

ABSTRACT
High-throughput small interfering RNA (siRNA) screening is a useful methodology to identify cellular factors required for virus replication. Here we utilized a high-throughput siRNA screen based on detection of a viral antigen by microscopy to interrogate cellular protein kinases and phosphatases for their importance during human cytomegalovirus (HCMV) replication and identified the class II phosphatidylinositol 3-kinase class II alpha (PI3K-C2A) as being involved in HCMV replication. Confirming this observation, infected cells treated with either pooled or individual siRNAs targeting PI3K-C2A mRNA produced approximately 10-fold less infectious virus than the controls. Western blotting and quantitative PCR analysis of infected cells treated with siRNAs indicated that depletion of PI3K-C2A slightly reduced the accumulation of late but not immediate early or early viral antigens and had no appreciable effect on viral DNA synthesis. Analysis of siRNA-treated cells by electron microscopy and Western blotting indicated that PI3K-C2A was not required for the production of viral capsids but did lead to increased numbers of enveloped capsids in the cytoplasm that had undergone secondary envelopment and a reduction in the amount of viral particles exiting the cell. Therefore, PI3K-C2A is a factor important for HCMV replication and has a role in the production of HCMV virions.
IMPORTANCE There is limited information about the cellular factors required for human cytomegalovirus (HCMV) replication. Therefore, to identify proteins involved in HCMV replication, we developed a methodology to conduct a high-throughput siRNA screen of HCMV-infected cells. From our screening data, we focused our studies on the top hit from our screen, the lipid kinase phosphatidylinositol 3-kinase class II alpha (PI3K-C2A), as its role in HCMV replication was unknown. Interestingly, we found that PI3K-C2A is important for the production of HCMV virions and is involved in virion production after secondary envelopment of viral capsids, the encapsidation of HCMV capsids by a lipid bilayer that occurs before virions exit the cell.
INTRODUCTION
Identification of factors encoded by the cell that are required for virus replication can illuminate important features of virus-host interactions and identify novel drug targets for therapeutic intervention. Stages of productive human cytomegalovirus (HCMV) replication take place in both the nucleus and the cytoplasm (1). After replication of the viral DNA genome in the nucleus, newly synthesized viral genomic DNA is packaged into nascent capsids in the nucleus. These capsids then bud through the nuclear membrane and, after accessing a viral assembly compartment in the cytoplasm (2), undergo a process of secondary envelopment in the cytoplasm in which capsids gain a lipid bilayer before exiting the cell (1). Many of these processes require the function of cellular factors, a number of which are unknown.
High-throughput small interfering RNA (siRNA) screening has been a successful strategy to identify cellular factors important for replication of several viruses (3–9). Many of the factors that have been identified by this strategy are kinases that are involved in a diverse range of cellular processes (3–9). Cellular protein, nucleotide, or lipid kinases are involved in many aspects of HCMV replication (1). For example, cellular protein kinases are involved in the intracellular signaling required for activation of viral transcription (10, 11) and many other processes (1). The roles of cellular nucleotide kinases in HCMV replication are less well characterized, but they are likely to be important for HCMV DNA synthesis, as they are involved in nucleotide metabolism. Another form of kinase protein that must be considered are lipid kinases, proteins that phosphorylate the inositol ring of phosphatidylinositol (PtdIns). Only a few investigations have examined whether lipid kinases are involved in HCMV replication. Class I phosphatidylinositol 3-kinase (PI3K) is important for the intracellular signaling involved in HCMV replication (12), and the class III PI3K Vps34 is required for secondary envelopment to occur (13). However, the role of class II PI3K proteins in HCMV replication is unknown.
High-throughput siRNA screens targeting human kinase and metabolic proteins have been performed and have highlighted the involvement of many cellular proteins and pathways in HCMV replication, notably, the cellular metabolic pathways involving 5′-AMP-activated protein kinase (AMPK) (8, 9). However, studies of multiple siRNA screens against a common target demonstrate that different siRNA screens can produce different outcomes due to false positives, false negatives, and the efficiency of siRNAs utilized in different experiments (7). Therefore, it is possible that any number of kinase proteins that are required for HCMV replication have yet to be identified.
To identify the cellular kinases required for HCMV replication, we developed a high-throughput siRNA screen based on the detection of a viral antigen in infected cells by microscopy. From this screen, we found that several unrelated kinase proteins scored as hits, including the phosphatidylinositol 3-kinase class II alpha (PI3K-C2A) protein, which we examined further.
MATERIALS AND METHODS
Cells and viruses.
Human foreskin fibroblasts (HFFs; clone Hs29) were obtained from the American Type Culture Collection (ATCC; CRL-1684; Manassas, VA). All cells were maintained in complete medium: Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 5% fetal bovine serum (FBS; Gibco) plus penicillin and streptomycin. Unless indicated otherwise, HCMV strain AD169 was used. HCMV strain Merlin (RCMV1111) (14) was a kind gift from Richard Stanton (Cardiff University).
Transfection and infection of cells for high-throughput screening.
The Dharmacon SMARTpool kinase-phosphatase collection (catalog number G-003500) comprises 789 siRNA targets (see Table S1 in the supplemental material) and was screened in triplicate at the Institute of Chemistry and Chemical Biology-Longwood facility at Harvard Medical School. Each pool contains 4 individual siRNAs. At 24 h before transfection, 1,000 HFFs were seeded in each well of each Corning 384-well plate in complete medium with no antibiotics. Unless stated otherwise, liquid was added to wells using a WellMate apparatus. At the time of transfection, an intermediate plate (384-well plate; catalog number 4309; Thermo Scientific) was prepared to mix siRNA with lipid. Thus, 8.5 μl of each siRNA at a concentration of 1 μM was added to the intermediate plate along with 7 μl of Opti-MEM medium (Gibco). The intermediate plate was incubated at room temperature for 5 min. During this incubation period a lipid–Opti-MEM medium mix was prepared (0.17 μl DharmaFECT 2 lipid [Dharmacon] plus 11 μl Opti-MEM medium per well) and incubated for no more than 5 min at room temperature. The lipid–Opti-MEM medium mix (11.17 μl) was added to each well of the intermediate plate, and the plate was incubated for a further 20 min at room temperature. Medium was removed from plates containing HFFs with a suction manifold, and 8 μl of the siRNA-lipid mix from the intermediate plate was added to triplicate plates using a liquid-handling robot. Thirty microliters of complete medium without antibiotics was added to the transfected cells in each well. In each plate, the wells used for 6 negative and 6 positive controls were transfected with SMARTpool nontargeting siRNA number 3 (control [Ctrl] siRNA; catalog number D-001810-03-05) or SMARTpool human polo-like kinase 1 (PLK-1; catalog number M-003290-01), respectively. Where indicated, individual PI3K-C2A SMARTpool siRNAs (PLK-1; catalog number LU-006771-00-0005) were used in 4 wells of a plate. Control and individual siRNAs were added to the intermediate plates by hand. Transfected cells were incubated for 72 h at 37°C. The cells were then infected with HCMV strain AD169 (multiplicity of infection [MOI], 1) in a total volume of 20 μl for 2 h at 37°C. The inoculum was removed, and the cells were incubated for a further 72 h at 37°C. The plates were then analyzed by microscopy to assess HCMV replication.
Preparation of screening plates for high-throughput microscopy analysis.
Cell culture medium was removed from infected cells and replaced with 20 μl Hoechst 33342 stain (Sigma) diluted in phosphate-buffered saline (PBS) to a final concentration of 10 μg/ml. After incubation for 1 h at 37°C, 20 μl of Deep Red CellMask (Invitrogen) (diluted in PBS to a concentration of 5 μg/ml) was added to each well. The cells were incubated for a further 5 min at 37°C. The cells were then fixed by removing the PBS containing Hoechst stain and CellMask and adding 50 μl of 3.5% formaldehyde (Sigma) in PBS to each well. After incubating at room temperature for 10 min, the fixative was removed and 50 μl of PBS containing 0.5% Triton X-100 was added per well to permeabilize the cells. After 10 min of incubation at room temperature, the PBS containing the detergent was removed and the cells were washed once with PBS. The PBS was removed and replaced with 20 μl monoclonal antibody P207 recognizing pp28 (dilution, 1:1,000; Virusys) and anti-mouse immunoglobulin secondary antibody conjugated to the fluorophore Alexa Fluor 488 (dilution, 1:1,000; Molecular Probes). The plates were incubated at 37°C for 1 h. After incubation, the PBS containing the antibodies was removed and replaced with 50 μl of PBS. The plates were then analyzed by microscopy.
