

Abstract
N-myc oncogene amplification is associated but not present in all cases of high-risk neuroblastoma (NB). Since oncogene expression could often modulate sensitivity to oncolytic viruses, we wanted to examine if N-myc expression status would determine virotherapy efficacy to high-risk NB. We showed that induction of exogenous N-myc in a non-N-myc-amplified cell line background (TET-21N) increased susceptibility to oncolytic vesicular stomatitis virus (mutant VSVΔM51) and alleviated the type I IFN-induced antiviral state. Cells with basal N-myc, on the other hand, were less susceptible to virus-induced oncolysis and established a robust IFN-mediated antiviral state. The same effects were also observed in NB cell lines with and without N-myc amplification. Microarray analysis showed that N-myc overexpression in TET-21N cells downregulated IFN-stimulated genes (ISGs) with known antiviral functions. Furthermore, virus infection caused significant changes in global gene expression in TET-21N cells overexpressing N-myc. Such changes involved ISGs with various functions. Therefore, the present study showed that augmented susceptibility to VSVΔM51 by N-myc at least involves downregulation of ISGs with antiviral functions and alleviation of the IFN-stimulated antiviral state. Our studies suggest the potential utility of N-myc amplification/overexpression as a predictive biomarker of virotherapy response for high-risk NB using IFN-sensitive oncolytic viruses.
Introduction
Neuroblastoma (NB) is the most common cancer in the first years of life, and the most common solid tumor of childhood. Patients are risk-stratified using a combination of clinical, pathological, and molecular characteristics. The survival of patients with high-risk disease has not improved and remains less than 60%.1 Historically, standard therapy for high-risk disease includes chemotherapy, surgery, radiation, and bone marrow transplant, which appear to provide some control of disease progression, but is complicated by significant morbidity and mortality.2,3 Innovative approaches such as GD-2 antibody-mediated immune therapy have demonstrated the first improvements in survival for high-risk NB patients in over two decades, though mechanisms limiting its efficacy still occur.4 Therefore, novel approaches to this disease are necessary.
Viral oncolysis is a novel approach to NB that has shown promise in various preclinical cancer models.5,6 Despite their promise as therapeutics, oncolytic viruses (OVs) face application hurdles due to our incomplete understanding of the role of the tumor microenviroment and antiviral immune responses on virotherapy. In general, OVs can selectively kill tumor cells while leaving normal cells intact.7 They achieve this by exploiting the same cellular defects that promote tumor growth. One of such defects is the type I interferon (IFN) signaling, which sensitizes tumor cells to IFN-sensitive OVs such as vesicular stomatitis virus (VSV) and Newcastle disease virus.8–10 In this study, we used VSV based on its known efficacy as a potent oncolytic agent to several tumor types.11–13 The deletion of a single amino acid of the M-protein (VSVΔM51) increases safety by restricting its infection to cancer cells with defects in type I IFN response.13,14 However, tumors with functional type I IFN signaling can hamper its clinical application.12
N-myc amplification, although not present in all cases,15 is the best-characterized aberrant genetic alteration associated with poor prognosis in high-risk NB.16 The mechanisms whereby MYC proteins (c-myc, N-myc and L-myc) sensitize cancer cells to OVs remain unexplored. Previous studies have shown that some c-myc-amplified cancer cell lines are highly susceptible to VSV-induced cell killing.17 Though not studied in the context of oncolytic virotherapy, c-myc negatively regulates type I IFN signaling through STAT-1, which is one of the mechanisms of pathogenesis in Burkitt’s lymphoma and uveal melanoma.18,19
Since oncogenic expression often correlates with increased susceptibility of cancer cells to OVs20–22 and the effects of N-myc on virotherapy are unknown, we reasoned that N-myc overexpression, due to amplification, could be a clinically important biomarker of virotherapy efficacy to high-risk NB. We showed that N-myc-amplified NB cell lines and a non-N-myc-amplified cell line (TET-21N) induced to overexpress exogenous N-myc had augmented susceptibility to virus-induced cell killing and failed to establish a robust type I IFN-stimulated antiviral state. To study the effects of N-myc on susceptibility to OV, we performed microarray analysis in TET-21N cells expressing low and high levels of exogenous N-myc. Before infection, we found that several interferon-stimulated genes (ISGs), some with antiviral functions, were downregulated when N-myc levels increased. Furthermore, changes in global gene expression upon infection were nearly 10-fold higher in TET-21N (high N-myc) with respect to TET-21N (low N-myc).
Results
Effects of N-myc overexpression on virus replication and oncolysis
Since oncogene expression status often determines virotherapy response as shown in some preclinical studies,20–22 we hypothesized that N-myc overexpression, as a consequence of amplification, would further sensitize NB cells to OVs. To test this hypothesis, we first used human-derived high-risk NB cell lines consisting on N-myc-amplified neuroblastic (N) cells (IMR-5, IMR-32, and LAN-1) and non N-myc-amplified substrate-adherent (S) cells (SK-N-HS, SK-N-AS, and SH-EP). Previous studies have shown that N-myc expression status does not correlate to the N and S phenotypes.23,24
Cells were infected with VSVΔM51 at a multiplicity of infection (MOI) of 0.5 to study productive infection and virus spread. Productive infection and differences in virus spread varied among the analyzed cell lines with no apparent correlation with N-myc amplification/overexpression status (Figure 1a). We next examined the oncolytic effects of VSVΔM51 in these cell lines. Interestingly, virus-induced cell killing kinetics was faster in the N-myc-amplified cells than non N-myc-amplified cells (Figure 1b).
Figure 1.

Effects of N-myc overexpression on VSVΔM51 spread and oncolysis. Human-derived neuroblastoma (NB) cell lines consisting of N-myc-amplified cells (IMR-5, IRM-32 and LAN-1) and non-N-myc-amplified cells (SK-N-SH, SK-N-AS, and SH-EP) were seeded in 35 mm cell culture dishes. The number of cells for each cell line was calculated to achieve 80–90% confluency and the multiplicity of infection (MOI) (0.5) was calculated accordingly. After infection with VSVΔM51 (MOI of 0.5) for 1 hour at RT, cells were washed five times with serum-free medium followed by incubation in Dulbecco’s modified Eagle’s medium containing 5% fetal bovine serum. Aliquots of culture supernatants were collected at indicated time points for virus titration in BHK cells and generation of multi-step growth curves. Virus production was normalized and titers were expressed as numbers of pfu/1 × 105 cells (y axis) (a). To examine the virus-induced killing rates, the analyzed cells were seeded in 96-well plates and infected at MOI of 0.5. Viability was determined by Alamar Blue at indicated time points (b). Before infection, TET-21 cells were seeded in 35 mm cell culture dishes and treated or not with 1 μg/ml doxycycline (dox) for 48 hours. Subsequently, cells were infected with VSVΔM51 (MOI of 0.5) and cell culture supernatants were collected at indicated time points for virus titration in BHK cells and generation of multi-step growth curves. Virus titers were expressed as numbers of pfu/1 × 105 cells (y axis) (c,d). To study the differential cell killing kinetics, TET-21N and TET-21C cells were seeded in 96-well plates, treated or not with doxycycline for 48 hours and infected with VSVΔM51 at MOIs of 0.5 (e,f). Viability was determined with Alamar Blue at indicated time points. Error bars indicate mean ± standard deviation of the mean from three replicates. Dox (-), no doxycycline added. Dox (+), doxycycline added.
