

ABSTRACT
Kaposi's sarcoma-associated herpesvirus (KSHV) maintains two modes of life cycle, the latent and lytic phases. To evade the attack of the cell host's immune system, KSHV switches from the lytic to the latent phase, a phase in which only a few of viral proteins are expressed. The mechanism by which KSHV evades the attack of the immune system and establishes latency has not been fully understood. Major histocompatibility complex class II (MHC-II) molecules are key components of the immune system defense mechanism against viral infections. Here we report that HLA-DRα, a member of the MHC-II molecules, was downregulated by the replication and transcription activator (RTA) protein encoded by KSHV ORF50, an important regulator of the viral life cycle. RTA not only downregulated HLA-DRα at the protein level through direct binding and degradation through the proteasome pathway but also indirectly downregulated the protein level of HLA-DRα by enhancing the expression of MARCH8, a member of the membrane-associated RING-CH (MARCH) proteins. Our findings indicate that KSHV RTA facilitates evasion of the virus from the immune system through manipulation of HLA-DRα.
IMPORTANCE Kaposi's sarcoma-associated herpesvirus (KSHV) has a causal role in a number of human cancers, and its persistence in infected cells is controlled by the host's immune system. The mechanism by which KSHV evades an attack by the immune system has not been well understood. This work represents studies which identify a novel mechanism by which the virus can facilitate evasion of an immune system. We now show that RTA, the replication and transcription activator encoded by KSHV (ORF50), can function as an E3 ligase to degrade HLA-DRα. It can directly bind and induce degradation of HLA-DRα through the ubiquitin-proteasome degradation pathway. In addition to the direct regulation of HLA-DRα, RTA can also indirectly downregulate the level of HLA-DRα protein by upregulating transcription of MARCH8. Increased MARCH8 results in the downregulation of HLA-DRα. Furthermore, we also demonstrate that expression of HLA-DRα was impaired in KSHV de novo infection.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV), also called human herpesvirus 8 (HHV-8), is a Rhadinovirus belonging to the Gammaherpesvirinae subfamily. It was discovered in Kaposi's sarcoma (KS) lesions and is also associated with primary effusion lymphoma (PEL) and multicentric Castleman's disease (1–3). Almost 20% of all human malignancies are caused by viruses (4). The majority of infected individuals do not develop tumors, although immunosuppressive conditions like AIDS and organ transplantation can dramatically increase the risk of developing a malignancy. This suggests an active role for the host immune system in controlling KSHV infections (5, 6). During its coevolution with the host, KSHV hijacks and manipulates many of the host machineries to override the host immune surveillance (7, 8). The host ubiquitin system has fundamental roles in regulating cellular events, like protein degradation and trafficking, signal transduction, endocytosis, and the immune response (9). It has been exploited or mimicked by many viruses to establish persistence in the host cell, including that utilized by KSHV. There are many challenging facets yet to be unraveled to fully comprehend the mechanisms by which KSHV uses the ubiquitin system for immune evasion and viral proliferation during infection and pathogenesis. The present study provides additional insight into the ubiquitin-related mechanisms deployed by KSHV, which may serve as potential targets for future therapeutic interventions.
MHC (major histocompatibility complex) class I and class II (MHC-I and -II) are necessary for the induction and regulation of the adaptive immune responses to pathogenic agents such as viruses (10). The early K3 and K5 genes of KSHV are capable of modulating the MHC-I pathway (11). Both K3 and K5 can significantly induce internalization and degradation of MHC-I molecules during KSHV de novo infection (11–13). Although the mechanism by which KSHV interferes with MHC-II during KSHV primary infection is poorly understood, mechanisms by which other herpesviruses modulate MHC-II have been reported. US2, US3, and pp65 encoded by human cytomegalovirus (HCMV; a betaherpesvirus) disrupts the MHC-II pathway by targeting HLA-DRα for degradation (14–16). BZLF1 and gp42 encoded by Epstein-Barr virus (EBV; a gammaherpesvirus) inhibit the MHC-II pathway by blocking the interaction between MHC-II and the CD4 receptor (17, 18). Herpes simplex virus 1 (HSV-1; an alphaherpesvirus)-encoded gB manipulates the MHC-II pathway by perturbing the trafficking of HLA-DR (19).
The RING-CH domain has been identified in different viral-associated E3 ligases, which include KSHV K3 and K5, also referred to as modulators of immune recognition 1 (MIR1) and MIR2, respectively. Murine gammaherpesvirus 68 (MHV-68) mK3, myxoma virus M153R, HSV ORF12, and swinepox virus C7L also contain RING-CH motifs (20). The viral versions were found to be pirated from their host counterparts, which included 11 human homologs called membrane-associated RING-CH 1 (MARCH1) through 11. MARCH8/cMIR (cellular MIR) was the first to be characterized (21, 22). MIR1 and MIR2 can polyubiquitinate MHC-I molecules and mark them for degradation (21). MIR1 downregulates all MHC-I molecules, but MIR2 downregulates only two members of MHC-I molecules (HLA-A and HLA-B) (11, 21). In addition, mK3 downregulates MHC-I molecules through the endoplasmic reticulum-associated degradation (ERAD) pathway that includes Lys48-linked polyubiquitination which marks it for proteasomal degradation (23). The 11 MARCH proteins are expressed in a tissue-specific manner and portray different subcellular localizations (24). Various substrates have been found for the MARCH proteins, and further research on substrate recognition is still ongoing (21). MARCH8/cMIR is expressed at a generally low level in selected tissues, including lymph nodes and bone marrow, with subcellular localization in endocytic or lysosomal vesicles (24). Interestingly, induced expression of MARCH8 is known to downregulate B7-2, Fas, HLA-A, MHC-II, and the transferrin receptor (21, 25).
Lytic replication of KSHV is initiated by the expression of RTA encoded by KSHV ORF50, is the control switch for expression of many early lytic genes, such as the K3 and K5 genes, and is also important for mediating KSHV viral DNA replication (26). As a transcription factor, recent studies showed that RTA regulates cellular genes by direct binding to sequence-specific DNA binding sites named RTA-responsive elements (RRE) or via its associations with other transcription factors (27, 28). However, the mechanisms of regulating cellular genes by RTA is still not fully elucidated.
It has been suggested that RTA may play an important role in immune modulation and can perturb the production of type I interferon (IFN) (29). Interferon regulatory factor 7 (IRF-7) is essential for the induction of IFN-α/β genes upon infection of KSHV (30). RTA promotes the ubiquitination and degradation of IRF-7, which results in inhibition of IRF-7-mediated IFN-α and IFN-β mRNA production (29). The myeloid differentiation primary response 88 (MyD88) and TIR domain-containing adapter inducing IFN-β (TRIF) are the major adaptors that bind to the Toll-like receptors (TLRs). MyD88 and TRIF activation leads to expression of numerous cytokines. These include tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), IFN-α, IFN-β, and IFN-γ (31–36). RTA also promotes the ubiquitination and degradation of MyD88 and TRIF through the ubiquitin-proteasome pathway during KSHV primary infection, which results in disruption of the signal transmission of TLRs and the subsequent antiviral cascade (37, 38).
RTA is upregulated during the KSHV lytic phase and packaged into KSHV particles during virus release. Therefore, its synthesis is not needed from the beginning of virus infection. This suggests that RTA may play an important role in immune evasion. Here we demonstrate that RTA can promote the ubiquitination and degradation of HLA-DRα and reveal a potential new mechanism by which KSHV can evade attack by the innate immune system.