Microscopy analysis of screening plates.
Infected cells stained with antibody to detect pp28 were imaged on an Image Express Micro (IXM) microscope (Molecular Devices) at a ×10 magnification to detect 3 wavelengths: 488 nm to detect antibody recognizing pp28, 568 nm to detect Deep Red CellMask, and 350 nm to detect Hoechst 33342 stain bound to DNA. Three images were captured from each wavelength in each well of the 384-well plates. The number of cells positive for all 3 wavelengths and the percentage of pp28-positive cells in each well were determined by analyzing the presence of the signal from each wavelength using MetaMorph multiwavelength cell scoring software (Molecular Devices). Typically, the average proportion of pp28-positive cells found when cells were treated with nontargeting siRNA was 30 to 40% (data not shown).
Analysis of screening results.
To assess the quality of the data that could be returned from the screening protocol, we calculated the Z′ factor (15, 16) derived from the positive controls (PLK-1-specific siRNA-treated infected cells) and the negative controls (nontargeting siRNA number 3-infected cells). The screening controls returned Z′ factors of greater than or equal to 0.5, indicating a robust separation of the difference in the data derived from the positive and negative controls. Thus, the screening protocol could be reliably used to screen the siRNA collection.
After screening of the siRNA collection, the data were analyzed to address siRNA cytotoxicity. siRNAs were judged to have a cytotoxic effect when the number of cells stained with Hoechst 33342 in a well fell below 2-fold of the mean number of cells in each well of the plate. Data from those wells containing cytotoxic siRNA were discarded. The data from the remaining wells from each plate were converted to a Z-score (15, 16), and the average Z-score from data in triplicate plates was determined. Images chosen at random were visually inspected throughout image capture and analysis to ensure that the raw data were consistent with the Z-scores.
GESS and Haystack analysis of siRNA binding.
The on-line version of genome-wide enrichment of seed sequences (GESS) (17, 18) was used. For Haystack (19) analysis of top screening hits, Haystack was downloaded from http://rnai.nih.gov/haystack/. siRNA sequences were compared to the sequences of the 3′ untranslated regions (3′ UTRs) of human mRNA provided by Haystack. A list of viral 3′ UTRs (300 nucleotides) was generated from the HCMV AD169 genome, and their sequences were compared to the siRNA sequences. All siRNA sequences were supplied by Dharmacon.
Kinase inhibitors.
Compound C was purchased from Merck and resuspended in dimethyl sulfoxide (DMSO).
Preparation of cell lysates for Western blotting.
In experiments to detect PI3K-C2A, 1 × 105 HFFs were washed twice with ice-cold PBS and immediately scraped into 100 ml of lysis buffer (50 mM Trizma base, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, 10% glycerol, 1 mM sodium fluoride, 2.5 mM sodium pyrophosphate, 50 mM β-glycerophosphate, 1 mmol/liter sodium orthovanadate, and 40 mg/liter phenylmethylsulfonyl fluoride plus 1 Halt protease inhibitor tablet [catalog number 1860932; Thermo Scientific] per 100 ml). The lysate was incubated on ice for 30 min and then centrifuged for 5 min at 12,700 rpm. The supernatant was removed and diluted 1:1 in 2× Laemmli buffer containing 5% β-mercaptoethanol. Samples were incubated to 60°C for 10 min.
All other samples were prepared for Western blotting by washing cells once in PBS and then suspending the cells directly in 2× Laemmli buffer containing 5% β-mercaptoethanol before incubation at 95°C for 5 min.
Western blotting.
Western blotting of proteins separated on 8% or 10% polyacrylamide gels was carried out as described elsewhere (20), using antibodies recognizing IE1/2, UL44, and pp28 (1:1,000 dilution; all from Virusys), β-actin (1:5,000 dilution; Sigma), UL86 (1:1,000 dilution; a kind gift from Wade Gibson, Johns Hopkins University), gB (1:1,000 dilution; catalog number F3-11E; NIH AIDS Reagents Program,) and PI3K-C2A (1:1,000 dilution; catalog number 611046; BD Biosciences). All primary antibodies were incubated overnight at 4°C and detected using anti-mouse- or anti-rabbit immunoglobulin horseradish peroxidase (HRP)-conjugated antibodies (Southern Biotech). Chemiluminescence solution (GE Healthcare) (or, when assaying for PI3K-C2A, Femto detection reagent [Thermo]) were used to detect secondary antibodies on film. Where indicated below, relative band intensities were calculated using ImageJ software, provided by the National Institutes of Health, USA.
Transfection of siRNA into HFFs.
Briefly, 1 × 105 HFFs per well were seeded in 12-well plates 24 h before transfection in DMEM–5% FBS with no antibiotics. Per well, 113 μl of 1 μM siRNA and 2 μl Dharmafect2 (Dharmacon) were diluted in 93 μl and 146 μl Opti-MEM medium (Invitrogen), respectively. After 5 min at room temperature, both solutions were combined. After 20 min, medium was removed from each well and replaced with the siRNA-Dharmafect mixture and 500 μl of DMEM–5% FBS with no antibiotics was added to each well. Transfected cells were incubated at 37°C for 72 h and then either prepared for Western blotting or infected with 1 × 105 PFU/ml of AD169.
Determination of viral titer.
Titers were determined by serial dilution of the viral supernatant onto HFF monolayers, which were then covered by DMEM containing 5% FBS and 0.6% methylcellulose. The cultures were incubated for 14 days, the cells were stained with crystal violet, and the plaques were counted.
Real-time quantitative PCR analysis of viral DNA synthesis in siRNA-treated cells.
HFFs were treated with siRNA and infected as outlined above in triplicate. DNA was isolated from infected cells using a NucleoSpin tissue kit (Macherey-Nagel) according to the manufacturer's instructions. Viral genomes were quantified with a primer pair (pp549s and pp812as) specific for UL83 (21), and the number of viral genomes was normalized to the number of cellular copies of adipsin (22). Values for unknown samples were determined on the basis of standard curves of known copy numbers of UL83 (pcDNAUL83; a kind gift from Jeremy Kamil, Louisiana State University) and adipsin (from uninfected cell DNA). PCRs for UL83 and adipsin were carried out on a Bio-Rad CFX96 machine using SsoAdvanced Universal SYBR green Supermix (Bio-Rad) per the manufacturer's instructions. Linear regression analysis of UL83 and adipsin standards in triplicate yielded R2 values of 0.997 and 0.996, respectively. The mean number of copies of the viral gene UL83 per copy of the cellular gene adipsin was calculated from the triplicate samples assayed.
Electron microscopy (EM).
Cells were transfected with siRNA and infected as outlined above. Infected cells were incubated for 1 h at room temperature in fixative (2.5% glutaraldehyde, 1.25% paraformaldehyde, and 0.03% picric acid in 0.1 M sodium cacodylate buffer [pH 7.4]) at the time points postinfection (p.i.) indicated below. Cells were provided to the Harvard Medical School Electron Microscope Facility, washed in 0.1 M sodium cacodylate buffer (pH 7.4), and then postfixed for 30 min in 1% osmium tetroxide (OsO4)–1.5% potassium ferrocyanide (KFeCN), washed in water 3 times, and incubated in 1% aqueous uranyl acetate for 30 min, followed by 2 washes in water and subsequent dehydration in ethanol (50%, 70%, 95%, and twice in 100% ethanol for 5 min each). Cells were removed from the dish and placed in propylene oxide, pelleted at 3,000 rpm for 3 min, and infiltrated for 2 h in a 1:1 mixture of propylene oxide and TAAB Epon (Marivac Canada Inc., St. Laurent, Canada). The samples were subsequently embedded in TAAB Epon and polymerized at 60°C for 48 h. Ultrathin sections (about 60 nm) were cut on a Reichert Ultracut-S microtome, and the sections were placed on copper grids and stained with lead citrate. All samples were examined and images were recorded using a JEOL 1200EX transmission electron microscope and an AMT 2k charge-coupled-device camera, respectively.