NB cell lines are well known to differ genetically, phenotypically, biochemically and in their tumorigenic capacity.24,25 Since the effects of N-myc on virus replication may have been obscured by cell-type specific differences, we used an isogenic cell system (TET-21N) that allows the exquisite regulation of N-myc by tetracycline addition/removal.26 TET-21N cells, derived from the non-N-myc amplified SH-EP cell line, express nearly undetectable levels of N-myc in the presence of doxycycline (low N-myc), while withdrawal of this antibiotic induces its overexpression (high N-myc). To rule out any effects of doxycycline, we used TET-21 control cells (TET-21C), which are isogenic to TET-21N cells without exogenous N-myc gene.26 Expression of N-myc in cells, either untreated or treated with doxycycline (dox -/+), was monitored by RT-PCR and western blot assays.
Virus spread as shown in multistep growth curves (MOI of 0.5) was slightly faster in TET-21N (high N-myc), although virus titers did not differ significantly to those observed in TET-21N (low N-myc) (P > 0.05, Figure 1c). TET-21C cells (dox -/+) did not show significant differences in virus spread (Figure 1d). Virus replication as determined by one-step growth curves (MOI of 5) was slightly higher in TET-21N (high N-myc) with no significant differences in virus titers to those observed in TET-21N (low N-myc) (P > 0.05, Supplementary Figure S1a). TET-21C cells (dox -/+) equally supported virus replication (Supplementary Figure S1b). Therefore, these observations collectively suggest that N-myc does not affect the rates of virus replication and spread in NB cells.
Consistent to our observations in the NB cell lines, TET-21N (high N-myc) had significantly faster cell killing kinetics than that of TET-21N (low N-myc) (P < 0.05, Figure 1e). Cell killing kinetics did not change in TET-21C (dox +, Figure 1f). These data suggest that N-myc expression can further sensitize NB cells to virus-induced cell killing.
Effects of N-myc on induction of type I IFNs
Since VSV infection induces the expression of type I IFNs,27 we hypothesized that N-myc-driven regulation of these cytokines promoted differential susceptibility of all NB cells to virus-induced cell killing. As positive controls for these assays, cells were treated with polyinosinic-polycytidylic acid (poly I:C) for 12 hours. Poly I:C is a strong inducer of type I IFNs when delivered intracellularly (transfection) or extracellularly (in cell culture medium). Intracellular and extracellular poly I:C mainly activate the retinoic acid-inducible gene (RIG-I) and toll-like receptor-3 (TLR-3) signaling, respectively, for the expression of type I IFNs.28 Cells were either infected at MOI of 0.1 or treated with poly I:C (intracellular and extracellular) and cells and cell culture supernatants were harvested at indicated time points. Secreted IFN-β was detected and quantified by ELISA while RT-PCR was used to detect the transcription of IFNs-α and -β.
Detection and quantitation of secreted IFN-β was performed at 6, 12, and 18 hpi. This cytokine was best detected at 18 hpi and thus our analysis was focused at this time point (Figure 2a). IFN-β levels increased in the TET-21 cells in response to infection, except for TET-21C (dox -). However, the increased levels of this cytokine did not differ significantly to basal levels (P > 0.05), except for, surprisingly, TET-21C (dox +). IFN-β levels increased upon stimulation with poly I:C, either intracellular or extracellular (Figure 2b). Interestingly, the increased levels of this cytokine in TET-21N (high N-myc) did not significantly differ to basal levels (P > 0.05). In contrast, endogenous levels of IFN-β significantly increased in TET-21N (low N-myc) and TET-21C (dox -/+) upon poly I:C treatment (P < 0.05). IFN-β levels in virus-infected and poly I:C-treated TET-21N (high N-myc) did not differ significantly (P > 0.05). In contrast, levels of this cytokine in poly I:C-treated TET-21N (low N-myc) and TET-21C (dox -/+) were significantly higher than levels observed in virus-infected cells (P < 0.05). These observations collectively suggest that, regardless of N-myc expression status, virus infection does not seem to induce enough levels of IFN-β to stimulate the antiviral state in cells, while N-myc seems to have negative effects in the expression of this cytokine when stimulated with poly I:C. At the transcriptional level, significant increases of IFN-β was observed upon infection and intracellular poly I:C. Interestingly, IFN-β transcript levels were lower in TET-21N (high N-myc) than those in TET-21N (low N-myc) (Supplementary Figure S2a). Transcription of IFN-α was detected upon infection and stimulation with extracellular poly I:C in TET-21N (high and low N-myc) but not in TET-21C cells (Dox-/+). Transcription of type I IFNs were also detected in all NB cells upon infection and treatment with poly I:C but their levels were cell-type specific regardless of N-myc amplification status (Supplementary Figure S2b).
Figure 2.

Expression of type I IFNs and activation of Jak/STAT pathway upon infection and poly I:C treatment. TET-21N or TET-21C cells were seeded in 24-well plates and either treated or not with 1 μg/ml doxycycline (dox) for 48 hours, infected with VSVΔM51 at multiplicity of infection of 0.1 for 6, 12, and 18 hpi. IFN-β was best detected at 18 hpi and thus the analysis at this time point is shown (a). Alternatively, cells were treated with extracellular and intracellular poly I:C (10 μg/ml and 10 ng/well in a 24-well plate format, respectively) for 12 hours (b). Detection of IFN-β in cell culture supernatants was performed using Verikine Human Interferon beta kit (R&D systems) according to the manufacturer’s instructions. To examine the Jak/STAT signaling, cells were infected with VSVΔM51 at MOI of 0.1 for 12, 18, and 24 hours. Alternatively, cells were treated with exogenous IFN-β (200 U/ml) or 10 μg/ml extracellular poly I:C (in reduced serum medium) for 12 hours. Cell lysates were used to examine the expression of master regulators of IFN signaling such as STATs 1 and 2 and IRF-9. Activation of STAT-1 and -2 were analyzed examining the phosphorylation status of STAT-1 (Y701 and S727) and STAT-2 (Y690) (c). The analyzed NB cell lines were either infected with VSVΔM51 at MOI of 0.1 for 12 hours or treated with exogenous β (200 U/ml) for 16 hours (d). Error bars indicate mean ± standard deviation of the mean from three replicates. Dox (-), no doxycycline added. Dox (+), doxycycline added. Ext PIC, extracellular poly I:C. Int PIC, intracellular poly I:C.