MATERIALS AND METHODS
Plasmid and antibodies.
pCDNA3-RTA, PGEX-RTA, His-Uba1, His-Ubc5a, and His-ubiquitin were described previously (39, 40). The coding sequences (CDS) of MHC-II molecules were generated by using PCR and cloned into pA3M vector using the following primers: for HLA-DRα, 5′-ATAAGAATGCGGCCGCCAGAGGCCCCCTGCGTTCTG-3′ and 5′-CGGGATCCATGGCCATAAGTGGAGTCCC-3′; for HLA-DRβ, 5′-CGGGATCCATGGTGTGTCTGAAGTTCCC-3′ and 5′-ATAAGAATGCGGCCGCGCTCAGGAATCCTGTTGGCT-3′; for HLA-DPα, 5′-CGGGATCCATGCGCCCTGAAGACAGAAT-3′ and 5′-ATAAGAATGCGGCCGCCAGGGTCCCCTGGGCCCGGG-3′; for HLA-DPβ, 5′-CGGGATCCATGATGGTTCTGCAGGTTTC-3′ and 5′-ATAAGAATGCGGCCGCTGCAGATCCTCGTTGAACTT-3′; for HLA-DQα, 5′-CGGGATCCATGATCACATTCCTGCCGCT-3′ and 5′-ATAAGAATCGGCCGCCAATGGCCCTTGGTGTCTGG-3′; for HLA-DQβ, 5′-CCCAAGCTTATGTCTTGGAAGAAGGCTTT-3′ and 5′-ATAAGAATGCGGCCGCGTGCAGAAGCCCTTTCTGAC-3′. The anti-Myc (9E10), anti-hemagglutinin (anti-HA; 12CA5), and anti-RTA antibodies were generated from hybridomas and column purified. Rabbit anti-HLA-DRα was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Mouse anti-Flag (M2) antibody was purchased from Sigma-Aldrich Biotechnology (St. Louis, MO, USA).
Expression and purification of recombinant proteins.
Overnight cultures (2 ml) of Escherichia coli BL21(DE3) were transferred to and incubated in 200 ml of culture medium and grown at 30°C to an optical density of about 0.6 at 600 nm. After IPTG (isopropyl-β-d-thiogalactopyranoside) induction (0.5 mM, 2 h at 30°C), the bacteria were collected and sonicated in lysis buffer containing 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 0.5% NP-40, 1 mM EDTA, 1 M dithiothreitol (DTT), 5% Sarkosyl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin. His-tagged proteins were purified by Ni2+-nitrilotriacetic acid (Ni2+-NTA) agarose chromatography (Qiagen, Valencia, CA, USA), and glutathione S-transferase (GST)-tagged proteins were purified by glutathione Sepharose 4B (GE Healthcare Life Science, Piscataway, NJ, USA). The purified proteins were then dialyzed with dialysis buffer containing 40 mM HEPES (pH 8.0), 60 mM potassium acetate, 1 mM MgCl2, 0.5 mM EDTA, 10% glycerol, and 1 mM DTT.
In vitro GST pulldown assay.
An in vitro GST pulldown assay was performed as previously described (41). Briefly, purified GST-tagged RTA proteins (5 μg) expressed in bacteria were incubated with 10 μl glutathione Sepharose 4B for 4 h at 4°C, followed by the addition of 2 μg purified His-tagged HLA-DRα or His-tagged HLA-DRβ protein, and then incubated at 4°C overnight. The mixture was washed four times with ice-cold phosphate-buffered saline (PBS). The pellet was then subjected to SDS-PAGE and stained with Coomassie brilliant blue G250.
Generation and purification of Bac16-KSHV viral particles.
Purified Bac16-KSHV plasmid was transfected into HEK293T cells by Ca3(PO4)2 to yield HEK293T-Bac16-KSHV cells. Hygromycin B (100 μg/ml) was added after 24 h of transfection for selection. Homogenous populations of cells harboring Bac16-KSHV were identified by GFP expression after 3 weeks of selection. Butyric acid at a final concentration of 3 mM and tetradecanoyl phorbol acetate (TPA) (Sigma Inc., St. Louis, MO, USA) at 20 ng/ml were used for lytic induction. Cell suspensions were centrifuged at 3,000 rpm for 20 min, and the supernatant was filtered through a 0.45-μm-pore-size filter. The viral particles were concentrated by ultracentrifugation at 70,000 × g at 4°C.
Cell culture of KSHV cells.
The KSHV-positive cell lines BCBL1, BC3, and TRExBCBL1-RTA and the KSHV-negative cell line TRExBJAB-RTA were grown in RPMI 1640 medium (Invitrogen Inc., Carlsbad, CA, USA), with 7% bovine growth serum (BGS), supplemented with l-glutamine, penicillin, and streptomycin. The HEK293T, HEK293T-Bac16-KSHV, and SAOS-2 cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen Inc., Carlsbad, CA, USA) with 5% BGS, supplemented with penicillin and streptomycin. The TRExBCBL1-RTA and TRExBJAB-RTA cells were kindly provided by Jae Jung (University of Southern California). Lytic reactivation of the KSHV-positive cells, BCBL1, BC3, and HEK293T-Bac16-KSHV, was induced using butyric acid at a final concentration of 3 mM and TPA at a final concentration of 20 ng/ml. TRExBCBL1-RTA and TRExBJAB-RTA cells were induced by adding tetracycline at a final concentration of 8 μg/ml.
IFA.
Immunofluorescence assays (IFA) were performed as previously described (39). Briefly, the cells were fixed with 4% paraformaldehyde and 0.1% Triton X-100 for 15 to 20 min. Fixed cells were incubated with the specific primary antibody in PBS overnight at 4°C. After subsequent washes with PBS, the cells were incubated with 1:1,000 secondary antibodies (Invitrogen Inc., Carlsbad, CA, USA) and 1:500 DAPI (4′,6-diamidino-2-phenylindole). Finally, the cells were washed and mounted in mounting medium and visualized using a FluoView FV300 confocal microscope (Olympus, Inc., Melville, NY, USA). The intensity of the IFA signal was analyzed using ImageJ software.
Transfection and WB.
The transfections were performed by electroporation using a Gene Pulser II RF module (Bio-Rad, Hercules, CA, USA). For SAOS-2 cells, electroporation was carried out at the following settings: voltage, 210 V; capacitance, 975 μF. For other cells, electroporation was carried out at the following settings: voltage, 230 V; capacitance, 975 μF. At 48 h posttransfection, cells were harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer, and the protein concentration was measured by the Bradford assay. Samples were applied to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred to nitrocellulose membranes. Western blotting (WB) was performed with appropriate antibody, and specific proteins were identified by specific secondary antibodies conjugated to IRDye 800 or 680 (Rockland Immunochemicals, Inc., Gilbertsville, PA, USA). The membranes were then washed, and the signals were visualized by using an Odyssey infrared image (LI-COR, Inc., Lincoln, NE, USA).
Quantitation by real-time PCR.
Total RNA was extracted from cells by treatment with TRIzol and purified with chloroform and isopropanol. cDNA was then made by using a high-capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA, USA) by following manufacturer's protocol. Quantitative real-time PCRs (RT-PCR) were performed with Power SYBR green mix (Applied Biosystems, Foster City, CA, USA). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as an internal control, and each sample was tested in triplicate. The primers used for MARCH8 were 5′-CGGGCCGAGCGGAACCAGGAAT-3′ and 5′-TGTGCACACTCAGGCCAAAGACGA-3′. The primers used for MARCH9 were 5′-AGTCTCGGCTCCGCATGTTTCTGA-3′ and 5′-CTCCGACCCGTAGTACTCCTCCTC-3′. The specific primers for MHC-II molecules were described previously (42). The RTA- and GAPDH-specific primers were described in earlier reports (43, 44). Data analyses were performed on the StepOnePlus RT-PCR platform (Applied Biosystems, Inc., Foster City, CA, USA) by using the 2−ΔΔCT method of relative quantitation.
Ubiquitination assays.
In vivo and in vitro ubiquitination assays were performed as described previously (40). Briefly, for the in vitro ubiquitination assay, 500 ng of purified His-tagged Uba1, 1.5 μg of purified His-tagged Ubc5a, 1.5 μg of GST-tagged RTA, 2.5 μg of purified His-tagged ubiquitin, and 2.5 μg of His-tagged HLA-DRα were mixed in a 50-μl reaction mixture containing 4 mM HEPES (pH 8.0), 6 mM potassium acetate, 5 mM MgCl2, 1 mM DTT, and 1.5 mM ATP. Reaction mixtures were incubated for 1 h at 30°C. Reaction products were fractionated by SDS-PAGE and analyzed by Western blotting with specific antibodies. For the in vivo ubiquitination assay, cells were transfected by electroporation with DNA vectors expressing a specific protein. Cells were incubated for 36 h and treated for an additional 12 h with 10 μM MG132 (Enzo Life Sciences, Farmingdale, NY, USA) before being harvested. Proteins were immunoprecipitated with specific antibodies and resolved by SDS-PAGE. The extent of ubiquitination of the immunoprecipitated complexes was detected by specific antibodies.