Preparation of virions from siRNA-treated cells for Western blotting.
HFFs were transfected with either Ctrl siRNA or siRNA targeting PI3K-C2A (PI3K-C2A siRNA) and infected as outlined above. At 96 h postinfection, viral supernatant from four Ctrl or four PI3K-C2A siRNA transfections was collected (4 ml in total). Supernatants were clarified by centrifugation (13,000 × g, 5 min, 4°C) to remove cells and cell debris. The virions were then pelleted from the supernatants by ultracentrifugation (20,000 rpm, 1 h, 4°C). The pellets were resuspended in 20 μl of phosphate-buffered saline (Gibco). To test for protection from protease digestion, 10 μl of each resuspended pellet was mixed with 10 μl of trypsin (Gibco) and incubated for 1 h at either 4°C or 37°C. After incubation, 20 μl of 2× Laemmli buffer containing 5% β-mercaptoethanol was added to each sample and the mixture was incubated at 95°C for 5 min. For each Western blot, 10 μl of each sample was analyzed.
RESULTS
Development of a high-throughput screening methodology.
To identify siRNAs that can affect HCMV replication, we sought to develop a high-throughput screening methodology. In our initial studies, we attempted to establish a screen in which plates of cells were treated with siRNAs and infected with viruses expressing reporters, such as green fluorescent protein (GFP) or luciferase (data not shown). However, we found that reporter expression was not sufficient to be readily detected in our assay (data not shown), and we found considerable well-to-well variation in the numbers of GFP-positive cells, which may have been due, in part, to the production of particles that initiate abortive infections. These issues were found in all cell lines tested, including human foreskin fibroblasts (HFFs) and U373-MG cells, where additional issues arose due to at least 10-fold lower titers (data not shown). Therefore, we chose not to pursue a screen based on detection of a reporter protein expressed from a recombinant HCMV. Instead, we decided to establish an automated high-throughput screening methodology based on the detection of a viral antigen by microscopy, similar to the siRNA screens created by Koyuncu and coworkers or Brass and coworkers to interrogate what cellular factors are required for replication of HCMV and a variety of other viruses (3–7, 9). In preliminary experiments, we found that antibody staining of HFFs to detect the viral antigen pp28 was a convenient, reproducible, and statistically robust methodology to detect HCMV replication, which, unlike in our preliminary experiments, could be readily detected and did not exhibit the well-to-well variation that would make data interpretation problematic. As pp28 is expressed late in infection, this allowed our screen to identify factors affecting all stages of virus replication through to late gene expression, including virus attachment, entry, transcriptional activation, and genome replication.
High-throughput screening of siRNAs.
We then carried out an siRNA screen, as outlined in Fig. 1A. Briefly, HFFs were transfected in triplicate with siRNAs from the Dharmacon kinase-phosphatase collection (the siRNAs are listed in Table S1 in the supplemental material). This collection of 779 pools of 4 siRNA targets mRNA encoding kinases (including nearly all protein kinases, 7 nucleoside kinases, and 15 lipid kinases) and kinase-related proteins (including 9 dual-specificity phosphatases and proteins that interact with kinases, such as CD4). As negative and positive controls for siRNA treatment, 6 wells in each plate were treated with either Dharmacon nontargeting siRNA number 3 (Ctrl siRNA) or siRNA targeting polo-like kinase 1 (PLK-1) mRNA, respectively. PLK-1 is routinely used as a positive control in siRNA screening, as depletion of PLK-1 induces apoptosis. Under the conditions used here, we found that treatment with PLK-1 siRNA inhibits HCMV replication without a statistically significant decrease in the number of cells per well. Thus, even though this treatment is cytotoxic, it fulfilled the conditions of the screen to serve as a positive control (see Materials and Methods and below). Each control and screened siRNA used contained a pool of 4 individual siRNAs. HFFs were incubated with siRNA for 72 h. In preliminary experiments, 72 h of incubation was required to deplete proteins to levels nearly undetectable by Western blotting after treatment with a number of siRNAs in the Dharmacon kinase-phosphatase collection (data not shown; see also Fig. 3A and C). After incubation, siRNA-transfected HFFs were infected with HCMV strain AD169 at a multiplicity of infection of 1. In preliminary experiments, this MOI provided the most statistically robust data (data not shown). At 72 h postinfection (p.i.), cells were stained with Hoechst 33342 to detect nuclear DNA and CellMask to identify the cell cytoplasm plus treated with antibodies to detect pp28. An automated microscopy system was then used to assay the number of cells in each well and the number of cells containing pp28. An image of infected cells treated as described above was captured using automated microscopy and is shown in Fig. 1B. DNA stained with Hoechst 33342 and cells stained with CellMask are shown in blue and red, respectively. The cytoplasmic localization of pp28 in viral assembly compartments is shown in green.
FIG 1.

High-throughput screening of siRNA. (A) Diagram of high-throughput screening process. (B) A representative example of a microscopy image from an Image Express Micro microscope of HFFs treated with Ctrl siRNA and infected with AD169. Cells were then treated with Hoechst 33342 (blue), Deep Red CellMask (red), and primary and secondary antibodies to detect HCMV pp28 (green). The large white box is an enlarged image of the area identified in the small white box. (C) Z-scores from the siRNA screen. The point that represents the Z-score for PI3K-C2A siRNA is indicated. A full list of siRNAs with Z-scores is shown in Table S1 in the supplemental material.
FIG 3.
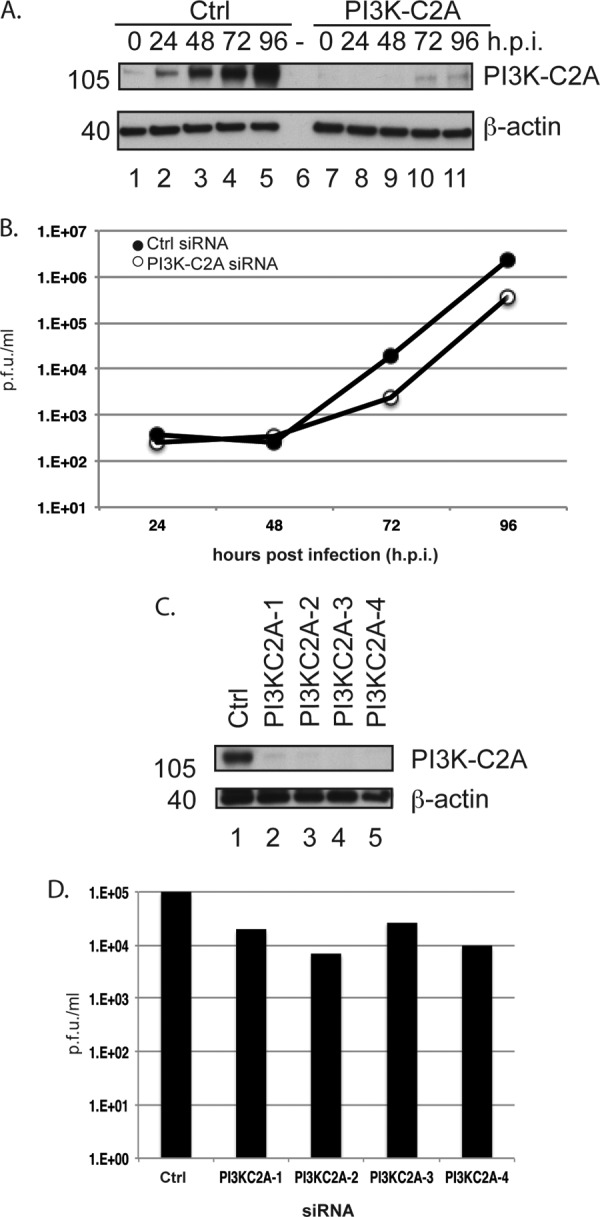
Production of HCMV from cells treated with PI3K-C2A siRNA. HFFs were treated with Ctrl siRNA, PI3K-C2A siRNA, or PI3KC2A-1 to -4 siRNAs and infected with HCMV. (A and B) At the time points (hours postinfection [h.p.i.]) indicated at the top (A) and on the x axis (B), lysates from infected cells treated with PI3K-C2A siRNA were prepared for Western blotting (A) and viral supernatant was titrated to quantify virus produced from siRNA-treated cells (B). The data in panel B are representative of those from two experiments. (C and D) Cells treated with Ctrl or PI3KC2A-1 to -4 siRNAs were infected with HCMV, and the viral supernatants and lysates were harvested at 72 h p.i. Uninfected cell lysate was prepared at the time of infection. Lysates were analyzed by Western blotting (C), and the supernatant was titrated to quantify virus production (D). In panels A and C, proteins recognized by the antibodies used in each experiment are indicated to the right. The positions of molecular mass markers (in kilodaltons) are indicated to the left. In panels B and D, the viral titer is expressed as PFU per milliliter.
