

Abstract
Recurrent hydatidiform mole (RHM) is defined by the occurrence of repeated molar pregnancies in affected women. Two genes, NLRP7 and KHDC3L, play a causal role in RHM and are responsible for 48–80% and 5% of cases, respectively. Here, we report the results of screening these two genes for mutations in one Iranian and one Indian patient with RHM. No mutations in NLRP7 were identified in the two patients. KHDC3L sequencing identified two novel protein-truncating mutations in a homozygous state, a 4-bp deletion, c.17_20delGGTT (p.Arg6Leufs*7), in the Iranian patient and a splice mutation, c.349+1G>A, that affects the invariant donor site at the junction of exon 2 and intron 2 in the Indian patient. To date, only four mutations in KHDC3L have been reported. The identification of two additional mutations provides further evidence for the important role of KHDC3L in the pathophysiology of RHM and increases the diversity of mutations described in Asian populations.
Introduction
A hydatidiform mole (HM, MIM no. 231090) is an aberrant human pregnancy in which there is nonexistent or abnormal embryonic development, excessive trophoblastic proliferation, and cystic degeneration of chorionic villi into grape-like structures. The common form of HM is sporadic, not recurrent, and affects 1 in 600 pregnancies in Western countries,1 but this incidence displays a gradient of increasing frequency from the West to the East, with the highest frequency in Southeastern Asia, where this condition affects 1 in 100 pregnancies.2,3 Common sporadic HM can be categorized into two main subtypes, complete HM (CHM) and partial HM (PHM), based on histopathological features and parental contribution to the molar tissues.4 Approximately 75% of clinically ascertained molar pregnancies are CHMs, mostly of diploid androgenetic origin, and the remaining are PHMs, mostly of triploid dispermic origin. Histologically, CHMs show excessive trophoblastic proliferation and no evidence of extraembryonic membranes, whereas PHMs exhibit mild trophoblastic proliferation and may contain extraembryonic membranes and embryonic tissues.4,5
Women who have experienced one molar pregnancy are at an increased risk of further HMs in subsequent pregnancies in comparison with other women in the general population. One to 6% of women with a prior mole experience a second mole, and ~10–20% will have a second nonmolar reproductive loss, most likely a spontaneous abortion.6–9 RHM may be non-familial and occurs in patients with no family history, or it may be familial, occurring in sisters or related women from the same family. The frequency of RHM varies among populations and countries. Again, RHM occurs at a higher frequency in patients originating from the Middle9 and Far East.7
To date, two maternal-effect genes have been found to be responsible for RHM, NLRP7 and KHDC3L,10,11 but their precise roles in the pathophysiology of molar pregnancies is not fully understood.
NLRP7 is a major gene for RHM and belongs to the NLRP family, which consists of 14 members with roles in apoptosis and inflammation. To date, 59 NLRP7 mutations in a recessive state have been reported.12 KHDC3L was identified in 2011 and is a minor gene for RHM. To date, four KHDC3L mutations in a recessive state have been reported.11,13 Although little is known about the function of KHDC3L, KHDC3L was demonstrated to colocalize with NLRP7 in lymphoblastic cell lines,13 human oocytes, and early preimplantation embryos.14 In addition, both genes, NLRP7 and KHDC3L, are believed to be involved, directly or indirectly, in setting or maintaining maternal imprints in the oocyte or in early embryonic development.15–20
To date, mutations in KHDC3L have been reported in two familial and four singleton cases of RHM who were found to be negative for NLRP7 mutations by conventional Sanger sequencing. Here, we report the identification of two additional novel protein-truncating mutations in KHDC3L in two patients.
Materials and methods
Patients and controls
Patient 1448 is from a large Iranian family and has no history of molar pregnancies in her relatives or in previous generations (Supplementary Figure S1). She was 23 years old at the time of her first pregnancy and had had a total of 11 HMs over a 13-year period. There is consanguinity between the parents of the patient and between the patient and her partner. All of her pregnancies were evacuated by suction curettage in the first trimester, and she was followed up through serial measurements of β-human chorionic gonadotropin for 1 year after each pregnancy.