Chromatin immunoprecipitation (ChIP) assay.
Fifty million BCBL1 cells were fixed with 1% formaldehyde for 10 min at room temperature after treatment with TPA and butyric acid for 24 h, followed by the addition of glycine at a final concentration of 125 mM for 5 min to stop cross-linking. The cells were washed three times with ice-cold PBS and then lysed in cell lysis buffer containing 5 mM PIPES (pH 8.0), 85 mM KCl, 0.5% NP-40, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin for 10 min on ice. The nuclei were pelleted down by centrifugation at 5,000 rpm for 5 min and sonicated in nuclear lysis buffer (50 mM Tris [pH 8.0], 10 mM EDTA, 1% SDS) supplemented with protease inhibitors to an average fragment of 500 to 1,000 bp. Samples were centrifuged for 10 min at 15,000 rpm to remove the cell debris and diluted 10-fold in dilution buffer (0.01% SDS, 1.1% Triton X-100, 16.7 mM Tris-HCl [pH 8.0], 167 mM NaCl, and 1.2 mM EDTA plus protein inhibitors). After preclearance with a salmon sperm DNA-protein A/G-Sepharose slurry for 30 min at 4°C with rotation, the samples were divided into two fractions. One fraction was incubated with RTA antibody and the other with control mouse IgG. The precipitated immune complex was washed subsequently with low-salt buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 0.1% SDS, 1.0% Triton X-100, 2 mM EDTA), high-salt buffer (20 mM Tris [pH 80], 500 mM NaCl, 0.1% SDS, 1.0% Triton X-100, 2 mM EDTA,), LiCl wash buffer (0.25 M LiCl, 1.0% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris[pH 8.0]), and twice in TE buffer (10 mM Tris [pH 8.0], 1 mM EDTA). Chromatin was then eluted with an elution buffer (1% SDS, 0.1 M NaHCO3). Elution was then reverse cross-linked by adding 1 μl of 10 mg/ml RNase and 5 M NaCl to reach a final concentration of 0.3 M and incubated at 65°C for 5 h. The reverse-cross-linked DNA was purified with a Zyppy spin column (Zymo Research, Irvine, CA, USA) after treatment with proteinase K.
The purified DNA was analyzed by RT-PCR for the presence of the MARCH8 promoter region. The primers used for MARCH8 promoter amplification were as follows: P1, 5′-GCTATCAAACGGGAGGAA-3′ and 5′-TAGGGAGCGAAAGCAAAC-3′; P2, 5′-GGTTTCACCGTGTTAGCC-3′ and 5′-GTATTCCCAGCACTTTGG-3′; P3, 5′-TGGCTCTACTGCTTCTGG-3′ and 5′-AACCCTTTGGCTCAACAA-3′; P4, 5′-GAGCTATGCAGGGCTGAT-3′ and 5′-CTCGGAAAGGTTAAGGAAAT-3′.
Statistical analysis.
Each experiment was repeated at least three times. The mean scores were examined by using Student's t test.
RESULTS
HLA-DRα protein levels are dramatically reduced in the presence of RTA.
Regulation of MHC-II molecules during the KSHV latent cycle by LANA has been well reported (42, 45). However, how KSHV regulates MHC-II molecules during KSHV de novo infection remains largely unknown. RTA is highly produced during the KSHV lytic phase and packaged into KSHV particles during virus release (46, 47). To examine whether RTA targeted the MHC-II pathway, a key component of the immunity system, we monitored changes in the MHC-II molecules by RT-PCR. We first investigated whether RTA can regulate MHC-II at the transcript level. TRExBJAB-RTA cells were treated with tetracycline (8 μg/ml). Forty-eight hours after treatment, RNA was isolated from these cells and RT-PCR analysis to determine levels of MHC-II molecules was performed. The results showed that there was no significant change in the transcription levels of MHC-II molecules (Fig. 1A). To investigate whether ectopic expression of RTA could affect the protein levels of MHC-II molecules, we transfected increasing amounts of the RTA expression plasmid together with the same amount of different MHC-II molecules into BJAB cells. Inclusion of RTA showed a reduction in the level of HLA-DRα protein in a dose-dependent manner. The levels of the other MHC-II molecules remained relatively unchanged (Fig. 1B). This result was further confirmed in another two KSHV-negative B-cell lines, DG-75 and Ramos (Fig. 1C). To validate the reduction of HLA-DRα by RTA, TRExBJAB-RTA cells were induced by tetracycline to induce the expression of RTA, and reduced levels of endogenous HLA-DRα were detected by immunoblotting 48 h after induction (Fig. 1D). Similarly, the protein levels of HLA-DRα were also reduced as expected in tetracycline-treated TRExBCBL1-RTA cells (Fig. 1E). These results suggest that RTA can contribute to the downregulation of HLA-DRα at the protein levels but not the transcript levels.
FIG 1.

RTA mediates the downregulation of HLA-DRα. (A) Quantitation of MHC-II transcripts by RT-PCR. The TRExBJAB-RTA cells were treated with 8 μg/ml of tetracycline for 48 h. RNA was isolated, and RT-PCR was performed. The relative levels of MHC-II were normalized to GAPDH. (B) BJAB cells were transfected with 0, 10, and 20 μg of pCDNA-RTA together with 10 μg of the indicated Myc-tagged MHC-II plasmid by electroporation. At 48 h posttransfection, the cells were lysed and subjected to WB analysis with RTA and Myc antibodies. (C) DG-75 and Ramos cells were cotransfected with 10 μg pCDNA-RTA and Myc–HLA-DRα by electroporation. At 48 h posttransfection, cells were lysed and subjected to WB analysis with RTA and Myc antibodies. (D) TRExBJAB-RTA cells were treated with 8 μg/ml of tetracycline. Forty-eight hours later, the cells were lysed and subjected to WB analysis using RTA and HLA-DRα antibodies. (E) TRExBCBL1-RTA cells were treated with 8 μg/ml of tetracycline, and 48 h later, the cells were lysed and subjected to WB analysis with RTA and HLA-DRα antibodies. Tet, tetracycline; RD, relative density.
RTA-mediated downregulation of HLA-DRα occurs through the proteasomal degradation pathway.
As described above, RTA did not regulate HLA-DRα at the level of transcription. We wondered whether RTA regulated HLA-DRα expression at the posttranslational level. For this purpose, cycloheximide (CHX) assays were performed in SAOS-2 cells. SAOS-2 cells were transfected with RTA expression plasmid or its control. At 24 h posttransfection, the cells were treated with 40 μg/ml CHX to stop the protein synthesis. Cell lysates harvested at the indicated times were subjected to Western blot analysis. The CHX assay revealed that ectopic expression of RTA is associated with decreased HLA-DRα half-life (Fig. 2A, compare lanes 3, 4, 7, and 8). Since protein degradation is usually related to ubiquitination, we wanted to further determine whether the downregulation of HLA-DRα is ubiquitin dependent. We treated SAOS-2 cells expressing RTA with increasing concentrations of proteasome inhibitor MG132 for 12 h (Fig. 2B). Cell lysates from these SAOS-2 cells were subjected to Western blot analysis. The results show that in the presence of MG132, the downregulation of HLA-DRα was inhibited and the reduction of HLA-DRα protein levels was almost completely reversed (Fig. 2B, compare lanes 1 and 8). These results strongly suggest that downregulation of HLA-DRα mediated by RTA is through the proteasomal degradation pathway.
FIG 2.