We then processed the screening data. Specifically, the mean number of cells in each well per plate was determined by counting the number of Hoechst-stained nuclei. Where the number of cells in any well was less than 2-fold below the mean number of cells for the plate, the siRNA in that well was judged to be grossly cytotoxic. Five hundred seventy-two siRNAs (listed in Table S2 in the supplemental material) did not pass this test and were not analyzed further. Data from the remaining 207 wells on triplicate plates were converted to Z-scores (the number of standard deviations from the mean of the data [15, 16]) to demonstrate the positive or negative effect of siRNA on the number of pp28-positive cells detected. These data were then combined to find the mean Z-score of each pool of siRNA (Fig. 1C; see also the list of siRNAs in Table S3 in the supplemental material).
Analysis of siRNAs that have positive and negative effects on HCMV replication.
We adjudged any siRNA that produced a Z-score of between 1 and −1 to have had little or no effect on HCMV replication, whereas siRNAs with Z-scores of −1 to −2 and 1 to 2 had modest negative or positive effects on HCMV replication, respectively. However, siRNAs with Z-scores of less than −2 or more than 2 were deemed to have strong negative or positive effects on HCMV replication, respectively. Twenty-five siRNAs were found to have modest positive effects on HCMV replication, and 26 siRNAs were found to have modest negative effects on HCMV replication. No siRNA was found to have a strong positive effect on HCMV replication, whereas 4 siRNAs (siRNAs targeting mRNA encoding PI3K-C2A, CD4, EXOSC10, and WNK4) had strong negative effects on HCMV replication (Fig. 1C; see also the list of siRNAs in Table S3 in the supplemental material).
Bioinformatics analysis of siRNA screening results to assay for off-target effects of siRNA treatment.
False-positive results can be found in siRNA screens due to off-target binding of siRNAs. Off-target siRNA binding is most likely to occur in the 3′ untranslated region (3′ UTR) of mRNA (24, 25), where siRNAs exhibit microRNA-like properties upon binding of a limited number of bases, akin to a microRNA seed region, to mRNA (24–26). Indeed, it has been proposed that the data returned from some screens are the result of unintentional screening of partial seed sequence matches and not on-target binding of siRNA (26). Therefore, using genome-wide enrichment of seed sequence (GESS) matches (17, 18), we investigated if the siRNAs screened in this study (all 4 siRNAs that constitute the pools of siRNAs used) were enriched with seed sequences that could bind either human 3′ UTRs or full human mRNA transcripts. We found no statistically significant evidence that the siRNAs screened here were enriched with seed sequences that would bind to any human mRNA sequence (data not shown). The binding of siRNA to the sequence of the entire HCMV AD169 genome was also assayed, and it was found that there was no statistically significant evidence for enrichment of the siRNAs assayed with seed sequences that would bind to any sequence in the viral genome (data not shown).
We sought to confirm the results of this analysis by analyzing the binding of siRNAs from the top 4 hits from our screen (siRNAs targeting mRNA encoding PI3K-C2A, CD4, EXOSC10, and WNK4; Fig. 1C) using Haystack (19), another bioinformatic analysis tool that searches for statistically significant matches of siRNA sequences with 3′ UTR transcripts. We found no statistically significant matches between the siRNAs tested and human or HCMV 3′ UTRs (data not shown).
Therefore, the effects of siRNAs judged to be toxic or to have negative or positive effects on HCMV replication in our analysis may not be due to off-target binding of siRNA seed matches to human or HCMV mRNA transcripts.
We then compared hits with Z-scores of less than −1 and greater than 1 to a proteomic data set that lists all viral and cellular proteins detected in HCMV-infected HFFs (27) (see Table S4 in the supplemental material). We found that a number of proteins purportedly depleted by siRNAs in our screen were not found in HCMV-infected HFFs (8 of 30 hits and 12 of 25 hits with negative and positive effects of HCMV replication, respectively.) Therefore, although GESS and Haystack indicate that off-target siRNA binding does not occur, several false positives could be identified in our screening data.
PI3K-C2A siRNAs deplete PI3K-C2A and specifically reduce the numbers of pp28-infected cells.
We then chose to focus our studies on one of the top hits in our screen. PI3K-C2A and EXOSC10, but not CD4 or WNK4, have been found in HFFs infected with HCMV (27) (see Table S4 in the supplemental material). Therefore, CD4 and WNK4 were excluded from further analysis. siRNAs targeting PI3K-C2A mRNA had the greatest negative effect in our screen, and a role for PI3K-C2A in HCMV replication had not, to our knowledge, been previously reported. We therefore decided to focus on the role of PI3K-C2A in HCMV replication. We first examined the effect of each of the 4 individual PI3K-C2A siRNAs (PI3K-C2A-1 to -4) from the PI3K-C2A siRNA pool, which was tested for its ability to inhibit HCMV replication by assaying the cell number and pp28 expression using the scheme shown in Fig. 1A. Compared to the effect of treatment of cells with Ctrl siRNA, treatment of cells with each PI3K-C2A siRNA had no statistically significant effect on cell number (an effect ±10% of that for the DMSO control; Fig. 2A) and resulted in a statistically significant decrease in the number of pp28-expressing cells by at least 45% (Fig. 2B). This confirmed that PI3K-C2A is involved in HCMV replication and implied that the effect of the PI3K-C2A siRNA pool used in our screen was due to the depletion of PI3K-C2A and was not the result of an off-target effect of any one siRNA in the siRNA pool.
FIG 2.
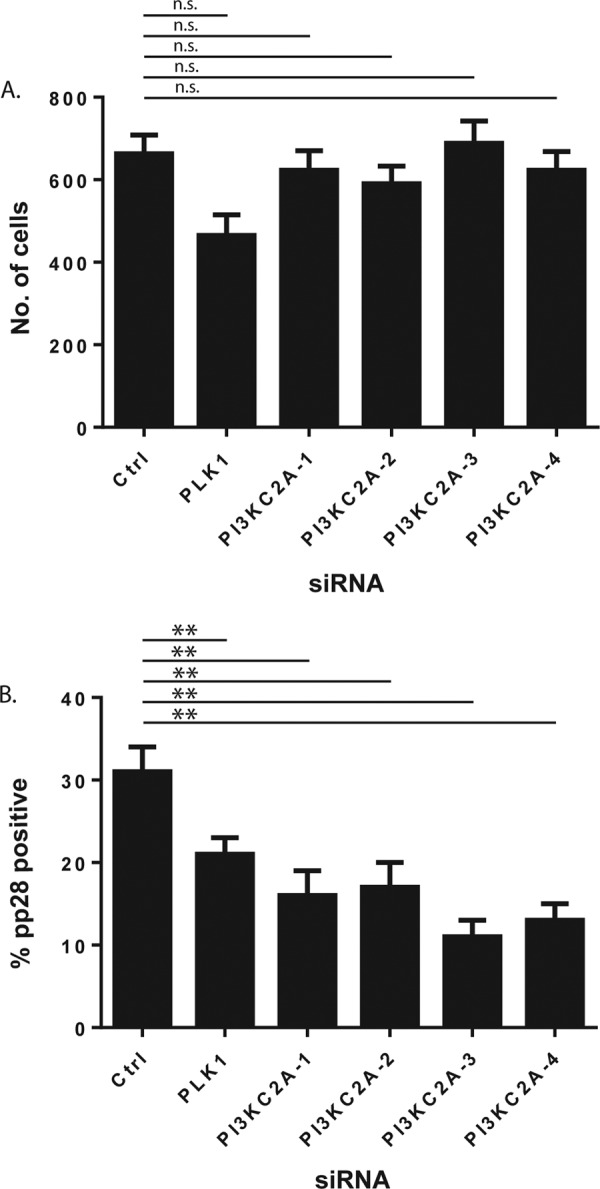
Analysis of individual PI3K-C2A siRNAs. (A and B) HFFs were treated with siRNA, infected, and then analyzed by automated microscopy. The number of cells treated with siRNA (number of Hoechst 33342-positive cells) (A) and the percentage of those cells that were pp28 positive (B) are shown. The mean and standard deviation of data from each of 4 wells treated with siRNA are shown. The significance derived from a two-tailed (unpaired) Student's t test, calculated using Prism software, is indicated at the top of the figure (**, P ≤ 0.05; n.s., no statistically significant difference).