Patient 1335 is a 35-year-old woman of Indian origin (Supplementary Figure S2). She had a total of six pregnancies in 7 years, all of which were diagnosed as HMs. The parents of the patient are first cousins, and there is also consanguinity between the patient and her partner. The patient and her partner had normal karyotypes. There is no known history of infertility or reproductive losses in the patient’s family.
Neither patient had experienced a normal pregnancy. The diagnosis of the HMs from the two patients was based on histopathological examination according to previously described standards.4,5 Only one tissue sample was available for histopathological review; this sample was from the eleventh HM of patient 1448. The tissue was sectioned, stained, and examined. Based on the presence of circumferential trophoblastic proliferation around most chorionic villi and the absence of embryonic tissues, this tissue was diagnosed as a complete HM (Figure 1).
Figure 1.
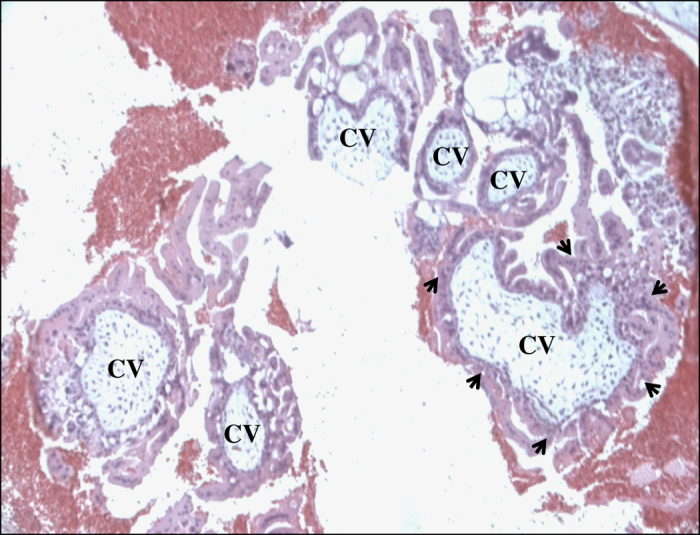
Photomicrograph of a complete hydatidiform mole (CHM) from patient 1448 showing several chorionic villi (CV). Circumferential trophoblastic proliferation around one villous on the right bottom is indicated by arrows.
DNA from 50 subjects from the general Iranian population was used as a control.
Mutation analysis
The study protocol was approved by both the Institutional Review Board and the ethics committee of Shiraz University of Medical Sciences and McGill University. Patients and participating family members gave written consent to participate in the study. Genomic DNA was extracted from peripheral blood samples using the Cinnagen DNA Extraction Kit (Cinnaclon, Iran) according to the manufacturer's protocol.
For both patients, mutation analysis was performed by PCR amplification of the 11 NLRP7 and 3 KHDC3L exons, as well as of the adjacent intronic sequences (GenBank accession no. NG_008056.1 and NG_031942.1, respectively) using specific primers designed by the AllelID v.7.5 software (PREMIER Biosoft, Palo Alto, CA, USA). Primer sequences are provided in Supplementary Table SI online. The PCR amplifications were performed in a 50-μl final reaction volume containing 1 μl of each primer (20 pmol/μl), 1 μl DNA template (50–200 ng), 25 μl Taq DNA Polymerase 2x Master Mix Red (Ampliqon, Odense M, Denmark), and 22 μl dH2O. The mixture was initially denatured at 95 °C for 4 min, and the PCR was carried out for 35 cycles in an ABI 96-well thermocycler (Applied Biosystems Instruments, Foster City, CA, USA) under the following conditions: denaturation at 94 °C for 30 s, annealing at 58–61 °C (depending on the pair of primers) for 30 s, extension at 72 °C for 30 s, and a final extension cycle of 72 °C for 7 min. The PCR products were separated on a 2% agarose gel in 1× TAE (Tris-acetate-EDTA) buffer and detected with 0.5 μg/ml ethidium bromide. The size of each amplicon was determined using a 100-bp ladder. The purified PCR products were directly sequenced using an ABI 3730xl DNA Analyzer (Applied Biosystems Instruments). The sequences were analyzed using DNASTAR (Madison, WI, USA) and compared with the reference sequence NM_001127255.1 for NLRP7 and NM_001017361.2 for KHDC3L.