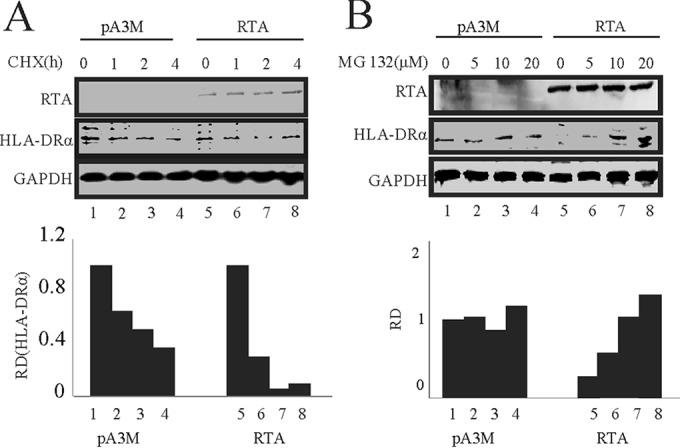
RTA induces degradation of HLA-DRα through the ubiquitin-proteasome degradation pathway. (A) SAOS-2 cells were transfected with RTA by electroporation, treated with 40 μg/ml cycloheximide (CHX), and collected at 0, 1, 2, and 4 h after treatment. RTA, HLA-DRα, and GAPDH antibodies were used for WB analysis. (B) SAOS-2 cells were transfected with RTA by electroporation and then treated with 0, 5, 10, and 20 mM MG132 for 12 h. RTA, HLA-DRα, and GAPDH antibodies were used for Western blotting. RD, relative density.
The observations that RTA decreases the half-life of HLA-DRα and that the proteasome inhibitor MG132 can reverse the downregulation of HLA-DRα mediated by RTA allow us to reach the potential conclusion that RTA can degrade HLA-DRα through the proteasome degradation pathway.
RTA interacts with HLA-DRα and mediates its downregulation via the ubiquitin-proteasome degradation pathway.
The studies described above showed that ectopic expression of RTA affects the levels of HLA-DRα protein. RTA possesses ubiquitin E3 ligase activity and promotes IRF7 degradation via the ubiquitin-proteasome pathway. This results in disruption of the antiviral pathway induced by KSHV infection (29), Therefore, we wanted to further examine whether RTA can physically bind to HLA-DRα, which can result in reduction of HLA-DRα protein levels. For this purpose, SOAS-2 cells were cotransfected with the RTA plasmid together with the Myc-tagged HLA-DRα plasmid. Cell lysates were harvested and immunoprecipitated with RTA specific antibody, followed by Western blotting with anti-Myc and anti-RTA antibodies to detect the association between RTA and HLA-DRα (Fig. 3A). The results show that RTA antibodies coimmunoprecipitated with the Myc-tagged HLA-DRα from the cell lysate in complex with RTA but not from the cell lysate without RTA (Fig. 3A). To further validate the result, a reverse coimmunoprecipitation assay was also performed in SAOS-2 cells that were cotransfected with RTA and HLA-DRα. Cell lysates were harvested, and relevant antigens were coimmunoprecipitated with the anti-Myc specific antibody (Fig. 3B). The results show that Myc antibodies coimmunoprecipitated RTA from the cell lysate with HLA-DRα but not from the lysate without HLA-DRα as expected (Fig. 3B). To further visualize the interaction of these two proteins under physiological conditions, we used KSHV-positive BCBL1 cells (Fig. 3C). Endogenous coimmunoprecipitation between RTA and HLA-DRα supported our result above which showed that RTA can associate with HLA-DRα. We further investigated whether RTA bound to HLA-DRα directly. E. coli-expressed GST-tagged RTA proteins, His-tagged HLA-DRα, and His-tagged HLA-DRβ proteins were purified, and an in vitro GST pulldown assay was performed (Fig. 3D). RTA directly bound to HLA-DRα instead of HLA-DRβ in the absence of any other protein (Fig. 3D, lane 4). Since RTA functioned as an E3 ubiquitin ligase and can directly bind to HLA-DRα, we wanted to investigate whether RTA can promote the polyubiquitination of HLA-DRα. For this purpose, we performed in vitro ubiquitination assays to test whether RTA can function as an E3 ubiquitin ligase to add a polyubiquitin chain to HLA-DRα (Fig. 3E). Purified bacterially expressed His-DRα (substrate), GST-RTA (E3 ubiquitin ligase), His-Uba1 (E1 ubiquitin activating enzyme), and His-Ubc5a (E2 ubiquitin conjugating enzyme), together with His-ubiquitin, were mixed and incubated at 30°C for 1 h in the presence of ATP in vitro. We observed that RTA along with E1 and E2 can promote the addition of polyubiquitin chains to HLA-DRα (Fig. 3E, lane 3). However, in the absence of E1 or E2, RTA failed to add the polyubiquitin chain to HLA-DRα (Fig. 3E, lanes 1 and 2, respectively). These results indicate that RTA can function as an E3 ubiquitin ligase to add the polyubiquitin chain to HLA-DRα and were responsible for HLA-DRα degradation through the proteasomal degradation pathway.
FIG 3.

RTA binds to and promotes polyubiquitination of HLA-DRα. (A) SAOS-2 cells were either transfected with 10 μg pA3M-HLA-DRα alone or cotransfected with 10 μg pCDNA3-RTA by electroporation. Thirty-six hours later, the cells were treated with 10 μM MG132 for another 12 h. Cell lysates were then immunoprecipitated with RTA antibodies. Ten percent of the lysates was loaded as input; lysates and immunoprecipitation (IP) complexes were resolved by SDS-PAGE and subjected to WB with the indicated antibodies. (B) SAOS-2 cells were either transfected with 10 μg pCDNA3-RTA alone or cotransfected with 10 μg pA3M-HLA-DRα by electroporation. Thirty-six hours later, the cells were treated with 10 μM MG132 for another 12 h. Cell lysates were then immunoprecipitated with Myc antibodies. Ten percent of the lysates was loaded as input; lysates and IP complexes were resolved by SDS-PAGE and subjected to WB with the indicated antibodies. (C) Thirty million BCBL1 cells were treated with TPA and butyric acid for 24 h. Cells were harvested and lysed with RIPA buffer. Cell lysates were then immunoprecipitated with mouse IgG (mIgG) and RTA antibodies. Ten percent of the lysates was loaded as input. Lysates and IP complexes were resolved by SDS-PAGE and subjected to WB with the indicated antibodies. (D) In vitro GST pulldown assays were performed as described in Materials and Methods. The interactions between RTA and HLA-DRα were resolved by SDS-PAGE and detected by Coomassie brilliant blue staining. (E) In vitro ubiquitination assays were carried out as described in Materials and Methods. The mixture was subjected to SDS-PAGE and WB analysis using specific antibodies. Ub, ubiquitin.
RTA expression leads to induction of MARCH8 levels.
It is well documented that HLA-DRα is a substrate of MARCH8 and MARCH9, members of the membrane-associated RING-CH (MARCH) family of proteins, and is degraded by MARCH8 and MARCH9 through the ubiquitin-proteasome degradation pathway (48). To investigate whether RTA can also affect MARCH8- or MARCH9-mediated HLA-DRα degradation, TRExBJAB-RTA and TRExBCBL1-RTA cells were treated with tetracycline (Fig. 4A and B). RTA expression was induced by tetracycline in TRExBJAB-RTA cells, and the cell lysates were harvested at the indicated days. As expected, the RTA protein expression steadily increased. Importantly, a significant increase in the transcription level as well as the protein level of MARCH8 was observed in induced cells, compared to the uninduced cells, which correlated with the observed increase in RTA protein levels. Detection of GAPDH confirmed that similar amounts of protein were loaded in each lane (Fig. 4A, right). Interestingly, the mRNA level of MARCH9 showed no significant change (Fig. 4A, middle). These results were further validated in TRExBCBL1-RTA cells. Similar results were obtained for the protein and transcript levels of MARCH8. A significant increase in MARCH8 protein expression, which correlated with the increase in RTA protein levels, was seen (Fig. 4B). Interestingly, although the patterns of increase of MARCH8 in protein and mRNA levels were similar, the MARCH8 mRNA levels increased about 40-fold at day 3 in TRExBCBL1-RTA cells treated with tetracycline, compared to a 7-fold increase in MARCH8 mRNA levels in TRExBJAB-RTA cells having the same treatment. This phenomenon was also observed at the protein level; the protein levels of MARCH 8 were upregulated about 10-fold at day 3 in TRExBCBL1-RTA cells induced with tetracycline. This was compared to an observation of a 4-fold increase of MARCH8 protein levels in TRExBJAB-RTA cells with the same treatment (Fig. 4A and B). This difference may be due to the difference in genomic background between BCBL-1 and BJAB cells. BCBL-1 is a KSHV-positive cell line, so RTA expression in this cell line will result in KSHV lytic activation. It is also possible that other KSHV genes may transcriptionally upregulate MARCH8.