Effect of PI3K-C2A siRNA on production of infectious HCMV.
We then assayed the effects of PI3K-C2A siRNA on the production of infectious virus. HFFs were treated with either Ctrl siRNA, PI3K-C2A siRNA, or the 4 individual PI3KC2A siRNAs from the PI3K-C2A siRNA pool (PI3K-C2A-1 to -4) and infected with HCMV. At various time points, supernatants were collected for titration of infectious virus and infected cells were prepared for Western blotting. Compared to the result of treatment of cells with Ctrl siRNA, treatment of cells with PI3K-C2A siRNA or the individual PI3KC2A siRNAs resulted in a clear decrease in the accumulation of PI3K-C2A in infected cells (Fig. 3A and C) and a decrease in the production of infectious virus over time (9- and 7-fold with PI3K-C2A siRNA at 72 and 96 h p.i., respectively, and 3- to 12-fold with the individual siRNAs; Fig. 3B and D, respectively). In these and subsequent Western blot analyses, the amount of β-actin in each sample was also assayed, and the equivalent loading of samples in each lane was demonstrated. Therefore, depletion of PI3K-C2A in infected cells was associated with a defect in the production of infectious HCMV. Also, a decrease in the production of virus from siRNA-treated cells similar to that seen in Fig. 3B was observed from HFFs treated with either Ctrl or PI3K-C2A siRNA and infected with HCMV strain Merlin (data not shown). Thus, PI3K-C2A is required for the replication of at least two different HCMV strains.
Examination of viral protein and DNA production in cells treated with siRNA.
We next sought to understand how HCMV replication was inhibited by the loss of PI3K-C2A. Therefore, Western blotting was used to analyze the accumulation of immediate early (IE1/IE2), early (UL44), and late (pp28) HCMV proteins and PI3K-C2A in lysates of HFFs treated with either Ctrl or PI3K-C2A siRNA and infected with HCMV (Fig. 4). Treatment of HFFs with PI3K-C2A siRNA resulted in a loss of PI3K-C2A accumulation in HCMV-infected cells (Fig. 4A). However, we found no obvious difference in the accumulation of any viral protein (Fig. 4A). We reasoned that this may be due to high levels of protein produced late in virus replication saturating the signal from our Western blots. Therefore, we created dilution series of the samples taken at 72 and 96 h p.i. from the assay whose results are presented in Fig. 4A and subjected these samples to Western blotting (Fig. 4B). We found no obvious difference in the accumulation of either IE1/2 or UL44. However, consistent with the data in Fig. 1 and 3, we observed an ∼2-fold difference in the accumulation of pp28 in cells treated with PI3K-C2A siRNA from that in cells treated with Ctrl siRNA (Fig. 4B). A similar reduction in the expression of the late viral antigen UL86 was observed (data not shown). However, the loss of viral production (Fig. 3) in the presence of PI3K-C2A siRNA was greater than the modest decrease in pp28 expression that we observed in Fig. 4B. Therefore, depletion of PI3K-C2A likely inhibits the production or function of viral or cellular factors important for productive replication other than pp28.
FIG 4.
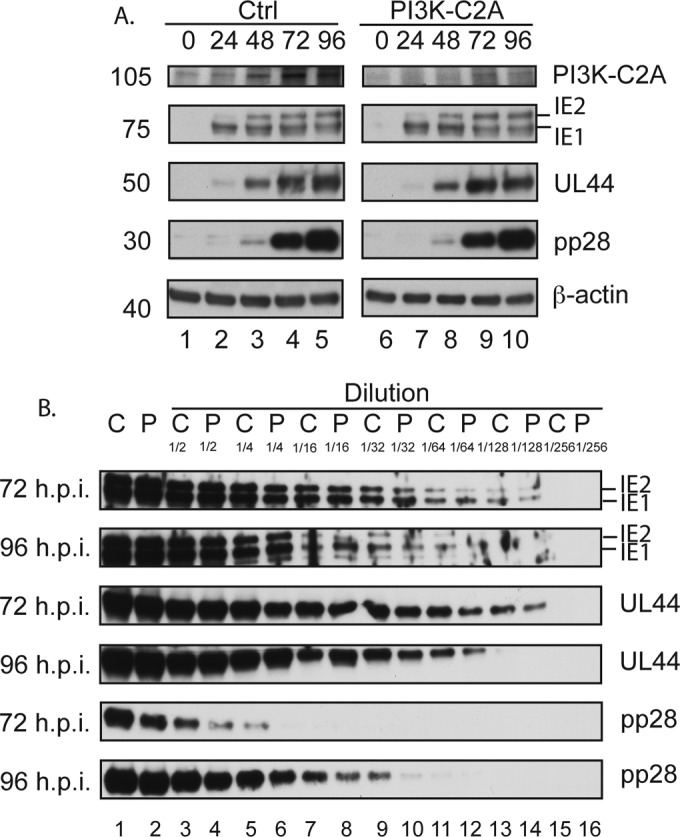
Western blotting of viral and cellular proteins from siRNA-treated cells. (A) HFFs were treated with either Ctrl or PI3K-C2A siRNA and then infected with AD169. Cell lysates were prepared for Western blotting at the time points (hours postinfection) indicated at the top. Uninfected cells harvested at the time of infection are shown as 0 h p.i. Panels showing signals from Ctrl or PI3K-C2A siRNA-treated cells are from the same exposure of the same blot. The positions of molecular mass markers (in kilodaltons) are indicated to the left. (B) A 2-fold dilution series was created from samples of infected cells from the assay whose results are presented in panel A that had been treated with either Ctrl siRNA (lanes C) or PI3K-C2A siRNA (lanes P) and harvested at 72 or 96 h p.i. The siRNA and dilution factor are indicated at the top. In both panels, proteins recognized by the antibodies used in each experiment are indicated to the right.
As HCMV late gene expression is related to viral DNA synthesis (1), we hypothesized that a decrease in the pp28 protein level might reflect a defect in genome replication. Therefore, we treated HFFs with Ctrl or PI3K-C2A siRNA and at 72 h p.i. used quantitative real-time PCR to assay the number of HCMV genomes in infected cells. The number of viral genomes present in each Ctrl or PI3K-C2A siRNA-treated sample was determined by normalizing the copy number of a viral locus (UL83) to the copy number of a cellular locus (adipsin). We found no obvious decrease in the accumulation of viral DNA between infected cells that had been treated with either Ctrl or PI3K-C2A siRNA (1,100 and 1,200 copies UL83 per copy of adipsin for cells treated with Ctrl or PI3K-C2A siRNA, respectively). Therefore, the defect in virus replication and the loss of pp28 expression in the absence of PI3K-C2A were unlikely due to a defect in viral DNA replication.
Analysis of HCMV capsid and virion production in cells treated with PI3K-C2A siRNA.
We then employed electron microscopy (EM) analysis to further investigate how PI3K-C2A is involved in HCMV replication. We treated cells with either Ctrl or PI3K-C2A siRNA and after infection submitted cells for EM analysis and counted the number of viral capsids present in the nucleus and cytoplasm in whole-cell sections of five cells (Fig. 5A and B, respectively). HCMV capsids can be found in 3 forms: A capsids, which are nonproductive forms thought to result from failed packaging of viral genomes; B capsids, which are forms that contain a scaffolding protein but no DNA; and C capsids, which are assembled forms in which the scaffolding protein has been removed and replaced with viral DNA. We therefore also counted the number of each form of HCMV capsid found in siRNA-treated cells (Fig. 5A and B). In both Ctrl and PI3K-C2A siRNA-treated cells approximately 3-fold more capsids were found in the nucleus than in the cytoplasm, and we found no obvious difference in the number of any form of the HCMV capsid in either the nucleus or the cytoplasm. Therefore, the loss of PI3K-C2A did not affect the production of capsids, including genome-containing C capsids that lead to infectious virions or the movement of capsids from the nucleus to the cytoplasm.