A simple amplification refractory mutation system PCR (ARMS-PCR) was established to study the mutation found in patient 1448 from family MoIr440. To discriminate the mutant and wild-type alleles by ARMS-PCR, three primers specific for exon 1 were designed; the four deleted nucleotides for the mutation and normal variant were localized at the 3′ end of the primers (Supplementary Table SII). The PCR amplifications were carried out in two separate reactions, and the products obtained from these primers were 260 and 264 bp for the mutant and normal alleles, respectively. In this ARMS-PCR system, a heterozygote mutation would be amplified with both the normal and mutant primers, but a homozygote mutation would be amplified only with the primer specific for the mutation. The PCR reaction protocol and cycling program were the same as those described above. To confirm the ARMS-PCR results, heterozygote and homozygous samples identified by this method were also subjected to direct DNA sequencing. The DNA mutation numbering is based on the cDNA sequence, with a 'c' symbol before the number, and +1 corresponding to the A of the ATG initiation codon in the reference sequence NM_001017361.2.
Results
Case 1
No mutation in NLRP7 was found in patient 1448. PCR amplification and sequencing of KHDC3L revealed a homozygous novel 4-bp deletion, c.17_20delGGTT, in the first exon, resulting in a frameshift and protein truncation, p.Arg6Leufs*7 (Figure 2a). This 4-bp deletion was not found in 100 chromosomes from ethnically matched control individuals who were screened by ARMS-PCR amplification and 2% agarose gel electrophoresis (Figure 2b). In addition, this mutation is not reported in 60,706 subjects in ExAC (http://exac.broadinstitute.org/) or in 2,535 subjects in 1000 Genomes Browser (http://www.1000genomes.org/1000-genomes-browsers). Because the parents of the patient are both deceased, we tested the DNA of her two paternal aunts for the c.17_20delGGTT mutation by ARMS-PCR and found that one is a heterozygous carrier of the mutation because both the normal and mutant PCR products were amplified when her DNA was used as the template (Figure 2a). Another variant in exon 2 was detected in a homozygous state in the patient. This variant, c.602C>G, in which a glycine is substituted for an alanine, p.Ala201Gly, is a common polymorphism reported in public databases. We also found it in a significant number of the control individuals.
Figure 2.

Chromatograms of the novel mutations in patients 1448 and 1335 and amplification refractory mutation system PCR (ARMS-PCR) gel electrophoresis result for c.17_20delGGTT mutation. (a) Sequence chromatograms showing the novel 4-bp deletion in the first exon of KHDC3L gene in patient 1448 (homozygous) and her paternal aunt (heterozygous) along with control subject with the normal sequence. (b) Agarose gel electrophoresis results of ARMS-PCR. Top panel shows the amplification of a 260-bp PCR product with mutant primers only in patient 1448 (lane 1) and not in six normal individuals (lanes 2–7). Lower panel shows the absence of PCR amplification with normal primers in patient 1448 (lane 1), but the amplification of a 264-bp product in normal individuals (lanes 2–7). The 100-bp DNA ladder is depicted on the right. (c) Sequence chromatogram showing the novel mutation affecting the donor splice site of KHDC3L intron 2 in patient 1335.