FIG 4.

RTA upregulated MARCH8. (A) TRExBJAB-RTA cells were induced with tetracycline for 0, 1, 2, and 3 days. RNA was isolated, and RT-PCR was performed. The relative levels of MARCH8 and MARCH9 were normalized to that of GAPDH (left and middle panels). Western blot analysis was used to detect the RTA and MARCH8 expression (right panel). Values represent the intensities of proteins normalized to that of GAPDH and compared to the signal obtained at day 0. (B) TRExBCBL1-RTA cells were induced by tetracycline for the indicated times. RNA was isolated, and RT-PCR was performed. The relative levels of MARCH8 and MARCH9 were normalized to that of GAPDH (left and middle panels). Western blot analysis was used to detect the RTA and MARCH8 expression levels (right panel). Values represent the intensities of MARCH8 proteins and were normalized against that of GAPDH and compared to the signal obtained at day 0. (C) BC3 and BCBL1 cells were reactivated using chemical inducer TPA and butyric acid for 0, 1, 2, and 3 days. RNA was isolated, and RT-PCR was performed. The relative levels of MARCH8 and RTA were normalized to that of GAPDH (top and lower right panels). Western blot analysis was used to detect RTA and MARCH8 expression (bottom panel). Values which represent the intensities of MARCH8 proteins were normalized against that of GAPDH and compared to the signal obtained at day 0. (D) RTA binds to the MARCH8 promoter. The schematic at the top shows the region of the MARCH8 promoter and the specific region targeted by the designed primers. Fifty million BCBL1 cells were treated with TPA and butyric acid for 24 h. Cells were harvested, and ChIP assays were performed as described in Materials and Methods using mouse IgG (mIgG) and anti-RTA antibodies; the cells were subjected to RT-PCR analysis using the primers described in Materials and Methods (P1 to P4). The DNA fragments after sonication and the immunoprecipitated RTA are shown to the right of the bar graph. 1K represents 1,000. (E) SAOS-2 and BJAB cells were cotransfected with 0, 10, and 20 μg RTA and 10 μg Flag-MARCH8. At 48 h posttransfection, the cells were lysed and subjected to WB analysis with RTA, Flag, and GAPDH antibodies.
To further confirm the role of RTA in MARCH8 upregulation, BCBL1 and BC3 cells were induced by TPA and butyric acid to lytic replication. BCBL1 and BC3 cells were harvested at the indicated days after induction (Fig. 4C). Expression levels of RTA and MARCH8 transcripts were quantitated by RT-PCR. MARCH8 expression at the mRNA and protein levels was induced upon increased RTA expression due to reactivation (Fig. 4C). To further confirm that RTA can transcriptionally upregulate MARCH8, a ChIP assay was performed (Fig. 4D). The chromatin was sonicated to an average length of 500 bp, as confirmed by DNA agarose gel electrophoresis (Fig. 4D, lower right). RTA protein from lysates was immunoprecipitated using the RTA specific antibody (Fig. 4D, lower right), and DNA fragments associated with the immunoprecipitated complex were purified and analyzed by RT-PCR using the designed primers (Fig. 4D). The results of the ChIP assay showed that RTA can bind to the promoter region between the region from bp −1,000 (−1K) to bp −500 (−0.5K) upstream from the TSS (transcription start site) of MARCH8 (Fig. 4D). To investigate whether ectopic expression of RTA could also affect the expression of MARCH8 at the protein level, SAOS-2 and BJAB cells were cotransfected with increasing amounts of Myc-RTA plasmid together with a constant amount of Flag-MARCH8 plasmid. The increased ectopic expression of RTA did not affect the expression of MARCH8 at the protein level (Fig. 4E). Together, these results showed that RTA upregulates MARCH8 through association with the MARCH8 promoter to transcriptionally activate MARCH8.
RTA downregulates HLA-DRα through the MARCH8 pathway.
The coimmunoprecipitation and reverse coimmunoprecipitation assays performed in SAOS-2 cells showed a complex formed between HLA-DRα and MARCH8 (Fig. 5A). These data further confirmed previous results showing that HLA-DRα is the substrate of MARCH8 (48). Our above-described data showed that RTA can transcriptionally upregulate MARCH8. Therefore, we hypothesized that RTA may indirectly mediate the downregulation of HLA-DRα through the MARCH8 E3 ligase. To test this hypothesis, we performed a ubiquitination assay in BJAB shMARCH8 cells and its control BJAB shCT cells (Fig. 5B). We found that ubiquitination of HLA-DRα mediated by RTA is reduced in a MARCH8 knockdown cell line (Fig. 5B, compare lanes 2 and 4). These results suggest that MARCH8 can partially contribute to the ubiquitination of HLA-DRα. Since the ubiquitination of proteins typically leads to protein degradation through the proteasome degradation pathway, we wondered whether the reduction in ubiquitination of HLA-DRα in the MARCH8 knockdown cells can also partially rescue the protein levels of HLA-DRα in the presence of RTA (Fig. 5C). Western blot results showed that knockdown of MARCH8 partially rescued the protein level of HL-DRα in the presence of RTA (Fig. 5C, compare lanes 1, 3, and 4). These results suggest that RTA can downregulate the HLA-DRα level through induction of the MARCH8 E3 ligase.
FIG 5.
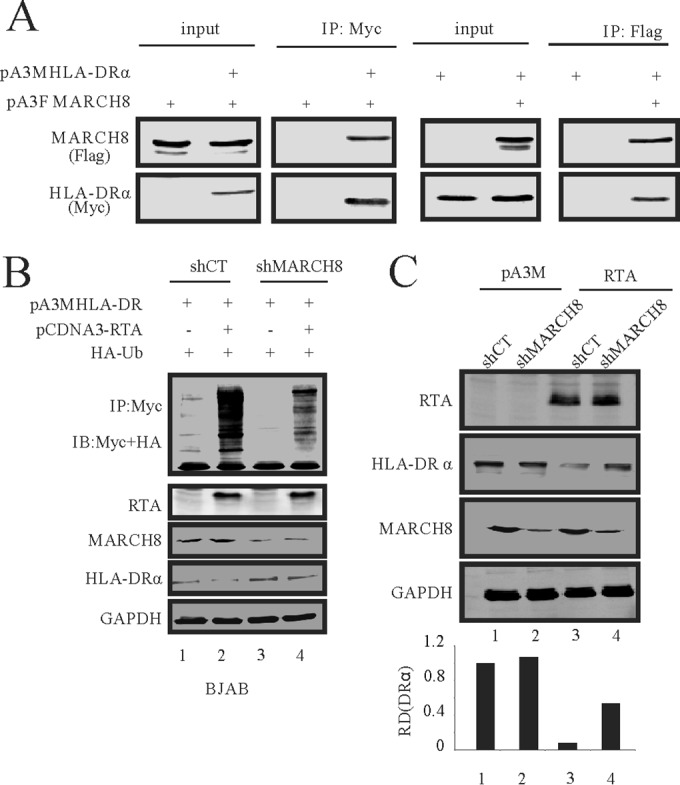
RTA-mediated HLA-DRα downregulation through the MARCH8 pathway. (A) SAOS-2 cells were either transfected with 10 μg pA3M-HLA-DRα alone or cotransfected with 10 μg of pA3F-MARCH8 by electroporation. Thirty-six hours later, the cells were treated with 10 μM MG132 for another 12 h. Cell lysates were then immunoprecipitated with Myc antibodies. Ten percent of the lysates was loaded as input; lysates and IP complexes were resolved by SDS-PAGE and subjected to WB with the indicated antibodies (left panel). SAOS-2 cells were either transfected with 10 μg pA3F-MARCH8 alone or cotransfected with 10 μg pA3M-HLA-DRα by electroporation. Thirty-six hours later, the cells were treated with 10 μM MG132 for another 12 h. Cell lysates were then immunoprecipitated with Flag antibodies. Ten percent of the lysates was loaded as input; lysates and IP complexes were resolved by SDS-PAGE and subjected to WB with the indicated antibodies (right panel). (B) In vivo ubiquitination assays were carried out in BJAB shMARCH8 cells in comparison to a control. Fifteen million cells were transfected with RTA by electroporation. At 36 h posttransfection, the cells were treated with 10 μM MG132. Twelve hours later, the cells were lysed and immunoprecipitated with HLA-DRα. Lysates and IP complexes were resolved by SDS-PAGE and subjected to WB with the indicated antibodies. (C) RTA was transfected into stable MARCH8 knockdown BJAB cells by electroporation. Forty-eight hours later, the cells were lysed and subjected to WB with the indicated antibodies. Ub, ubiquitin.