Type I IFN signaling through Jak/STAT
IFN-α/β secreted by virus infected- or poly I:C-treated cells interacts with the IFN (α, β, and ϵ) receptor 1 (IFNAR1) to activate the Jak/STAT pathway. Activation of this pathway results in phosphorylation of STAT-1 and STAT-2 at tyrosine 701 and 690, which allows them to heterodimerize and translocate into the nucleus to associate with IRF-9 and form the ISGF-3 complex. ISGF-3 activates promoters for the expression of several IFN-stimulated genes (ISGs). The collective contribution of ISGs renders cells in the antiviral state.29 We therefore studied the IFN signaling through Jak/STAT by analyzing the expression of the ISGF-3 complex and activation of STATs 1 and 2 by phosphorylation. We analyzed the expression of RIG-I as readout for the induction of the Jak/STAT signaling. Cells were infected at MOI of 0.1 and harvested at indicated time points before cytopathic effects were apparent, and components of the ISGF-3 complex were analyzed by western blot. As controls for these assays, cells were treated with exogenous IFN-β (200 U/ml) and extracellular poly I:C. Intracellular poly I:C was not included in the analysis due to its poor induction of the Jak/STAT signaling in TET-21 cells, based on STAT-1 and RIG-I expression (Supplementary Figure S3).
Basal levels of STATs 1 and 2 and their phosphorylated forms, before infection, were similar between TET-21N (high N-myc) and TET-21N (low N-myc). Levels of IRF-9, on the other hand, were slightly lower in TET-21N (high N-myc) (Figure 2c). Virus infections in TET-21 cells did not induce significant changes in the levels of the ISGF-3 members. Levels of phospho-STAT-2 (Y690) slightly increased upon infection in TET-21N (low and high N-myc) at 12 hpi. However, at 18 and 24 hpi, its levels slightly increased and decreased in TET-21N (high N-myc) and TET-21N (low N-myc), respectively. These observations suggest that VSVΔM51 does not induce robust changes in the Jak/STAT signaling in TET-21 cells, probably due to insufficient levels of IFN-β expression.
Treatments with IFN-β or extracellular poly I:C upregulated the expression of IRF-9, RIG-I and STAT-1 and significantly increased the levels of phospho-STAT-1 (Y701 and S727) and STAT-2 (Y690) in TET-21 cells regardless of N-myc expression status (Figure 2c). Furthermore, the level of these proteins were similar between cells with low and high N-myc. Relative to TET-21N (low N-myc) and TET-21C (dox-/+), RIG-I levels were slightly lower in TET-21N (high N-myc) after treatment with IFN-β or poly I:C (Figure 2c). These observations suggest that the Jak/STAT signaling is functional regardless of N-myc expression status.
Consistent to the observations in TET-21 cells, virus infection did not cause significant changes in the expression of STATs 1 and 2 and their phosphorylated forms in the analyzed NB cells (Figure 2d). Levels of IRF-9 slightly increased in IMR-32 (N-myc-amplified) and SH-EP (non-N-myc-amplified) in response to infection. Treatment of all cells with IFN-β upregulated the expression of ISGF-3 members and RIG-I, though their expression and phosphorylation status of STATs 1 and 2 varied among cells (Figure 2d). Consistent to our observations in the TET-21 system, the upregulation of these ISGs by IFN-β suggests a functional Jak/STAT signaling regardless of cell type and N-myc amplification/overexpression status.
Effects of N-myc on the establishment of the antiviral state
Since the Jak/STAT signaling is functional in all cells, we hypothesized that treatment with IFN-β prior to virus infection would induce the antiviral state in all NB cells. Cells were either treated or not with poly I:C and IFN-β (200 U/ml) for 12 and 6 hours, respectively, followed by virus infection (MOI of 5) at indicated time points. As expected, infection of control cells resulted in cell death. Interestingly, treatment of TET-21N (high N-myc) with exogenous IFN-β induced a weak antiviral state insufficient to protect cells from virus-induced cell killing during the course of the experiment (Figure 3a). In contrast, TET-21N (low N-myc) and TET-21C (dox -/+) established a robust antiviral state that fully protected cells from virus-induced cell killing (Figure 3a,b). Similar effects on the antiviral state in TET-21 cells were observed when treated with poly I:C (Figure 3c,d). Consistent to the observations in the TET-21 cell system, N-myc-amplified cell lines failed to establish the antiviral state when treated with poly I:C or IFN-β as opposed to those with no N-myc amplification (Figure 3e,f). These data collectively suggest N-myc-dependent alleviation of the IFN-induced antiviral state.
Figure 3.

Effects of N-myc on the establishment of the antiviral state. TET-21N and TET-21C cells were either pretreated or not with 1 μg/ml doxycycline (Dox +/-) and incubated for 48 hours. Cells were then treated with either 200 U/ml IFN-β (a,b) or extracellular poly I:C (c,d) for 6 and 12 hours, respectively, followed by virus infection at multiplicity of infection (MOI) of 5. Viability was determined with Alamar Blue at indicated time points. NB cell lines were transfected with 2.5 ng (in a 24-well plate format) or treated with 10 μg/ml poly I:C for 12 hours. Alternatively, cells were treated with exogenous IFN-β (200 U/ml) for 6 hours. Cells were subsequently infected with VSVΔM51 at MOI of 5 (e,f). Viability was determined with Alamar Blue. Error bars correspond to mean ± standard deviation of the mean from three replicates. Dox (-), no doxycycline added. Dox (+), doxycycline added.