Case 2
NLRP7 mutation analysis of the DNA of patient 1335 did not reveal any mutations. Sequencing of KHDC3L revealed that the patient has a novel splicing mutation that affects the invariant guanine at the first base of the donor splice site of intron 2, c.349+1G>A; this variant was present in a homozygous state (Figure 2c). Again, this mutation is not reported in 60,706 subjects in ExAC (http://exac.broadinstitute.org/) or in 2,535 subjects in 1000 Genomes Browser (http://www.1000genomes.org/1000-genomes-browsers). To predict the effect of the mutation on splicing, we used three splice prediction programs, GenScan (http://genes.mit.edu/GENSCAN.html), Human Splicing Finder (http://www.umd.be/HSF/), and Net2Gene (http://www.cbs.dtu.dk/services/NetGene2/). These three programs predicted that the c.349+1G>A variant would disrupt the splice donor site of exon 2. GenScan predicted that it would result in the activation of an intronic splice site and lead to the inclusion of the first 117 bases of intron 2 in the mRNA and an in-frame insertion of 39 amino acids between exons 2 and 3. Human Splicing Finder predicted three cryptic splice sites with high confidence. These sites were located very close to each other, and one was identical to the splice site predicted by GenScan.
Discussion
In this report, we describe two mutations in KHDC3L in two patients of Iranian and Indian origin with a history of RHM. The two mutations were found in a homozygous state, one in each patient. In the first case, patient 1448, a c.17_20delGGTT (p.Arg6Leufs*7), which results in a frameshift in exon 1, was identified. In the second case, patient 1335, a novel splicing mutation, c.349+1G>A, was detected. These two mutations have not been previously described in patients and are not present in public databases. Similar to previously reported cases of women with RHM, the pattern of inheritance of the defect in the two families is consistent with an autosomal recessive condition.11,13
To date, four different mutations in KHDC3L, including two missense variants and two 4-bp deletions, have been reported previously.11,13 In this study, we describe a third novel 4-bp deletion, c.17_20delGGTT. The two previously reported 4-bp deletions, c.322_325delGACT and c.299_302delTCAA, are in exon 2, whereas the novel deletion described in this report, c.17_20delGGTT, is in exon 1 (Figure 3).
Figure 3.
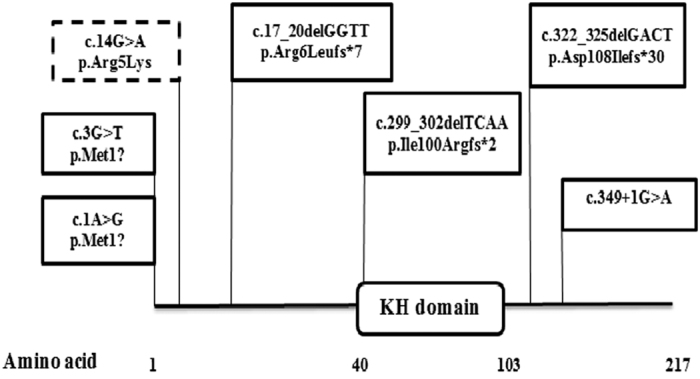
Schematic representation of KHDC3L protein structure and all identified mutations in patients with recurrent hydatidiform moles (RHMs). KH stands for K homology domain. Mutations found in patients with two defective alleles are in rectangles with continuous border. The dashed border rectangle corresponds to a single heterozygous variant found in one patient and not seen in controls. The c.17_20delGGTT and c.349+1G>A mutations were identified in the present study.
The presence of three 4-bp deletions out of six known mutations in KHDC3L, which is a small gene (654 bp), is intriguing and raises questions about the presence of small direct and inverted repeats that could have mediated or promoted the occurrence of these deletions via a slipped-strand mispairing mechanism, as reported previously.21,22 We therefore used GeneQuest, a software program included in the LaserGene package (DNASTAR), and looked for short repeats within KHDC3L cDNA. We found several small direct and inverted repeats. The inverted repeats were distributed throughout the cDNA, with a higher concentration in exons 1 and 2, whereas the direct repeats were mainly concentrated in exon 3 (Figure 4a). Our analysis showed that the c.17_20delGGTT novel deletion in exon 1 is flanked by a direct repeat of seven bases, whereas the c.299_302delTCAA and c.322_325delGACT deletions in exon 2 are flanked by inverted repeats of five bases and share the middle repeat (Figure 4b). These data suggest that these small repeats may have mediated the 4-bp deletions, but this hypothesis remains to be validated in future studies of more small deletions. In addition, using Tandem Repeat Finder (https://tandem.bu.edu/trf/trf.html), we found in exon 3 a consensus sequence of ~34 bp (5′- AGGCCGAGACCCAGCGGTCTTCAATAGAAGTCCGGG-3′) that is repeated four times (three times over its full length and one time over the first 19 bp) (Figure 4a). Tandem repeats have been found to be more frequent in primate-specific genes that are located in segmental duplications; in such cases, these tandem repeats are believed to have driven the recent duplication, fast evolution, and divergence of the genes containing them.22 This belief is in line with our observation of the tandem repeats in KHDC3L and that KHDC3L is located very close to three other related genes (KHDC1/DPPA5/OOEP) on chromosome 6.23
Figure 4.

