DNA-binding protein. DNA Repair (Amst), 1, 601–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Q.E., et al. (2013) p38 MAPK- and Akt-mediated p300 phosphorylation regulates its degradation to facilitate nucleotide excision repair. Nucleic Acids Res., 41, 1722–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pfeifer G.P. (1997) Formation and processing of UV photoproducts: effects of DNA sequence and chromatin environment. Photochem. Photobiol., 65, 270–283. [DOI] [PubMed] [Google Scholar]
- 28. Mitchell D.L., et al. (1990) Nonrandom induction of pyrimidine-pyrimidone (6-4) photoproducts in ultraviolet-irradiated human chromatin. J. Biol. Chem., 265, 5353–5356. [PubMed] [Google Scholar]
- 29. Gale J.M., et al. (1990) UV induced (6-4) photoproducts are distributed differently than cyclobutane dimers in nucleosomes. Photochem. Photobiol., 51, 411–417. [DOI] [PubMed] [Google Scholar]
- 30. Bubley G.J., et al. (1994) Differences in in vivo and in vitro sequence-specific sites of cisplatin-DNA adduct formation and detection of a dose-response relationship. Biochem. Pharmacol., 48, 145–153. [DOI] [PubMed] [Google Scholar]
- 31. Foka M., et al. (1986) Interaction of cis-diamminedichloroplatinum (II) to chromatin. Specificity of the drug distribution. Biochem. Pharmacol., 35, 3283–3291. [DOI] [PubMed] [Google Scholar]
- 32. Vaquero A., et al. (2007) NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene, 26, 5505–5520. [DOI] [PubMed] [Google Scholar]
- 33. Taddei A., et al. (2001) Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat. Cell Biol., 3, 114–120. [DOI] [PubMed] [Google Scholar]
- 34. Zheng H., et al. (2014) p53 promotes repair of heterochromatin DNA by regulating JMJD2b and SUV39H1 expression. Oncogene, 33, 734–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fujiwara Y., et al. (1999) Characterization of DNA recognition by the human UV-damaged DNA-binding protein. J. Biol. Chem., 274, 20027–20033. [DOI] [PubMed] [Google Scholar]
- 36. Payne A., et al. (1994) Xeroderma pigmentosum group E binding factor recognizes a broad spectrum of DNA damage. Mutat. Res. Fundam. Mol. Mech. Mutagen., 310, 89–102. [DOI] [PubMed] [Google Scholar]
- 37. Reardon J.T., et al. (1993) Comparative analysis of binding of human damaged DNA-binding protein (XPE) and Escherichia coli damage recognition protein (UvrA) to the major ultraviolet photoproducts: T[c,s]T, T[t,s]T, T[6-4]T, and T[Dewar]T. J. Biol. Chem., 268, 21301–21308. [PubMed] [Google Scholar]
- 38. Wittschieben B.O., et al. (2005) DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. J. Biol. Chem., 280, 39982–39989. [DOI] [PubMed] [Google Scholar]
- 39. Hwang B.J., et al. (1998) p48 Activates a UV-damaged-DNA binding factor and is defective in xeroderma pigmentosum group E cells that lack binding activity. Mol. Cell. Biol., 18, 4391–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moser J., et al. (2005) The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA Repair (Amst)., 4, 571–582. [DOI] [PubMed] [Google Scholar]
- 41. Tang J.Y., et al. (2000) Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol. Cell, 5, 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scrima A., et al. (2008) Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell, 135, 1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luijsterburg M.S., et al. (2012) DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol., 197, 267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhu Q., et al. (2011) BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature, 477, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]