FIG 5.

Analysis of capsid localization and virion production. (A to D) Electron microscopy analysis. HFFs were treated with either Ctrl or PI3K-C2A siRNA and then infected with AD169. At 96 h postinfection, cells were prepared for analysis by electron microcopy. Images that cover the entire area of five infected cells from each condition chosen at random were captured at a magnification of ×9,600. The total number of A, B, and C capsids in the nuclei (A) and cytoplasm (B) of infected cells was counted, as were the number of C capsids undergoing secondary envelopment (C) and the number of virions leaving the cell (D). Thin horizontal black bars indicate the mean value for each group. The significance derived from a two-tailed (unpaired) Student's t test, calculated using Prism software, is indicated at the top (**, P < 0.05; ns, no statistically significant difference). (E) (i) Examples of capsids undergoing secondary envelopment (arrows) in a PI3K-C2A siRNA-treated cell; (ii) a virion leaving a cell treated with Ctrl siRNA. (F) Western blotting of viral proteins in the supernatant of infected cells treated with siRNA. HFFs were treated with either control Ctrl or PI3K-C2A siRNA and infected with AD169. Cell supernatant was collected at 96 h postinfection. The supernatant was clarified by low-speed centrifugation, and then the virions were pelleted by ultracentrifugation. Pelleted virions were treated with trypsin at 4°C or 37°C and prepared for Western blotting. Proteins recognized by the antibodies used in each experiment are indicated to the right. The positions of molecular mass markers (in kilodaltons) are indicated to the left. *, a nonspecific band recognized by the gB antibody; arrow, the position of gB.
However, compared to cells treated with Ctrl siRNA, we noted that in cells treated with PI3K-C2A siRNA there was an ∼3-fold increase in the number of cytoplasmic capsids surrounded by lipid bilayer envelopes in the cytoplasm (Fig. 5C). Such particles are thought to have undergone secondary envelopment, a late step in virion maturation prior to egress from the cell. Examples of such enveloped capsids in cells treated with PI3K-C2A siRNA are shown in Fig. 5Ei). Therefore, depletion of PI3K-C2A produced an accumulation of capsids that had undergone secondary envelopment in the cytoplasm, which may reflect a defect in the ability of these particles to egress from the cell.
We noted in our EM analysis that more extracellular virions leaving the cell were found in cells treated with Ctrl siRNA than in those treated with PI3K-C2A siRNA (Fig. 5D). An example of a virion leaving a cell treated with Ctrl siRNA is shown in Fig. 5Eii. Although this difference was not statistically significant, there was a clear trend toward a defect in virion production upon PI3K-C2A depletion. A confounding factor in this analysis could be that it does not account for the number of virions that have left infected cells.
To further investigate virion production from infected cells, we used Western blotting to assay supernatant from infected cells that had been treated with either Ctrl or PI3K-C2A siRNA for the presence of virion proteins (Fig. 5F). Viral supernatants were collected and were first clarified by low-speed centrifugation to remove cells and cellular debris. The virions were then pelleted using ultracentrifugation, and the pellets were treated with trypsin to remove protein not protected by virion membranes. Thus, HCMV glycoprotein B (gB), which is found on the exterior of virions, on cells, or in cell debris or shed into the supernatant could be found in the samples treated with trypsin at 4°C (Fig. 5F, lanes 1 and 3) but not 37°C (Fig. 5F, lanes 2 and 4). The similar amounts of gB found in the supernatants of cells treated with either Ctrl or PI3K-C2A siRNA is consistent with our observations from Fig. 4, wherein depletion of PI3K-C2A has only a modest effect on the production of late viral proteins. The major HCMV virion protein UL86, which is a component of virions and protected from trypsin digestion by the virion membrane, was found in all samples. However, notably less UL86 was found in pellets from cells treated with PI3K-C2A siRNA (Fig. 5F, lanes 3 and 4) than in pellets from cells treated with Ctrl siRNA (Fig. 5F, lanes 1 and 2). Using ImageJ software, we found 4-fold (Fig. 5F, lanes 1 and 3) and 9-fold (Fig. 5F, lanes 2 and 4) decreases in the relative band intensities of UL86 blots of samples treated at 4°C and 37°C, respectively. Therefore, consistent with the data presented in Fig. 5A to E, depletion of PI3K-C2A was associated with a defect in virion production from infected cells.
The accumulation of capsids that have undergone secondary envelopment and a lack of virion production inferred that there was an accumulation of infectious intracellular virus in infected cells treated with PI3K-C2A siRNA. We examined this possibility by treating cells with Ctrl or PI3K-C2A siRNA and assaying the amount of infectious virus released from infected cells and the amount of intracellular virus at 96 h p.i. To release intracellular virus, infected cells were lysed by three sequential freeze-thaw cycles at −80°C. In cells treated with Ctrl or PI3K-C2A siRNA, we observed a 10-fold decrease in the titer of infectious virus released from infected cells (8 × 105 and 8 × 104 PFU/ml, respectively) and a less than a 2-fold difference in the amount of infectious intracellular virus produced (1 × 103 and 2 × 103 PFU/ml, respectively). Similar results were observed when infected cells were lysed by passing cells three times through a needle (data not shown). Therefore, we found no obvious increase in the amount of infectious intracellular virus in infected cells treated with PI3K-C2A siRNA, which implied that the capsids that had undergone secondary envelopment in infected cells treated with PI3K-C2A siRNA were not infectious.
DISCUSSION
Here we provide a further demonstration that high-throughput screening of siRNAs is a valid methodology to identify cellular factors required for viral replication. We identified a range of cellular kinase proteins involved in HCMV replication and focused our studies on how PI3K-C2A might be required for HCMV replication. These studies indicated that PI3K-C2A is involved in the production of late viral proteins and the production of infectious virus. However, in infected cells treated with PI3K-C2A siRNA, we observed a greater defect in virion production than viral protein production. Thus, PI3K-C2A most likely plays a more prominent role in the production of infectious HCMV virions than the production of HCMV proteins. Our data indicate that PI3K-C2A is involved in processes that lead to egress from the cell of capsids that have undergone secondary envelopment.
Our screening methodology allowed us to survey siRNAs that affect nearly all facets of HCMV replication, and as we demonstrate here, we could identify siRNAs that have a fairly modest effect on pp28 expression but a greater effect on virus replication. An added advantage was that the cytoplasmic localization of pp28 in viral assembly compartments could be dictated by the virally encoded kinase UL97 (28), which is required for the function of known anti-HCMV drugs (29) and is itself a major drug target (30). Therefore, in future experiments, our methodology can be converted to a high-throughput/high-content screening approach wherein the number of pp28-positive cells and localization of pp28 within infected cells can identify factors affecting UL97 function and assembly compartment morphology. Conversely, this screening approach did not allow us to assess to what degree any protein is depleted by siRNA within the screen, and the use of antibodies to detect viral replication limited the screen to the detection only of HCMV replication. Furthermore, the infections at an MOI of 1 used during the screening process typically resulted in only 30 to 40% of cells being infected in wells treated with Ctrl siRNA (data not shown). Therefore, the transfection process may have limited HCMV infection, and further optimization of our methodology may be required.
In our screen, depletion of a range of kinase proteins had positive or negative effects on HCMV replication. Each of these proteins may act alone or in concert with other factors to facilitate HCMV replication. No phosphatase proteins were found to have any effect. The identification of pathways involved in viral replication has been possible from the results of whole-genome siRNA screens. Bioinformatics analysis of our screening results using the STRING database, a bioinformatics application that identifies known and predicted protein-protein associations (31), did not identify obvious pathways that might be involved in HCMV replication (data not shown). These pathways may reveal themselves only in the context of a whole-genome siRNA screen. However, these approaches must be applied cautiously, as bioinformatics analysis may draw together protein-protein associations that may be the result of false-positive or false-negative screening hits or that have only very modest effects on viral replication.