Inverted, direct, and repeats identified in KHDC3L cDNA. (a) Distribution of inverted, direct, and tandem repeats along KHDC3L cDNA using GeneQuest (DNASTAR, Madison, WI) with the following parameters (a minimum number of 5-bp that are <50-bp apart) and Tandem Repeat Finder (https://tandem.bu.edu/trf/trf.html). An aqua box indicates an inverted repeat; a pink box indicates a direct repeat; and green boxes indicate tandem repeats. (b) Direct and inverted repeats flanking the 4-bp deletions in exons 1 and 2 are indicated by asterisks in (a) and brackets in (b). Inverted repeats flanking c.299_302delTCAA are indicated by brackets below exon 2 and those flanking c.322_325delGACT are indicated by brackets above exon 2.
Among patients with recessive NLRP7 mutations, variable reproductive outcomes have been reported, including live birth, early neonatal death, stillbirth, spontaneous abortion, blighted ovum, partial HM, and complete HM. In addition, two studies have implicated NLRP7 in genetic susceptibility to recurrent spontaneous abortions.24,25 A summary of the reproductive outcomes of women with recessive mutations in KHDC3L is given in Table 1. Among the 10 listed patients, only 2 had spontaneous abortions, and these abortions accounted for only 3 out of 65 conceptions (4.6%). In contrast, 27 out of 154 (18%) of the conceptions of patients with two defective alleles in NLRP7 led to SA.24 This difference is statistically significant (two-sided Fisher's exact test, P-value=0.0097). In addition, 5 out of a total of 131 patients (3.8%) with NLRP7 recessive mutations had six live births out of a total of 612 pregnancies,26 whereas to date, no live births have been reported among the 65 conceptions of patients with two defective KHDC3L alleles. These data suggest that mutations in KHDC3L may be more severe than those in NLRP7 and may not be associated with other forms of reproductive loss or with live births. Again, this observation remains to be validated in a larger cohort of patients with two defective KHDC3L alleles.
Table 1. Recapitulation of the reproductive outcomes and ethnic origin of all described women with recessive KHDC3L mutations.
| Family ID | Patient ID | Origin |
Mutation |
Reproductive outcome | References | |
|---|---|---|---|---|---|---|
| cDNA level | Protein level | |||||
| Family T | T1 | Tunisian | c.322_325delGACT hom | p.Asp108Ilefs*30 | 7 CHMs, 1 SA, 1VTP | 11 |
| Family W | W1 | Asian | c.1A>G; c.322_325delGACT | p.?; p.Asp108Ilefs*30 | 4 CHMs | 11 |
| Family L | V:3 | Pakistani | c.3G>T hom | 7 CHMs, 2 SAs | 11 | |
| IV:5 | As above | 4 CHMs | ||||
| MoUs70 family | 481 | African-American | c.299_302delTCAA hom | p.Ile100Argfs*2 | 4 CHMs, 3 HMs | 13 |
| MoTu98 family | 654 | Tunisian | c.322_325delGACT hom | p.Asp108Ilefs*30 | 5 CHMs, 2 failed IVF | 13 |
| MoIn355 family | 1096 | Indian | c.322_325delGACT hom | p.Asp108Ilefs*30 | 8 HMs | 13 |
| 1094 | As above | p.Asp108Ilefs*30 | 3 HMs | |||
| MoIr440 family | 1448 | Iranian | c.17_20delGGTT hom | p.Arg6Leufs*7 | 11 HMs | This study |
| MoIn494 family | 1335 | Indian | c.349+1G>A hom | p.? | 6 HMs | This study |
Abbreviations: CHM, complete hydatidiform mole; HM, undefined hydatidiform mole; IVF, in vitro fertilization; SA, spontaneous abortion; VTP, voluntary termination of pregnancy.