To further investigate the negative effects of N-myc expression on the establishment of the antiviral state, we studied the antiviral activity of conditioned media from poly I:C-treated TET-21 cells. We also included conditioned media from TET-21 cells infected with VSVΔM51 at 18 hpi, which was UV treated for 5 minutes to inactivate virus. SK-N-AS cells (non-N-myc-amplified) were used as indicators due to their ability to establish a robust antiviral state as demonstrated in this work and by others.30 Since extracellular poly I:C activated the Jak/STAT signaling more efficiently than intracellular poly I:C (Supplementary Figure S3), TET-21 cells were incubated in Dulbecco’s modified Eagle’s medium (DMEM) (reduced serum) with or without poly I:C (10 μg/ml) for 6 hours. After removing media, cells were washed four times with serum-free medium to eliminate any residual poly I:C. Cells were then incubated in fresh DMEM medium without serum for another 12 hours. Cell culture supernatants were harvested, centrifuged and fetal bovine serum (FBS) was added to 5% final concentration. Alternatively, cell culture supernatants were fourfold concentrated and FBS was added. Both concentrated and nonconcentrated supernatants from poly I:C- and virus-infected cells were used as conditioned media. SK-N-AS cells were incubated in either conditioned media or regular medium (5% FBS) for 24 hours followed by infection (MOI of 5) for another 48 hours. Nonconcentrated conditioned media, from controls, virus-infected or poly I:C-treated TET-21 cells, did not protect SK-N-AS from virus-induced cell killing (not shown). Treatment of SK-N-AS with concentrated conditioned media from poly I:C-treated TET-21N (low N-myc) and TET-21C (dox -/+) established a robust antiviral state (Supplementary Figure S4a,b). Interestingly, concentrated conditioned media from poly I:C-treated TET-21N (high N-myc) did not protect SK-N-AS from virus-induced cell killing (Supplementary Figure S4a). Conditioned media from infected TET-21 cells did not protect SK-N-AS from virus-induced cell killing (not shown). Therefore, these findings further support the negative role of N-myc on the establishment of the antiviral state.
Microarray analysis and identification of ISGs
Since N-myc levels correlated to the differential susceptibility to virus-induced cell killing, we then sought to perform microarray assays in TET-21N cells to study the N-myc-driven global changes in gene expression in response to infection. For these assays, we used VSVΔM51-GFP to monitor virus infection based on GFP expression. Upon optimization, we found that infections at MOI of 0.5 for 18 hours were the most suitable for microarray analysis for the following reasons: (i) Earlier and later time points showed limited infection to TET-21N (low N-myc) and extensive cytopathic effects in TET-21N (high N-myc), respectively; (ii) infections, based on GFP expression, reached between 50–60% of cells with limited cytopathic effects in TET-21N (high N-myc) (not shown); and (iii) infections with RNA viruses at high MOIs are known to interfere with the microarray analysis.31
Comparisons were made as follows: group 1, TET-21N (low N-myc, infected/uninfected); group 2, TET-21N (high N-myc, infected/uninfected); and group 3, TET-21N controls (high N-myc/low N-myc). Figure 4 illustrates the comparison groups and the numbers of identified probe sets (numbers in black). A total of 60 and 670 probe sets were identified in TET-21N (low N-myc) and TET-21N (high N-myc), respectively, in response to infection. These data suggest that the higher number of changes in TET-21N (high N-myc) in response to infection seems to correlate to the increased susceptibility to virus-induced cell killing.
Figure 4.
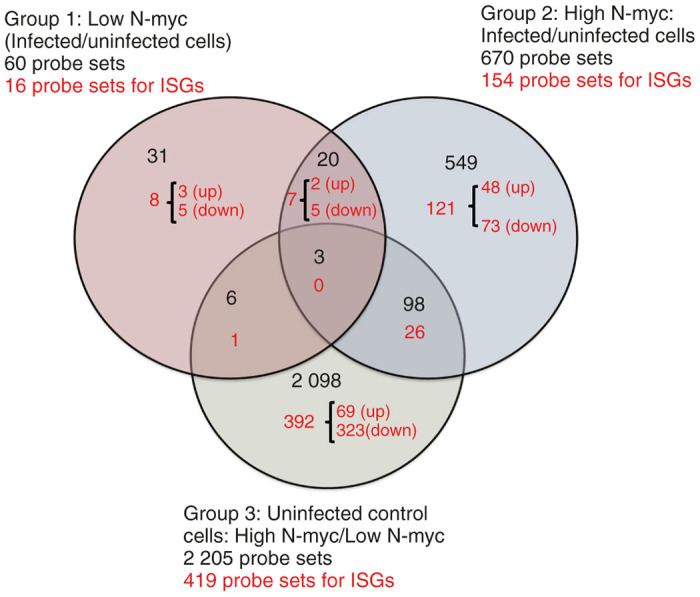
Comparison groups for microarray analysis. Comparison groups are illustrated in a Venn Diagram generated from Affymetrix GeneChip array. Analysis of variance, P < 0.05, was applied as statistically significant. Fold changes of ≥ 1.5 and ≤−1.5 were included in the analysis. The numbers in black correspond to the total probe sets. Numbers in red correspond to the total of probe sets for interferon-stimulated genes (ISGs) identified in the Interferome database.32 Up, upregulated ISGs; down, downregulated ISGs.
Some of the identified probe sets corresponded to ISGs, which were identified by the interferome database V2.32 Several ISGs are known for their antiviral functions and to be induced by IFN-dependent or independent mechanisms in response to virus infection.33 We therefore focused our analysis on these genes. A total of 8, 121, and 392 probe sets for ISGs were identified exclusively in groups 1, 2, and 3, respectively. 7, 1, and 26 ISGs were common between groups 1 and 2, 1 and 3, and 2 and 3, respectively. Interestingly, most identified ISGs in all groups were downregulated (Figure 4).
Regulation of ISGs by N-myc in uninfected cells
A total of 323 out of 392 ISGs (82%) were downregulated when N-myc was overexpressed (Figure 4). Some of these genes had known antiviral functions such as IL-6, AIM2, IFNAR1, STAT-2, IRF-9, and IFITM10 (Table 1). As shown in Figure 2c, basal IRF-9 was slightly lower in TET-21N (high N-myc) than TET-21N (low N-myc), which is consistent with the microarray data. Other ISGs that are negative regulators of IFN signaling with potential proviral functions were also found downregulated such as NMI and IFI-35 (Table 1). These observations suggest that N-myc-driven downregulation of ISGs with antiviral functions may further sensitize cells to virus-induced cell killing and contribute to the alleviation of the IFN-induced antiviral state.