Expression of HLA-DRα is impaired during KSHV de novo infection.
HLA-DRα is considered to be an intrinsically cytoplasmic protein that is delivered to the cell surface and other organelles after synthesis (49, 50). We queried whether the protein levels of HLA-DRα were reduced during KSHV primary infection, especially during the early stage. For this purpose, 20 million BJAB cells were infected with Bac16-KSHV virus particles and monitored at different days postinfection (Fig. 6A). Subsequently, 1 million cells were harvested and examined by immunofluorescence. The rest of the cells were subjected to Western blot analysis (Fig. 6B). Since RTA is packaged in the KSHV virion and released into host cells during primary infection, we monitored RTA signals during the early stages of KSHV de novo infection at day 1. RTA signals were increased in expression up to 7 days postinfection. This increase in RTA signals corresponded to a gradual decrease in HLA-DRα as well as upregulation of MARCH8 expression in Bac16-KSHV-infected BJAB cells (Fig. 6B). The HLA-DRα and RTA signals observed by immunofluorescence were quantitated using ImageJ software. Interestingly, the levels of HLA-DRα showed a striking reduction as infection proceeded up to 7 days (Fig. 6B). These results further support our hypothesis that RTA can contribute to downregulation of HLA-DRα in KSHV-infected cells during the early stage of de novo infection. Interestingly, the RTA protein expression at day 7 was slightly reduced but did not lead to a rescue in expression of HLA-DRα, as measured by immunofluorescence and Western blot analysis (Fig. 6B). The protein level of MARCH8, however, was depressed at day 7 compared to that at day 5 (Fig. 6B, lower right). This difference may be due to the fact that HLA-DRα is regulated not only by RTA but also by the KSHV antigen LANA, a latency-associated protein which can transcriptionally inhibit MHC-II molecules, including HLA-DRα (42, 45). Similar to previous studies, in this study we showed that exogenously expressed LANA can inhibit the MHC-II molecules, including HLA-DRα in BJAB cells (Fig. 6C, left panel). Furthermore, LANA knockdown in the KSHV PEL cell line BC3 showed rescue of the MHC-II molecules, including HLA-DRα (Fig. 6C, right panel). To further corroborate the role of RTA in the downregulation of HLA-DRα, a similar IFA was performed using TRExBJAB-RTA cells that were treated or mock treated with tetracycline (8 μg/ml) for 48 h (Fig. 6D). As expected, HLA-DRα expression was downregulated and MARCH8 was upregulated in RTA-expressed BJAB cells. Furthermore, quantitation of HLA-DRα signals demonstrated a clear reduction in the tetracycline-induced cells (Fig. 6D). We conclude from these results that RTA can contribute to the downregulation of HLA-DRα in KSHV-infected cells expressing the lytic transactivator RTA.
FIG 6.

Expression of HLA-DRα is impaired during KSHV primary infection. (A) BJAB cells were infected with Bac16-KSHV. All infections were monitored by GFP expression using a fluorescence microscope at 0, 1, 3, 5, and 7 days postinfection (dpi). (B) IFA for BJAB cells infected with Bac16-KSHV. Uninfected and infected cells were harvested and stained with RTA antibodies and HLA-DRα specific antibodies. Nuclei were counterstained by using DAPI. Western blot analysis was used to detect the RTA, HLA-DRα, and MARCH8 expression (lower right). Values represent the intensities of the indicated proteins and were normalized against that of GAPDH and compared to the signal obtained at day 0. (C) Quantitative RT-PCR analysis of MHC-II in BJAB cells transfected with LANA (left panel). Quantitative RT-PCR analysis of MHC-II in LANA stable knockdown BC3 cells (right panel). (D) IFA for TRExBJAB-RTA. Induced or mock-induced cells were harvested and stained with RTA antibodies and HLA-DRα antibodies. Nuclei were counterstained by using DAPI. Western blot analysis was used to detect RTA, HLA-DRα, and MARCH8 expression (right panel). Values represent the intensities of the indicated proteins and were normalized against that of GAPDH and compared to the signal obtained without treatment with tetracycline. (E) Model depicting the role of RTA in the downregulation of HLA-DRα. In KSHV-infected cells, the lytic transactivator RTA is bound to the MARCH8 promoter and transcriptionally upregulates MARCH8. Upregulated MARCH8 can bind to and degrade HLA-DRα. At the same time, RTA can promote ubiquitination of HLA-DRα and downregulate HLA-DRα through the ubiquitin-proteasome degradation pathway.
DISCUSSION
KSHV infection has multiple effects on the MHC-I and -II pathways. K3 and K5 encoded by KSHV induce the internalization and degradation of MHC-I molecules (11, 13, 25). LANA, the major latency antigen, inhibits the expression of MHC-II through binding to the RFX and IRF4 (42, 45). Recently, studies showed that primary effusion lymphoma (PEL) cells lost the capability to be recognized by specific CD4 T cell clones. This was in response to LANA, which is found abundantly in PEL cells. However, if MHC-II molecules can be reintroduced in the PEL cells, these cells can then be recognized by the CD4 T cells (51, 52). This finding strongly suggests that the regulation of MHC-II molecules is important for evasion of the immune surveillance system and might be an important strategy for persistent infection of KSHV. Here we demonstrate a new strategy in that RTA facilitates evasion of the adaptive immune system by KSHV through the degradation of HLA-DRα (Fig. 6E). RTA can bind directly to HLA-DRα and promote its ubiquitination and degradation. In addition to the direct downregulation of HLA-DRα, RTA can also indirectly downregulate HLA-DRα. RTA-induced transcriptional increase of the E3 ligase MARCH8 results in subsequent ubiquitination and degradation of HLA-DRα, a substrate of MARCH8 (48).
The role of RTA in binding and degrading HLA-DRα early after infection is advantageous, as it is already present in the virion particle and does not need to be synthesized before targeting HLA-DRα for degradation (46, 47). RTA can immediately inhibit the MHC-II pathway through degradation of HLA-DRα when released into the host cell after entry of the virus. Our IFA data showed that RTA signals can be abundantly found in the host cell during the early stages of KSHV infection of the B-cell line BJAB, which results in downregulation of HLA-DRα. Coimmunoprecipitation and in vitro GST pulldown assays clearly showed a direct interaction between RTA and HLA-DRα. The in vitro ubiquitination results showed that RTA can function as an E3 ligase to ubiquitinate and degrade HLA-DRα via the ubiquitin-proteasome degradation pathway. Furthermore, the degradation of HLA-DRα results in disruption of the MHC-II pathway.
Studies performed on different isotypes of MHC-II to determine the isotypes of MHC-II regulated by MARCH8 showed that MARCH8 was able to redirect HLA-DRα from exosomes into a degradation pathway in a cell type-dependent manner (53). HLA-DRα was ubiquitinated and degraded by MARCH8, and in the presence of MARCH8, the cell surface levels of HLA-DRα were dramatically reduced (53, 54). Upregulation of MARCH8 also led to impaired development of CD4+ T cells and downregulation of IL-1 receptor accessory protein (IL1RAP), thus preventing subsequent activation of NF-κB (55, 56). These findings strongly suggest that MARCH8 can regulate the adaptive immune system, especially through the MHC-II pathway. We showed that during KSHV infection, RTA can manipulate this pathway. Transcriptional increase of MARCH8 upon RTA expression resulted in the downregulation of its substrate, HLA-DRα, which results in perturbation of the MHC-II pathway.