Patients 1096 and 1094 are sisters; V:3 and IV:5 are first cousins. Variants in coding DNA and protein are annotated according to reference sequences NM_001017361.2 and NP_001017361.1, respectively.
The identification of two novel mutations in an Iranian and an Indian patient is in line with a previous observation regarding the increased frequency of patients with RHM and NLRP7 or KHDC3L mutations in Asian countries such as Turkey, Iran, Pakistan, and India.13 The identification of two novel protein-truncating mutations in the present study, in addition to a few previously reported mutations, confirms the causal role of KHDC3L as the second, albeit minor, gene responsible for RHM. This gene should be screened if no mutations are detected in NLRP7.
Acknowledgments
The authors thank the families for participating in this study. This project was financially supported by Shiraz University of Medical Sciences Grant No. 92-6747 and by the Canadian Institute of Health Research (MOP-130364 and PPP-122897 to RS). NMPN was supported by a Max E Binz Award from the Faculty of Medicine of the McGill University, an Award from the Center for Research in Reproduction and Development at the McGill University, and a fellowship from the McGill University Health Centre Research Institute.
The authors declare no conflict of interest.
Footnotes
Supplementary Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv)
References
- Savage P, Williams J, Wong SL, Short D, Casalboni S, Catalano K et al. The demographics of molar pregnancies in England and Wales from 2000–2009. J Reprod Med 2010; 55: 341–345. [PubMed] [Google Scholar]
- Grimes DA. Epidemiology of gestational trophoblastic disease. Am J Obstet Gynecol 1984; 150: 309–318. [DOI] [PubMed] [Google Scholar]
- Bracken MB. Incidence and aetiology of hydatidiform mole: an epidemiological review. Br J Obstet Gynaecol 1987; 94: 1123–1135. [DOI] [PubMed] [Google Scholar]
- Le-ming S, Robert JK. Molecular basis of gestational trophoblastic diseases. Curr Mol Med 2002; 2: 1–12. [DOI] [PubMed] [Google Scholar]
- Szulman AE, Surti U. The syndromes of hydatidiform mole. Am J Obstet Gynecol 1978; 132: 20–27. [DOI] [PubMed] [Google Scholar]
- Sebire NJ, Fisher RA, Foskett M, Rees H, Seckl MJ, Newlands ES. Risk of recurrent hydatidiform mole and subsequent pregnancy outcome following complete or partial hydatidiform molar pregnancy. BJOG 2003; 110: 22–26. [PubMed] [Google Scholar]
- Kim JH, Park DC, Bae SN, Namkoong SE, Kim SJ. Subsequent reproductive experience after treatment for gestational trophoblastic disease. Gynecol Oncol 1998; 71: 108–112. [DOI] [PubMed] [Google Scholar]
- Mosher R, Goldstein DP, Berkowitz R, Bernstein M, Genest DR. Complete hydatidiform mole. Comparison of clinicopathologic features, current and past. J Reprod Med 1998; 43: 21–27. [PubMed] [Google Scholar]
- Kronfol NM, Iliya FA, Hajj SN. Recurrent hydatidiform mole: a report of five cases with review of the literature. J Med Liban 1969; 22: 507–520. [PubMed] [Google Scholar]
- Murdoch S, Djuric U, Mazhar B, Seoud M, Khan R, Kuick R et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet 2006; 38: 300–302. [DOI] [PubMed] [Google Scholar]
- Parry DA, Logan CV, Hayward BE, Shires M, Landolsi H, Diggle C et al. Mutations causing familial biparental hydatidiform mole implicate C6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am J Hum Genet 2011; 89: 451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy R, Nguyen NM, Sarrabay G, Rezaei M, Rivas MC, Kavasoglu A et al. The genomic architecture of NLRP7 is Alu rich and predisposes to disease-associated large deletions. Eur J Hum Genet (e-pub ahead of print 9 March 2016; doi:10.1038/ejhg.2016.9).