Table 1. Interferon-stimulated genes with known pro- and antiviral functions in uninfected control cells (group 3): high N-myc/low N-myc.
| Gene symbol | Gene title | Fold change | Viral functions (pro-/antiviral) |
|---|---|---|---|
| IL-6 | Interleukin-6 | −4.6 | Antiviral48 |
| AIM2 | Absent in melanoma 2 | −3.2 | Antiviral, 50.25% reduction in VSV replication49 |
| IFITM10 | IFN-inducible transmembrane 10 | −3.2 | Antiviral50 |
| FAM46C | Family with sequence similarity 46, member C | −2.3 | Proviral51 |
| CXCL5 | Chemokine (C-X-C motif) ligand 5 | −2.2 | Antiviral52 |
| ABCA1 | ATP-binding cassette, subfamily A (ABC1), member 1 | −2.0 | Antiviral53 |
| EDEM-1 | ER degradation enhancer, mannosidase α-like 1 | −2.0 | Antiviral54 |
| IFI-35 | Interferon-induced protein 35 | −2.0 | Proviral55 |
| STAT-2 | Signal transducer and activator of transcription 2 | −2.0 | Antiviral10 |
| CASP-1 | caspase 1 | −1.8 | Antiviral,56 |
| IRF-9 | IFN regulatory factor 9 | −1.8 | Antiviral10 |
| NMI | N-myc (and STAT) interactor | −1.6 | Proviral57; antiviral58 |
| IFNAR1 | IFN (α, β, and ϵ) receptor 1 | −1.5 | Antiviral59 |
| IFI27L2 | Interferon, α-inducible protein 27-like 2 | −1.9 | Probably antiviral |
| c-myc | v-myc myelocytomatosis viral oncogene homolog (avian) | −18.8 | Antiviral60; proviral17 |
N-myc-independent regulation of ISGs upon infection
Infection of cells with VSVΔM51 up and downregulated the expression of 2 and 5 genes, respectively, regardless of N-myc expression status (Figure 4 and Table 2). Fold changes in the transcription of these genes were slightly higher in TET-21N (high N-myc) except for RAP1A. Interestingly, transcription of RAP1A, also known as KREV1, had the highest fold change in response to infection: 575.4- and 1 048-fold in TET-21N (high N-myc) and TET-21N (low N-myc), respectively. Though its role on virus replication is unclear, RAP1A has been shown to reverse the proliferative signal of polyomavirus middle T-antigen in transformed rat cells.34 We are currently investigating the role of RAP1A on VSV replication and oncolysis.
Table 2. Interferon-stimulated genes regulated upon infection regardless of N-myc expression: genes commonly regulated in groups 1 and 2.
| Gene symbol | Gene title | Fold-change TET-21N (high N-myc) | Fold-change TET-21N (low N-myc) | Viral functions (pro-/antiviral) |
|---|---|---|---|---|
| RAP1A | RAP1A, member of RAS oncogene family | 575.4 | 1,048.0 | Probably antiviral34 |
| JUP /// KRT17 | Junction plakoglobin /// Keratin 17 | 1.9 | 1.6 | Unknown for JUP. Unclear viral function for KRT1761 |
| BANF1 | Barrier to autointegration factor 1 | −1.8 | 1.5 | Proviral62 |
| BTN3A1 /// BTN3A2 | Butyrophilin, subfamily 3, member A1 /// butyrophilin, subfamily 3, member A2 | −1.8 | −1.6 | Unknown |
| STK35 | Serine/threonine kinase 35 | −1.9 | −1.7 | Unknown |
| DDR1 | Discoidin domain receptor tyrosine kinase 1 | −2.0 | −1.5 | Unknown |
| SPP1 | Secreted phosphoprotein 1 | −4.0 | −2.0 | Antiviral63 |
Virus-mediated regulation of ISGs in TET-21N (low N-myc)
Eight probe sets for ISGs were exclusively identified in TET-21N (low N-myc) in response to infection (Figure 4 and Table 3). Though pro- and antiviral functions for these ISGs have been reported, their contribution to VSV infection is unknown.
Table 3. IFN-stimulated genes identified in TET-21N (low N-myc) and TET-21N (high N-myc) in response to infection: infected/uninfected cells.
| TET-21N (low N-myc) | |||
|---|---|---|---|
| Gene symbol | Gene title | Fold change | Viral functions (Pro- or antiviral) |
| APOC2 /// APOC4 /// APOC4-APOC2 | Apolipoprotein C-II /// apolipoprotein C-IV /// APOC4-APOC2 readthrough | 1.5 | Probably proviral64 |
| PIK3IP1 | Phosphoinositide-3-kinase interacting protein 1 | 1.5 | Probably antiviral |
| PSG5 | Pregnancy specific β-1-glycoprotein 5 | 1.5 | Unknown |
| DNAJB14 | DnaJ (Hsp40) homolog, subfamily B, member 14 | −1.5 | Proviral65 |
| SH3BGRL2 | SH3 domain binding glutamic acid-rich protein like 2 | −1.5 | Unknown |
| ZNF230 | Zinc finger protein 230 | −1.5 | Unknown |
| PTCH1 | Patched 1 | −1.6 | Proviral66 |
| IDS | Iduronate 2-sulfatase | −1.7 | Unknown |
| TET21N (high N-myc) | |||
| DDIT-3/CHOP | DNA-damage-inducible transcript 3 | 7.2 | Antiviral35 and proviral36 |
| IFNA1 | IFN-α1 | 3.5 | Antiviral10 |
| IFNA13 | IFN-α13 | 3.5 | Antiviral10 |
| CREBRF | CREB3 regulatory factor | 2.7 | Likely proviral |
| RICTOR | RPTOR independent companion of MTOR, complex 2 | 1.7 | Proviral67 |
| IFNA21 | IFN-α21 | 1.6 | Antiviral10 |
| MAFG | v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog G | 1.6 | Antiviral 60.78% reduction in VSV replication49 |
| TRAF4 | TNF receptor-associated factor 4 | 1.5 | Proviral68 |
| UBE3A | Ubiquitin protein ligase E3A | 1.5 | Antiviral 62.72% reduction in VSV replication49 |
| TRIM-22 | Tripartite motif containing 22 | −1.5 | Antiviral69 |
| TNFSF10/TRAIL | Tumor Necrosis Factor (Ligand) Superfamily, Member 10 | −1.5 | Antiviral70 |
| TANK | TRAF family member-associated NFKB activator | −1.6 | Antiviral71 |
| BRCA1 | Breast cancer 1, early onset | −1.7 | Probably antiviral72 |
| RTN3 | Reticulon 3 | −1.6 | Proviral73 |
| CASP2 | Caspase 2 | −1.7 | Unclear antiviral function49 |
| TRIM-37 | Tripartite motif containing 37 | −1.7 | Antiviral74 |
| TRIM-32 | Tripartite motif containing 32 | −2.1 | Antiviral69 |
| TRIM-13 | Tripartite motif containing 13 | −2.3 | Proviral and antiviral75 |
| MAT2A | Methionine adenosyltransferase II, α | −3.7 | Proviral role on VSV replication76 |
Virus-mediated regulation of ISGs in TET-21N (high N-myc)
Infection of TET-21N (high N-myc) caused changes in the expression of 121 probe sets for ISGs (Figure 4, Table 3, and Supplementary Table S1). From these probe sets, 48 and 73 were up- and downregulated, respectively. Most of the identified ISGs have unknown roles on virus replication. Table 3 lists the identified ISGs with known pro- and antiviral functions. Upregulated ISGs with antiviral functions included IFN-α1, IFN-α13, IFN-α21, DDIT-3/CHOP, MAFG, and TRIMs 22 and 37. Downregulated ISGs with antiviral functions included TANK, TRIM-37, TRIM-32, and TNFSF10/TRAIL.