The present study has identified an important role for RTA in the downregulation of HLA-DRα expression and the upregulation of MARCH8. As an E3 ligase of HLA-DRα, RTA reduces the protein levels of HLA-DRα and also negatively regulates the MHC-II pathway. This property may be necessary for KSHV infection as well as for replication in KSHV-infected cells. Negative regulation of the MHC-II pathway especially through HLA-DRα provides a strategy for bypassing the immune response for escape and survival of KSHV. However, further investigation is needed to explore the detailed molecular mechanism for inhibition of HLA-DRα and upregulation of MARCH8 by RTA, as well as the other potential consequences due to upregulation of MARCH8.
ACKNOWLEDGMENTS
This project was supported by public health service grants R01-CA-171979, R01-CA-177423, P30-DK-050306, and P01-CA-174439 (to Erle S. Robertson). Erle S. Robertson is a scholar of the Leukemia and Lymphoma Society of America.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 3.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280. [PubMed] [Google Scholar]
- 4.McLaughlin-Drubin ME, Munger K. 2008. Viruses associated with human cancer. Biochim Biophys Acta 1782:127–150. doi: 10.1016/j.bbadis.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung MC, Pantanowitz L, Dezube BJ. 2005. AIDS-related malignancies: emerging challenges in the era of highly active antiretroviral therapy. Oncologist 10:412–426. doi: 10.1634/theoncologist.10-6-412. [DOI] [PubMed] [Google Scholar]
- 6.Engels EA, Pfeiffer RM, Fraumeni JF Jr, Kasiske BL, Israni AK, Snyder JJ, Wolfe RA, Goodrich NP, Bayakly AR, Clarke CA, Copeland G, Finch JL, Fleissner ML, Goodman MT, Kahn A, Koch L, Lynch CF, Madeleine MM, Pawlish K, Rao C, Williams MA, Castenson D, Curry M, Parsons R, Fant G, Lin M. 2011. Spectrum of cancer risk among US solid organ transplant recipients. JAMA 306:1891–1901. doi: 10.1001/jama.2011.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee HR, Lee S, Chaudhary PM, Gill P, Jung JU. 2010. Immune evasion by Kaposi's sarcoma-associated herpesvirus. Future Microbiol 5:1349–1365. doi: 10.2217/fmb.10.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coscoy L. 2007. Immune evasion by Kaposi's sarcoma-associated herpesvirus. Nat Rev Immunol 7:391–401. doi: 10.1038/nri2076. [DOI] [PubMed] [Google Scholar]
- 9.Hershko A, Ciechanover A. 1998. The ubiquitin system. Annu Rev Biochem 67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 10.Waldburger JM, Suter T, Fontana A, Acha-Orbea H, Reith W. 2001. Selective abrogation of major histocompatibility complex class II expression on extrahematopoietic cells in mice lacking promoter IV of the class II transactivator gene. J Exp Med 194:393–406. doi: 10.1084/jem.194.4.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishido S, Wang C, Lee BS, Cohen GB, Jung JU. 2000. Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J Virol 74:5300–5309. doi: 10.1128/JVI.74.11.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sirianni MC, Libi F, Campagna M, Rossi D, Capello D, Sciaranghella G, Carbone A, Simonelli C, Monini P, Gaidano G, Ensoli B. 2005. Downregulation of the major histocompatibility complex class I molecules by human herpesvirus type 8 and impaired natural killer cell activity in primary effusion lymphoma development. Br J Haematol 130:92–95. doi: 10.1111/j.1365-2141.2005.05581.x. [DOI] [PubMed] [Google Scholar]
- 13.Brulois K, Toth Z, Wong LY, Feng P, Gao SJ, Ensser A, Jung JU. 2014. Kaposi's sarcoma-associated herpesvirus K3 and K5 ubiquitin E3 ligases have stage-specific immune evasion roles during lytic replication. J Virol 88:9335–9349. doi: 10.1128/JVI.00873-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomazin R, Boname J, Hegde NR, Lewinsohn DM, Altschuler Y, Jones TR, Cresswell P, Nelson JA, Riddell SR, Johnson DC. 1999. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat Med 5:1039–1043. doi: 10.1038/12478. [DOI] [PubMed] [Google Scholar]
- 15.Hegde NR, Tomazin RA, Wisner TW, Dunn C, Boname JM, Lewinsohn DM, Johnson DC. 2002. Inhibition of HLA-DR assembly, transport, and loading by human cytomegalovirus glycoprotein US3: a novel mechanism for evading major histocompatibility complex class II antigen presentation. J Virol 76:10929–10941. doi: 10.1128/JVI.76.21.10929-10941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Odeberg J, Plachter B, Branden L, Soderberg-Naucler C. 2003. Human cytomegalovirus protein pp65 mediates accumulation of HLA-DR in lysosomes and destruction of the HLA-DR alpha-chain. Blood 101:4870–4877. doi: 10.1182/blood-2002-05-1504. [DOI] [PubMed] [Google Scholar]
- 17.Zuo J, Thomas WA, Haigh TA, Fitzsimmons L, Long HM, Hislop AD, Taylor GS, Rowe M. 2011. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathog 7:e1002455. doi: 10.1371/journal.ppat.1002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ressing ME, van Leeuwen D, Verreck FA, Gomez R, Heemskerk B, Toebes M, Mullen MM, Jardetzky TS, Longnecker R, Schilham MW, Ottenhoff TH, Neefjes J, Schumacher TN, Hutt-Fletcher LM, Wiertz EJ. 2003. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc Natl Acad Sci U S A 100:11583–11588. doi: 10.1073/pnas.2034960100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Temme S, Eis-Hubinger AM, McLellan AD, Koch N. 2010. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J Immunol 184:236–243. doi: 10.4049/jimmunol.0902192. [DOI] [PubMed] [Google Scholar]
- 20.Ohmura-Hoshino M, Goto E, Matsuki Y, Aoki M, Mito M, Uematsu M, Hotta H, Ishido S. 2006. A novel family of membrane-bound E3 ubiquitin ligases. J Biochem 140:147–154. doi: 10.1093/jb/mvj160. [DOI] [PubMed] [Google Scholar]
- 21.Boname JM, Lehner PJ. 2011. What has the study of the K3 and K5 viral ubiquitin E3 ligases taught us about ubiquitin-mediated receptor regulation? Viruses 3:118–131. doi: 10.3390/v3020118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goto E, Ishido S, Sato Y, Ohgimoto S, Ohgimoto K, Nagano-Fujii M, Hotta H. 2003. c-MIR, a human E3 ubiquitin ligase, is a functional homolog of herpesvirus proteins MIR1 and MIR2 and has similar activity. J Biol Chem 278:14657–14668. doi: 10.1074/jbc.M211285200. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Ye Y, Lencer W, Hansen TH. 2006. The viral E3 ubiquitin ligase mK3 uses the Derlin/p97 endoplasmic reticulum-associated degradation pathway to mediate down-regulation of major histocompatibility complex class I proteins. J Biol Chem 281:8636–8644. doi: 10.1074/jbc.M513920200. [DOI] [PubMed] [Google Scholar]