Previously, a screen of siRNA targeting the human kinome based on detection of virus produced from siRNA-treated cells was conducted by Terry and coworkers and implicated AMPK as being important in HCMV replication (8). In agreement with the findings of Terry et al. (8), we found that treatment of infected cells with the AMPK inhibitor compound C decreases the accumulation of HCMV proteins and the production of infectious HCMV (data not shown). However, we observed very little overlap in the identification of siRNAs that had either a positive or a negative effect on HCMV replication when comparing the results of Terry et al. (8) and our own. For example, in the work by Terry et al. (8), siRNA targeting PI3K-C2A had little or no effect on HCMV replication and the siRNA targets involved or implicated in AMPK function during HCMV replication differed from those found in our study. It is likely that differences in screening methodologies and siRNA reagents reflect the differences in our data sets. It is widely accepted that different siRNA reagents display different false positives and false negatives in different screens against the same pathogen (7, 32). To combat this issue, several siRNA data sets using orthologous RNA interference (RNAi) reagents must be integrated and refined to identify with a high certainty factors that have positive or negative effects (7, 32). Therefore, it is likely that several more siRNA data sets will have to be generated before we can fully understand the effects of siRNAs on HCMV replication.
Moreover, screening data can be further interrogated with the use of bioinformatics tools, such as GESS and Haystack, to eliminate the off-target effects of siRNA seed sequence binding to mRNAs. However, it must be stressed that the statistical power of these analyses increases with the number of siRNAs assayed. Therefore, siRNA collections of the size used here are not often assayed to find siRNA seed matches (Eugen Buehler, personal communication). Thus, while the GESS and Haystack analysis used here suggests that no obvious off-target binding effects occurred in our screen, combining the data presented here with other siRNA screening data sets may reveal as yet unappreciated off-target effects that influence our interpretation of siRNA function in HCMV-infected cells.
Furthermore, several of the targets of siRNA in our screening data were not found in a proteomic study of HFFs infected with HCMV (27). Therefore, analysis of siRNA off-target binding alone may not be sufficient to exclude siRNAs from analysis. Indeed, analysis of siRNA screening data can include a comparison of screening hits with gene expression profiles to identify false positives (7). Further analysis of siRNA screening data for HCMV-infected cells should benefit from a comparison of siRNA screening hits with proteins known to be expressed in HCMV-infected cells (27), as we have performed here. It remains unknown what RNAs are targeted by the siRNAs that we judged to be false positives in our screening data. Further analysis of this question should identify viral or cellular RNA transcripts involved in HCMV replication.
We chose to focus our study on the involvement of PI3K-C2A in HCMV replication. The role of at least one class I PI3K protein and at least one class III PI3K protein has been investigated (12, 13); however, the role of class II PI3K proteins in HCMV replication is unknown. PI3K-C2A is found in endosomes, the trans-Golgi network, and clathrin-coated vesicles (33, 34). Like other PI3K proteins, PI3K-C2A phosphorylates PtdIns at the D3 position of the inositol ring, producing 3-phosphorylated PtdIns (PtdIns3P), lipids involved in membrane specification and dynamics (33–36). Other phosphorylated lipids involved in membrane trafficking can be produced by this lipid kinase (37). PI3K-C2A and the phosphorylated lipids that it produces are associated with a number of processes that involve intracellular membranes, including exocytosis, endocytosis, and autophagy (33, 34, 38), although the function of the PtdIns3P produced by PI3K-C2A is largely unclear. Also, PI3K-C2A can be found in the nucleus, where it appears to associate with cellular RNA splicing factors (39, 40).
Our observations point to a role for PI3K-C2A in the production of HCMV virions at a step after secondary envelopment of capsids. HCMV secondary envelopment is very poorly defined, and our understanding of this process largely relies on observations made using related viruses, such as herpes simplex virus. Briefly, secondary envelopment occurs upon budding of capsids into cytoplasmic membranes related to the Golgi apparatus (including the cis-Golgi and trans-Golgi networks and endosomes) and is associated with changes in cytoplasmic membranes that occur during development of viral assembly compartments (2, 41). PI3K-C2A is known to be enriched in the aforementioned cytoplasmic membranes (33, 34, 38), and in preliminary immunofluorescence experiments, we observed PI3K-C2A in the cytoplasm of HCMV-infected cells (data not shown). Therefore, it is plausible that PI3K-C2A could be associated with secondary envelopment. Furthermore, PI3K-C2A is reported to be involved in exocytosis (33, 34, 38), which is thought to be involved in the movement of enveloped herpesvirus particles to the plasma membrane (41). Therefore, we suggest that PI3K-C2A functions in exocytosis in HCMV-infected cells, possibly linking the completion of secondary envelopment with virion egress. Interestingly, in HCMV-infected cells, the function of the class III PI3K protein Vps34 appears to be separable from that of PI3K-C2A, as these proteins are required for processes before and after secondary envelopment, respectively (13). A further point to consider is our observation that intracellular virus that accumulates in infected cells treated with PI3K-C2A may not be infectious. This would imply that the secondary envelopment of capsids that we observe in cells depleted of PI3K-C2A is somehow defective or that there are further, as yet unrecognized functions of PI3K-C2A that are required for the production of infectious virus.
It is worth considering whether PI3K-C2A might be a future anti-HCMV drug target. To our knowledge, there is currently no compound that potently and specifically inhibits PI3K-C2A function or the function of any other class II PI3K protein. Also, it has been reported that well-characterized inhibitors of class I PI3K proteins, wortmannin and LY294002, have little or no effect on PI3K-C2A function (42). Therefore, an original approach is required to discover inhibitors of PI3K-C2A.
Supplementary Material
ACKNOWLEDGMENTS
We express our thanks to Richard Bethell and Peter Seither of Boehringer Ingelheim for their great advice and support of this project. Thanks go to members of the D. M. Coen and B. L. Strang laboratories, especially Jean Pesola for assistance with statistical and PCR analyses. Our thanks also go to Ken Laing and Eugen Buehler for support with quantitative PCR and siRNA binding analysis, to several colleagues (Jeremy Kamil, Wade Gibson, Nathanael Gray, and Richard Stanton) and to the NIH AIDS Reagent Program for the kind gifts of reagents, and to Laura Hudson and Lisa Rickelton for assistance in the preparation of figures. We also acknowledge Maria Ericsson and the staff of the Harvard Medical School Electron Microscope Facility for all their invaluable assistance. Special thanks go to all members of the Institute of Chemistry and Chemical Biology-Longwood at Harvard Medical School for all of their help with all aspects of the screening process.
Funding Statement
This work, including the efforts of Donald M. Coen, was funded by HHS | National Institutes of Health (NIH) (R01 AI019838, R01 AI026077). This work was supported by a Boehringer Ingelheim RNAi Screening Collaboration between Boehringer Ingelheim and Harvard Medical School award to B.L.S. and D.M.C., grants from the National Institutes of Health to D.M.C. (R01 AI019838 and R01 AI026077), and a studentship from the Kingdom of Saudi Arabia (to H.F.M.) plus New Investigator funds from St. George's, University of London, a St. George's Impact & Innovation Award, and a PARK/WestFocus Award (all to B.L.S.). The funders had no role in data collection, interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01134-16.