From all probe sets, DDIT-3/CHOP was the most highly upregulated ISG in response to infection (7.2-fold), suggesting its possible implication on differential susceptibility to VSVΔM51 determined by N-myc. DDIT-3/CHOP, known for its pro- or antiviral functions,35,36 is a transcription factor upregulated during cellular stress including glucose deprivation, amino acid starvation, endoplasmic reticulum (ER) stress, and virus infections.37 The direct role of DDIT-3/CHOP on VSV replication is yet to be determined. To validate our findings, cells were infected with VSVΔM51 at MOI of 0.5 for 12, 18, and 24 hours. As controls for DDIT-3/CHOP expression, cells were either treated or not with 1 μg/ml tunicamycin (an ER poison) for 18 and 24 hours. Upon virus infection, DDIT-3/CHOP protein was detected at 24 hpi in TET-21N (high N-myc) while undetected in TET-21N (low N-myc) and TET-21C (dox -/+). Tunicamycin treatment robustly induced DDIT-3/CHOP in all cells regardless of N-myc expression status (Figure 5a). We further validated our findings in the analyzed NB cell lines. At the protein level, DDIT-3/CHOP could not be detected despite several attempts. At the transcriptional level, DDIT-3/CHOP in response to infection varied among the analyzed NB cells regardless of N-myc expression status (Figure 5b). For example, slight upregulation of DDIT-3/CHOP was observed in LAN-1 (N-myc-amplified) and SK-N-AS (non-N-myc-amplified) while no changes were observed in IMR-32 (N-myc-amplified) and SH-EP (non-N-myc-amplified). These findings suggest that the link between N-myc and DDIT-3/CHOP may be restricted to the TET-21 cell system or, perhaps, certain NB cell populations.
Figure 5.

Differential expression of DDIT-3. (a) TET21 cells were either treated or not with doxycycline for 48 hours and infected with VSVΔM51 at multiplicity of infection (MOI) of 0.5 for 12, 18, and 24 hours. Alternatively, cells were treated with 1 μg/ml tunicamycin (an ER poison) as controls for DDIT-3/CHOP expression. Expression of DDIT-3/CHOP was analyzed by western blot (a). NB cell lines were either infected at MOI of 0.1 or treated with tunicamycin for 15 hours. RNA was extracted and the expression of DDIT-3/CHOP was analyzed by RT-PCR (b). Dox (-), no doxycycline added. Dox (+), doxycycline added.
Discussion
Oncolytic viruses (OVs) are promising new therapies for many cancer types including high-risk NB.38 However, their application in the clinic could be hampered due to the complexity of the tumor microenviroment and the presence of host immune responses to OVs. It is now understood that the interaction of cancer cells and stroma contribute to cancer progression, metastasis, resistance to chemo drugs, recurrence after treatment,39,40 and resistance to virotherapy.41 Due to the highly heterogeneous nature of NB, it is not surprising if some subpopulations of tumor cells with differences in permissiveness to virus infection or functional IFN signaling would hamper the virotherapy efficacy. We performed in vitro assays to study N-myc expression, virus susceptibility, and innate immunity in the context of virotherapy for NB. In this study, we used TET-21N cells, which have been a powerful system to study N-myc in NB biology.26 This cell system allows the overexpression of N-myc at will by adding and removing tetracycline from the cell culture medium (Tet-OFF system). However, limitations of this system include the use of tetracycline, known to alter the cellular gene expression and thus confound microarray analysis,42 and the lack of tumorigenic capacity of TET-21N (low N-myc) to perform in vivo studies in mice.43 Tetracycline seemed to have had a major effect on IFN-β expression in TET-21C (Dox +) in response to infection, though the expression of this cytokine by poly I:C treatment, virus replication, virus-induced cell killing and establishment of the antiviral state did not seem to have been affected by this antibiotic. Furthermore, SK-N-AS cells were not protected by conditioned media from infected TET-21C (Dox +) cells (not shown) suggesting limited effects of doxycycline in virus-induced cell killing. Although cautious interpretation of data from the Tet-system (ON and OFF) is paramount, many of the microarray hits from TET-21N cells (ArrayEpress, accession number E-MEXP-2340) have been validated.26,44 Some validated hits such as c-myc45 and some Wnt signaling pathway genes46 were consistent with our microarray data. Although we did not include TET-21C in our microarray analysis and the effects of tetracycline could not be ruled out, validation of some hits (STAT-2, IRF-9, c-myc and IFITM10) by semiquantitative RT-PCR in both TET-21N and TET-21C confirmed our microarray analysis (not shown).
Little is known about the role of MYC oncoprotein family on susceptibility of cancer cells to OVs. Some cell lines transformed with c-myc are susceptible to VSV in vivo,17 but its role on promoting virus-induced oncolysis in such transformed cells remains unknown. In this study, we found that N-myc did not cause significant changes in the levels of virus replication, either in the analyzed NB or TET-21 cells. However, N-myc-amplified cells and TET-21N (high N-myc) had increased susceptibility to virus-induced cell killing and failed to establish a robust antiviral state induced by exogenous IFN or poly I:C (either extracellular or intracellular). Virus infection did not cause serious changes in the Jak/STAT signaling, probably due to insufficient levels of secreted IFN-β. Treatments with exogenous IFN-β or poly I:C, on the other hand, caused significant changes in the expression and phosphorylation levels of these proteins regardless of N-myc expression status. Overall, our western blot and microarray data suggest that N-myc overexpression does not affect the IFN signaling through Jak/STAT pathway directly but rather the expression of some ISGs stimulated by this pathway. Therefore, these findings suggest that the increased susceptibility of NB cells to VSVΔM51 and alleviation of type I IFN-induced antiviral state by N-myc at least involves the downregulation of ISGs with antiviral functions.