- 24.Bartee E, Mansouri M, Hovey Nerenberg BT, Gouveia K, Fruh K. 2004. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J Virol 78:1109–1120. doi: 10.1128/JVI.78.3.1109-1120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehner PJ, Hoer S, Dodd R, Duncan LM. 2005. Downregulation of cell surface receptors by the K3 family of viral and cellular ubiquitin E3 ligases. Immunol Rev 207:112–125. doi: 10.1111/j.0105-2896.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 26.Haque M, Chen J, Ueda K, Mori Y, Nakano K, Hirata Y, Kanamori S, Uchiyama Y, Inagi R, Okuno T, Yamanishi K. 2000. Identification and analysis of the K5 gene of Kaposi's sarcoma-associated herpesvirus. J Virol 74:2867–2875. doi: 10.1128/JVI.74.6.2867-2875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang H, Gwack Y, Kingston D, Souvlis J, Liang X, Means RE, Cesarman E, Hutt-Fletcher L, Jung JU. 2005. Activation of CD21 and CD23 gene expression by Kaposi's sarcoma-associated herpesvirus RTA. J Virol 79:4651–4663. doi: 10.1128/JVI.79.8.4651-4663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehrlich ES, Chmura JC, Smith JC, Kalu NN, Hayward GS. 2014. KSHV RTA abolishes NFkappaB responsive gene expression during lytic reactivation by targeting vFLIP for degradation via the proteasome. PLoS One 9:e91359. doi: 10.1371/journal.pone.0091359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu Y, Wang SE, Hayward GS. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59–70. doi: 10.1016/j.immuni.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Jones M, McCabe A, Winslow GM, Avram D, MacNamara KC. 2013. MyD88 signaling in CD4 T cells promotes IFN-gamma production and hematopoietic progenitor cell expansion in response to intracellular bacterial infection. J Immunol 190:4725–4735. doi: 10.4049/jimmunol.1203024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colpitts SL, Stoklasek TA, Plumlee CR, Obar JJ, Guo C, Lefrancois L. 2012. Cutting edge: the role of IFN-alpha receptor and MyD88 signaling in induction of IL-15 expression in vivo. J Immunol 188:2483–2487. doi: 10.4049/jimmunol.1103609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Briere F. 2005. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol 175:6723–6732. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]
- 34.Hirano H, Yoshioka T, Yunoue S, Fujio S, Yonezawa H, Niiro T, Habu M, Oyoshi T, Sugata S, Kamezawa T, Arimura H, Hanaya R, Tokimura H, Tokudome M, Arita K. 2012. TLR4, IL-6, IL-18, MyD88 and HMGB1 are highly expressed in intracranial inflammatory lesions and the IgG4/IgG ratio correlates with TLR4 and IL-6. Neuropathology 32:628–637. doi: 10.1111/j.1440-1789.2012.01310.x. [DOI] [PubMed] [Google Scholar]
- 35.Xu P, Sun Z, Wang Y, Miao C. 2015. Long-term use of indomethacin leads to poor prognoses through promoting the expression of PD-1 and PD-L2 via TRIF/NF-kappaB pathway and JAK/STAT3 pathway to inhibit TNF-alpha and IFN-gamma in hepatocellular carcinoma. Exp Cell Res 337:53–60. doi: 10.1016/j.yexcr.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Gais P, Tiedje C, Altmayr F, Gaestel M, Weighardt H, Holzmann B. 2010. TRIF signaling stimulates translation of TNF-alpha mRNA via prolonged activation of MK2. J Immunol 184:5842–5848. doi: 10.4049/jimmunol.0902456. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Q, Liang D, Sun R, Jia B, Xia T, Xiao H, Lan K. 2015. Kaposi's sarcoma-associated herpesvirus-encoded replication and transcription activator impairs innate immunity via ubiquitin-mediated degradation of myeloid differentiation factor 88. J Virol 89:415–427. doi: 10.1128/JVI.02591-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahmad H, Gubbels R, Ehlers E, Meyer F, Waterbury T, Lin R, Zhang L. 2011. Kaposi sarcoma-associated herpesvirus degrades cellular Toll-interleukin-1 receptor domain-containing adaptor-inducing beta-interferon (TRIF). J Biol Chem 286:7865–7872. doi: 10.1074/jbc.M110.191452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun Z, Jha HC, Robertson ES. 2015. Bub1 in complex with LANA recruits PCNA to regulate Kaposi's sarcoma-associated herpesvirus latent replication and DNA translesion synthesis. J Virol 89:10206–10218. doi: 10.1128/JVI.01524-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Z, Xiao B, Jha HC, Lu J, Banerjee S, Robertson ES. 2014. Kaposi's sarcoma-associated herpesvirus-encoded LANA can induce chromosomal instability through targeted degradation of the mitotic checkpoint kinase Bub1. J Virol 88:7367–7378. doi: 10.1128/JVI.00554-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Z, Ren H, Liu Y, Teeling JL, Gu J. 2011. Phosphorylation of RIG-I by casein kinase II inhibits its antiviral response. J Virol 85:1036–1047. doi: 10.1128/JVI.01734-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai Q, Banerjee S, Cervini A, Lu J, Hislop AD, Dzeng R, Robertson ES. 2013. IRF-4-mediated CIITA transcription is blocked by KSHV encoded LANA to inhibit MHC II presentation. PLoS Pathog 9:e1003751. doi: 10.1371/journal.ppat.1003751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu J, Verma SC, Cai Q, Saha A, Dzeng RK, Robertson ES. 2012. The RBP-Jkappa binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog 8:e1002479. doi: 10.1371/journal.ppat.1002479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu J, Jha HC, Verma SC, Sun Z, Banerjee S, Dzeng R, Robertson ES. 2014. Kaposi's sarcoma-associated herpesvirus-encoded LANA contributes to viral latent replication by activating phosphorylation of survivin. J Virol 88:4204–4217. doi: 10.1128/JVI.03855-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thakker S, Purushothaman P, Gupta N, Challa S, Cai Q, Verma SC. 2015. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen inhibits major histocompatibility complex class II expression by disrupting enhanceosome assembly through binding with the regulatory factor X complex. J Virol 89:5536–5556. doi: 10.1128/JVI.03713-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bechtel JT, Winant RC, Ganem D. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol 79:4952–4964. doi: 10.1128/JVI.79.8.4952-4964.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lan K, Kuppers DA, Verma SC, Sharma N, Murakami M, Robertson ES. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J Virol 79:7453–7465. doi: 10.1128/JVI.79.12.7453-7465.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bartee E, Eyster CA, Viswanathan K, Mansouri M, Donaldson JG, Fruh K. 2010. Membrane-associated RING-CH proteins associate with Bap31 and target CD81 and CD44 to lysosomes. PLoS One 5:e15132. doi: 10.1371/journal.pone.0015132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gruhler A, Fruh K. 2000. Control of MHC class I traffic from the endoplasmic reticulum by cellular chaperones and viral anti-chaperones. Traffic 1:306–311. doi: 10.1034/j.1600-0854.2000.010403.x. [DOI] [PubMed] [Google Scholar]
- 50.Joyce S. 1997. Traffic control of completely assembled MHC class I molecules beyond the endoplasmic reticulum. J Mol Biol 266:993–1001. doi: 10.1006/jmbi.1996.0822. [DOI] [PubMed] [Google Scholar]
- 51.Sabbah S, Jagne YJ, Zuo J, de Silva T, Ahasan MM, Brander C, Rowland-Jones S, Flanagan KL, Hislop AD. 2012. T-cell immunity to Kaposi sarcoma-associated herpesvirus: recognition of primary effusion lymphoma by LANA-specific CD4+ T cells. Blood 119:2083–2092. doi: 10.1182/blood-2011-07-366476. [DOI] [PubMed] [Google Scholar]
- 52.Fan W, Bubman D, Chadburn A, Harrington WJ Jr, Cesarman E, Knowles DM. 2005. Distinct subsets of primary effusion lymphoma can be identified based on their cellular gene expression profile and viral association. J Virol 79:1244–1251. doi: 10.1128/JVI.79.2.1244-1251.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lapaque N, Jahnke M, Trowsdale J, Kelly AP. 2009. The HLA-DRalpha chain is modified by polyubiquitination. J Biol Chem 284:7007–7016. doi: 10.1074/jbc.M805736200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohmura-Hoshino M, Matsuki Y, Aoki M, Goto E, Mito M, Uematsu M, Kakiuchi T, Hotta H, Ishido S. 2006. Inhibition of MHC class II expression and immune responses by c-MIR. J Immunol 177:341–354. doi: 10.4049/jimmunol.177.1.341. [DOI] [PubMed] [Google Scholar]
- 55.Toyomoto M, Ishido S, Miyasaka N, Sugimoto H, Kohsaka H. 2011. Anti-arthritic effect of E3 ubiquitin ligase, c-MIR, expression in the joints. Int Immunol 23:177–183. doi: 10.1093/intimm/dxq470. [DOI] [PubMed] [Google Scholar]
- 56.Chen R, Li M, Zhang Y, Zhou Q, Shu HB. 2012. The E3 ubiquitin ligase MARCH8 negatively regulates IL-1beta-induced NF-kappaB activation by targeting the IL1RAP coreceptor for ubiquitination and degradation. Proc Natl Acad Sci U S A 109:14128–14133. doi: 10.1073/pnas.1205246109. [DOI] [PMC free article] [PubMed] [Google Scholar]