- Reddy R, Akoury E, Phuong Nguyen NM, Abdul-Rahman OA, Dery C, Gupta N et al. Report of four new patients with protein-truncating mutations in C6orf221/KHDC3L and colocalization with NLRP7. Eur J Hum Genet 2013; 21: 957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akoury E, Zhang L, Ao A, Slim R. NLRP7 and KHDC3L, the two maternal–effect proteins responsible for recurrent hydatidiform moles, co-localize to the oocyte cytoskeleton. Hum Reprod 2015; 30: 159–169. [DOI] [PubMed] [Google Scholar]
- El-Maarri O, Seoud M, Coullin P, Herbiniaux U, Oldenburg J, Rouleau G et al. Maternal alleles acquiring paternal methylation patterns in biparental complete hydatidiform moles. Hum Mol Genet 2003; 12: 1405–1413. [DOI] [PubMed] [Google Scholar]
- Hayward BE, De Vos M, Talati N, Abdollahi MR, Taylor GR, Meyer E et al. Genetic and epigenetic analysis of recurrent hydatidiform mole. Hum Mutat 2009; 30: E629–E639. [DOI] [PubMed] [Google Scholar]
- Sanchez-Delgado M, Martin-Trujillo A, Tayama C, Vidal E, Esteller M, Iglesias-Platas I et al. Absence of maternal methylation in biparental hydatidiform moles from women with NLRP7 maternal-effect mutations reveals widespread placenta-specific imprinting. PLoS Genet 2015; 11: e1005644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou YC, Shao L, Peng HH, Rosetta R, del Gaudio D, Wagner AF et al. A recurrent intragenic genomic duplication, other novel mutations in NLRP7 and imprinting defects in recurrent biparental hydatidiform moles. Mol Hum Reprod 2008; 14: 33–40. [DOI] [PubMed] [Google Scholar]
- Judson H, Hayward BE, Sheridan E, Bonthron DT. A global disorder of imprinting in the human female germ line. Nature 2002; 416: 539–542. [DOI] [PubMed] [Google Scholar]
- Ito M, Sferruzzi-Perri AN, Edwards CA, Adalsteinsson BT, Allen SE, Loo TH et al. A trans-homologue interaction between reciprocally imprinted miR-127 and Rtl1 regulates placenta development. Development 2015; 142: 2425–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczak M, Cooper DN. Gene deletions causing human genetic disease: mechanisms of mutagenesis and the role of the local DNA sequence environment. Hum Genet 1991; 86: 425–441. [DOI] [PubMed] [Google Scholar]
- De Grassi A, Ciccarelli FD. Tandem repeats modify the structure of human genes hosted in segmental duplications. Genome Biol 2009; 10: R137–R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Pascal G, Fouchecourt S, Pontarotti P, Monget P. Gene birth, death, and divergence: the different scenarios of reproduction-related gene evolution. Biol Reprod 2009; 80: 616–621. [DOI] [PubMed] [Google Scholar]
- Messaed C, Chebaro W, Roberto RBD, Rittore C, Cheung A, Arseneau J et al. NLRP7 in the spectrum of reproductive wastage: rare non-synonymous variants confer genetic susceptibility to recurrent reproductive wastage. J Med Genet 2011; 48: 540–548. [DOI] [PubMed] [Google Scholar]
- Huang J-Y, Su M, Lin S-H, Kuo P-L. A genetic association study of NLRP2 and NLRP7 genes in idiopathic recurrent miscarriage. Hum Reprod 2013; 28: 1127–1134. [DOI] [PubMed] [Google Scholar]
- Akoury E, Gupta N, Bagga R, Brown S, Déry C, Kabra M et al. Live births in women with recurrent hydatidiform mole and two NLRP7 mutations. Reprod BioMed Online 2015; 31: 120–124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.