Our findings contrast previous studies showing the negative effects of c-myc on IFN signaling through Jak/STAT pathway in the context of pathogenesis of Burkitt’s lymphoma and uveal melanoma.18,19 Those studies demonstrated that ISGF-3 protein members and IFN signaling were downregulated by c-myc in a dose-dependent manner.18 Our western blot analyses, on the other hand, showed no drastic differences in the levels of ISGF-3 proteins and activation of Jak/STAT signaling through phosphorylation of STATs 1 and 2 when N-myc was overexpressed (Figure 2c). We and others45 have observed elevated levels of c-myc in non-N-myc-amplified NB cells, and its expression significantly decreases when N-myc is overexpressed (not shown). Despite the elevated c-myc levels in these cells, treatments with IFN-β or poly I:C established a robust antiviral state. Therefore, our observations and those previously reported for c-myc18 suggest differential functions of MYC proteins on IFN signaling and, perhaps, susceptibility to virus infection and establishment of the antiviral state.
Relative to TET-21N (low N-myc), significant changes in global gene expression were observed in TET-21N (high N-myc). Though only one time point was examined (18 hpi), differential changes in global gene expression driven by N-myc are likely to occur during the whole replication cycle and virus spread. Most of the identified ISGs have unknown antiviral functions, but it is highly likely that the collective contribution of ISGs and non-ISGs further sensitizes cells to virus infection and alleviates the IFN-stimulated antiviral state. Interestingly, DDIT-3/CHOP, known for its proapoptotic function, was the most highly upregulated ISG in TET-21N (high N-myc) in response to infection. This finding, confirmed by western blot, suggested a possible link between N-myc and DDIT-3/CHOP. However, the expression of DDIT-3/CHOP in response to infection was cell line-dependent regardless of N-myc expression status. Since NB is a highly heterogeneous malignancy, it would not be surprising if this link were restricted to the TET-21 cell system or, perhaps, other NB cell populations. The same seems to be true for the link between N-myc and type I IFN transcription in response to infection and poly I:C treatment (Supplementary Figure S2b). Though our studies with the TET-21 cell system and NB cell lines suggest a link between N-myc and the observed effects on virus susceptibility and establishment of the IFN-induced antiviral state, such link may not be true for all NB cell populations. Studies on the frequency of these links in cells from various clinical specimens would help determine the utility of N-myc as biomarker for virotherapy response.
Our microarray data consisted on several hits that we classified between ISGs and non-ISGs. Non-ISGs consisted on genes that were not found in the Interferome database.32 Due to the high number of identified ISGs in our microarray data, known antiviral functions in some of these genes and the differential establishment of the antiviral state by exogenous IFN-β or poly I:C, we decided to focus our analysis on these genes. However, the observed differences in virus replication and virus-induced cell killing can also be attributed to non-ISGs with various cellular functions (e.g., cell cycle progression, activation/regulation of signaling pathways, enzymatic and apoptotic functions, etc). We are currently investigating the role of some non-ISGs on virus replication.
In summary, we show that N-myc-amplified cells and cells induced to overexpress N-myc were more susceptible to virus-induced cell killing and failed to establish a robust antiviral state by type I IFN. High N-myc levels correlated with increases in susceptibility to virus-induced cell killing and changes in global gene expression. Our data suggest that N-myc-driven downregulation of ISGs with antiviral functions may be some of the mechanisms promoting differential virus-induced cell killing. These studies suggest that N-myc amplification, a hallmark of high-risk neuroblastoma, can be a potential biomarker of VSVΔM51 response, while virotherapy efficacy in high-risk disease with non-N-myc amplification may be suboptimal. Therefore, oncolytic virotherapy for high-risk NB with no N-myc amplification would probably require sensitization of tumors with chemo drugs prior to infection. Development of syngeneic models with isogenic cell lines expressing low and high N-myc levels would provide insights into functional relationships between tumor and stroma, which may determine the virotherapy efficacy in vivo.
Materials and Methods
Cell lines
Neuroblastoma cell lines IMR-5, IMR-32, LAN-1, SK-N-SH, SK-N-AS, SH-EP, TET-21N, and TET-21 control cells (TET-21C) were maintained in DMEM tissue culture medium (Life Technologies, Grand Island, NY) supplemented with 10% FBS (Life Technologies), 1% L-glutamine (Life Technologies) at 37 °C in a humidified 5% CO2 incubator. Doxycycline at 1 μg/ml was added to culture medium for TET-21N cells. TET-21N and TET21C cells were provided by Dr. Manfred Schwab (Deutches Krebsforschungszentrum, division B030 Tumor Genetics). Cultures were routinely tested for mycoplasma contamination.
Viruses and infections
VSVΔM51, a genetically modified VSV strain derived from the Indiana serotype of VSV,14 was propagated on BHK cells. This mutant virus has a deletion of methionine 51 in the M protein and insertion of an extra cistron encoding green fluorescent protein (GFP) between the G and L sequences.14 Cells were infected at indicated multiplicity of infections (MOIs) and cell culture supernatants were collected at various time points for virus titration in BHK cells. Virus growth curves (one- and multi-step) and Alamar blue assays (Invitrogen, Frederik, MD) were carried out to study virus replication and spread and cell viability, respectively.
TET-21N cell line (derived from SH-EP) harbors a repressible control system to regulate the expression of exogenous N-myc at will.26 N-myc overexpression was induced by doxycycline withdrawal from culture medium for 48 hours. TET-21 control cells (TET-21C) harbor the same repressible system except that exogenous N-myc gene is not present. N-myc expression was assessed by RT-PCR and western blot.
Type I interferon (IFN) expression and IFN signaling through Jak/STAT
Cells treated or not with doxycycline were infected with VSVΔM51 at MOI of 0.1 for 6, 12, and 18 hpi or treated with intracellular or extracellular polyinosinic-polycytidylic acid (poly I:C; Sigma–Aldrich, St Louis, MO): 10 ng/well in a 24-well plate format and 10 μg/ml, respectively, for 12 hours. Transfections with poly I:C above 10 ng/well had toxic effects for TET-21N (low N-myc). Cell culture supernatants were harvested, pulsed to remove cell debris, and stored at −80 °C until ready to use. Detection and quantitation of secreted IFN-β were carried out using the Verikine Human Interferon beta kit (R&D systems, Minneapolis, MN) according to the manufacturer’s instructions.