REFERENCES
- 1.Mocarski ES, Shenk T, Griffiths PD, Pass RF. 2015. Cytomegaloviruses, p 1960–2015. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Alwine JC. 2012. The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog 8:e1002878. doi: 10.1371/journal.ppat.1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 4.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, Sultana H, Brass AL, Adametz R, Tsui M, Qian F, Montgomery RR, Lev S, Mason PW, Koski RA, Elledge SJ, Xavier RJ, Agaisse H, Fikrig E. 2008. RNA interference screen for human genes associated with West Nile virus infection. Nature 455:242–245. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, Elledge SJ. 2009. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A 106:16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu J, Davoli T, Perriera JM, Chin CR, Gaiha GD, John SP, Sigiollot FD, Gao G, Xu Q, Qu H, Pertel T, Sims JS, Smith JA, Baker RE, Maranda L, Ng A, Elledge SJ, Brass AL. 2014. Comprehensive identification of host modulators of HIV-1 replication using multiple orthologous RNAi reagents. Cell Rep 9:752–766. doi: 10.1016/j.celrep.2014.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terry LJ, Vastag L, Rabinowitz JD, Shenk T. 2012. Human kinome profiling identifies a requirement for AMP-activated protein kinase during human cytomegalovirus infection. Proc Natl Acad Sci U S A 109:3071–3076. doi: 10.1073/pnas.1200494109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koyuncu E, Purdy JG, Rabinowitz JD, Shenk T. 2013. Saturated very long chain fatty acids are required for the production of infectious human cytomegalovirus progeny. PLoS Pathog 9:e1003333. doi: 10.1371/journal.ppat.1003333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodems SM, Spector DH. 1998. Extracellular signal-regulated kinase activity is sustained early during human cytomegalovirus infection. J Virol 72:9173–9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson RA, Huong SM, Huang ES. 2000. Activation of the mitogen-activated protein kinase p38 by human cytomegalovirus infection through two distinct pathways: a novel mechanism for activation of p38. J Virol 74:1158–1167. doi: 10.1128/JVI.74.3.1158-1167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson RA, Wang X, Ma XL, Huong SM, Huang ES. 2001. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J Virol 75:6022–6032. doi: 10.1128/JVI.75.13.6022-6032.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharon-Friling R, Shenk T. 2014. Human cytomegalovirus pUL37x1-induced calcium flux activates PKCalpha, inducing altered cell shape and accumulation of cytoplasmic vesicles. Proc Natl Acad Sci U S A 111:E1140–E1148. doi: 10.1073/pnas.1402515111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanton RJ, Baluchova K, Dargan DJ, Cunningham C, Sheehy O, Seirafian S, McSharry BP, Neale ML, Davies JA, Tomasec P, Davison AJ, Wilkinson GW. 2010. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J Clin Invest 120:3191–3208. doi: 10.1172/JCI42955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, Santoyo-Lopez J, Dunican DJ, Long A, Kelleher D, Smith Q, Beijersbergen RL, Ghazal P, Shamu CE. 2009. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods 6:569–575. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 17.Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B, King RW. 2012. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Methods 9:363–366. doi: 10.1038/nmeth.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yilmazel B, Hu Y, Sigoillot F, Smith JA, Shamu CE, Perrimon N, Mohr SE. 2014. Online GESS: prediction of miRNA-like off-target effects in large-scale RNAi screen data by seed region analysis. BMC Bioinformatics 15:192. doi: 10.1186/1471-2105-15-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buehler E, Khan AA, Marine S, Rajaram M, Bahl A, Burchard J, Ferrer M. 2012. siRNA off-target effects in genome-wide screens identify signaling pathway members. Sci Rep 2:428. doi: 10.1038/srep00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strang BL, Stow ND. 2005. Circularization of the herpes simplex virus type 1 genome upon lytic infection. J Virol 79:12487–12494. doi: 10.1128/JVI.79.19.12487-12494.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gault E, Michel Y, Dehee A, Belabani C, Nicolas JC, Garbarg-Chenon A. 2001. Quantification of human cytomegalovirus DNA by real-time PCR. J Clin Microbiol 39:772–775. doi: 10.1128/JCM.39.2.772-775.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strang BL, Bender BJ, Sharma M, Pesola JM, Sanders RL, Spector DH, Coen DM. 2012. A mutation deleting sequences encoding the amino terminus of human cytomegalovirus UL84 impairs interaction with UL44 and capsid localization. J Virol 86:11066–11077. doi: 10.1128/JVI.01379-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reference deleted. [Google Scholar]
- 24.Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, Baskerville S, Maksimova E, Robinson K, Karpilow J, Marshall WS, Khvorova A. 2006. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods 3:199–204. doi: 10.1038/nmeth854. [DOI] [PubMed] [Google Scholar]
- 25.Ma Y, Creanga A, Lum L, Beachy PA. 2006. Prevalence of off-target effects in Drosophila RNA interference screens. Nature 443:359–363. doi: 10.1038/nature05179. [DOI] [PubMed] [Google Scholar]
- 26.Franceschini A, Meier R, Casanova A, Kreibich S, Daga N, Andritschke D, Dilling S, Ramo P, Emmenlauer M, Kaufmann A, Conde-Alvarez R, Low SH, Pelkmans L, Helenius A, Hardt WD, Dehio C, von Mering C. 2014. Specific inhibition of diverse pathogens in human cells by synthetic microRNA-like oligonucleotides inferred from RNAi screens. Proc Natl Acad Sci U S A 111:4548–4553. doi: 10.1073/pnas.1402353111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang EC, Aicheler R, Murrell I, Wilkinson GW, Lehner PJ, Gygi SP. 2014. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157:1460–1472. doi: 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azzeh M, Honigman A, Taraboulos A, Rouvinski A, Wolf DG. 2006. Structural changes in human cytomegalovirus cytoplasmic assembly sites in the absence of UL97 kinase activity. Virology 354:69–79. doi: 10.1016/j.virol.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan V, Talarico CL, Stanat SC, Davis M, Coen DM, Biron KK. 1992. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 358:162–164. doi: 10.1038/358162a0. [DOI] [PubMed] [Google Scholar]
- 30.Biron KK, Harvey RJ, Chamberlain SC, Good SS, Smith AA III, Davis MG, Talarico CL, Miller WH, Ferris R, Dornsife RE, Stanat SC, Drach JC, Townsend LB, Koszalka GW. 2002. Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole l-riboside with a unique mode of action. Antimicrob Agents Chemother 46:2365–2372. doi: 10.1128/AAC.46.8.2365-2372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. 2013. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, Yanguez E, Andenmatten D, Pache L, Manicassamy B, Albrecht RA, Gonzalez MG, Nguyen Q, Brass A, Elledge S, White M, Shapira S, Hacohen N, Karlas A, Meyer TF, Shales M, Gatorano A, Johnson JR, Jang G, Johnson T, Verschueren E, Sanders D, Krogan N, Shaw M, Konig R, Stertz S, Garcia-Sastre A, Chanda SK. 2015. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18:723–735. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Falasca M, Maffucci T. 2012. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem J 443:587–601. doi: 10.1042/BJ20120008. [DOI] [PubMed] [Google Scholar]
- 34.Mazza S, Maffucci T. 2011. Class II phosphoinositide 3-kinase C2alpha: what we learned so far. Int J Biochem Mol Biol 2:168–182. [PMC free article] [PubMed] [Google Scholar]
- 35.Di Paolo G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 36.Jean S, Kiger AA. 2012. Coordination between RAB GTPase and phosphoinositide regulation and functions. Nat Rev 13:463–470. doi: 10.1038/nrm3379. [DOI] [PubMed] [Google Scholar]
- 37.Posor Y, Eichhorn-Gruenig M, Puchkov D, Schoneberg J, Ullrich A, Lampe A, Muller R, Zarbakhsh S, Gulluni F, Hirsch E, Krauss M, Schultz C, Schmoranzer J, Noe F, Haucke V. 2013. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 499:233–237. doi: 10.1038/nature12360. [DOI] [PubMed] [Google Scholar]
- 38.Devereaux K, Dall'Armi C, Alcazar-Roman A, Ogasawara Y, Zhou X, Wang F, Yamamoto A, De Camilli P, Di Paolo G. 2013. Regulation of mammalian autophagy by class II and III PI 3-kinases through PI3P synthesis. PLoS One 8:e76405. doi: 10.1371/journal.pone.0076405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Didichenko SA, Fragoso CM, Thelen M. 2003. Mitotic and stress-induced phosphorylation of HsPI3K-C2alpha targets the protein for degradation. J Biol Chem 278:26055–26064. doi: 10.1074/jbc.M301657200. [DOI] [PubMed] [Google Scholar]
- 40.Didichenko SA, Thelen M. 2001. Phosphatidylinositol 3-kinase C2alpha contains a nuclear localization sequence and associates with nuclear speckles. J Biol Chem 276:48135–48142. [DOI] [PubMed] [Google Scholar]
- 41.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 42.Maffucci T, Brancaccio A, Piccolo E, Stein RC, Falasca M. 2003. Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J 22:4178–4189. doi: 10.1093/emboj/cdg402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.