For transcriptional analysis, RNA was extracted (Qiagen) followed by RT-PCR. Degenerate primers were used to detect the IFN-α subtypes47 and specific primers to detect IFN-β (forward, 5′-TGGCAATTGAATGGGAGGCT-3′; and reverse GTCTCATTCCAGCCAGTGCT) and N-myc (forward, 5′-TCCACCAGCAGCACAACTATG; and reverse, 5′-GTCTAGCAAGTCCGAGCGTGT 3′). As controls for these assays, cells were treated with either intracellular (transfection) or extracellular poly I:C in reduced serum medium (Life Technologies). Concentrations of intracellular and extracellular poly I:C were optimized for all cells. Except for SK-N-AS, NB cells were treated with 10–20 μg/ml extracellular poly I:C. SK-N-AS were treated with 0.1 μ/ml extracellular poly I:C because of toxicity beyond this concentration. Poly I:C transfections were performed in a six-well plate format. N-myc-amplified cells were transfected with 15 μg poly I:C using lipofectamin 2000 (Invitrogen, Carlsbad, CA). Non-N-myc-amplified cells were transfected with 0.1 μg poly I:C, as higher amounts had toxic effects on these cells. TET-21 cells were incubated with extracellular (10 μg/ml in reduced serum medium) or intracellular poly I:C (0.1 μg in six-well plate format) for 12 hours. Transfections beyond 0.1 μg poly I:C had toxic effects for TET-21N (low N-myc). To monitor transfection efficiency, cells were transfected with various amounts of pLL 3.7 carrying the GFP gene (Addgene, Cambridge, MA).
For analysis of the Jak/STAT signaling and N-myc detection, cells either treated or not with doxycycline were seeded at 5 × 105 cells per well in six-well plates and incubated at 37 °C in 5% CO2. Cells either infected or not were washed with ice-cold PBS, collected by scraping and lysed in 500 μl of lysis buffer (20 mmol/l Tris pH 8.0, 136 mmol/l NaCl, 10% glycerol, 1% NP40, 0.02% leupeptin, 0.5% aprotinin, and 1.5% sodium orthovanadate) for 20 minutes followed by sonication (three times, 10-second pulse, 10% amplitude) and centrifugation at 4 °C. All protein preparations were quantified (BCA protein assay, Biolynx, Brockville, ON, Canada), run in 10% polyacrilmyde gels and transferred onto nitrocellulose membranes. Membranes were incubated with monoclonal antibodies to N-myc (EMD Millipore, Darmstadt, Germany), STATs 1 and 2, phospho-STAT-1 Y701 (Abcam, Cambridge, MA), actin (EMD Millipore), RIG-I (Novous Biologicals, Littleton, CO); and polyclonal antibodies to IRF-9, phospho-STAT-1 (S727) and phospho-STAT-2 (Y690) (Abcam). Goat anti-mouse and -rabbit IgG-HRP (Santa Cruz Biotechnology, Dallas, TX) were used as secondary antibodies.
IFN protection assays
Cells treated or not with doxycycline were seeded in 96-well plates (5 × 103 cells/well) for 48 hours followed by pretreatment with 200 U/ml of exogenous human IFN-β (PBL Interferon Source, Piscataway, NJ) for 6 hours. Cells were then infected with VSVΔM51 at MOI of 5 and viability determined with Alamar Blue assay (Invitrogen, Frederik, MD) according to the manufacturer’s instructions. All experiments were done in triplicate. Phase-contrast and fluorescent images of cells were taken using a Carl Zeiss inverted microscope (Axiovert 200M) mounted with a Carl Zeiss digital camera (AxioCam MRc, Lake Success, NY).
Conditioned media protection assays
TET-21N and TET21C treated or not with 1 μg/ml doxycycline were cultured in 15 cm dishes and incubated for 48 hours. Cell culture supernatants were removed and replaced with 10 μg/ml poly I:C in reduced serum media (Life Technologies). Cells were incubated for another 12 hours. Supernatants were removed and cells were washed four times with serum-free medium (DMEM, Life Technologies) to eliminate any remaining poly I:C. Fresh serum-free DMEM was added onto cells followed by incubation for another 24 hours. Supernatants were harvested, centrifuged and either four times concentrated or not. FBS was added (5% final concentration) and supernatants were used as conditioned media. For protection assays, SK-N-AS was cultured in conditioned media or regular DMEM medium (10% FBS) overnight followed by infection (MOI of 5 for 48 hours) and viability assays with Alamar blue (Life Technologies).
Before use, conditioned media from virus-infected cells (18 hpi, MOI of 0.5) were UV-irradiated for 5 minutes to inactivate virus. UV-irradiated conditioned media were tested in IMR-5 cells to monitor virus inactivation.
Global gene expression analysis by microarray assays
Microarray assays using Affymetrix GeneChip Human Prime View was carried out. Cells either treated or not with doxycycline were infected at an MOI of 0.5 for 18 hours, time by which around 50–60% of cells expressed GFP encoded by VSVΔM51. RNA was extracted with RNeasy Plus Mini Kit (Qiagen). To assess RNA quality, RNA Integrity Number (RIN) was measured with Agilent RNA 6000 NanoChip on 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). The quantity was measured using NanoDrop 1000 (NanoDrop Technologies, Wilmington, DE). A total of 100 ng of RNA for each sample with a RIN higher than 9 was labeled with 3’ IVT Express Kit (Ambion) and hybridized to Affymetrix GeneChip Human PrimeView Arrays at 45 °C for 16 hours. Arrays were stained and washed using Affymetrix GeneChip Fluidics_450 following manufacturer’s protocol and scanned using the Affymetrix GeneChip Scanner 3000 7G System. For data analysis, Affymetrix GeneChip array data files were generated using GeneChip Command Console Software (AGCC) and statistical analysis was carried out using Partek Genomics Suite 6.0 (Partek Incorporated). Analysis of variance, P < 0.05, was applied as statistically significant. Fold changes of ≥ 1.5 and ≤−1.5 with P < 0.05 were included in the analysis.
ISGs were identified using the interferon database V2.32
Differential expression of DDIT-3/CHOP
Cells were infected at MOI of 0.5 for 12, 18, and 24 hours. As controls, cells were treated with 1 μg/ml tunicamycin (Sigma–Aldrich) for 18 and 24 hours. Cells were harvested for RNA and protein extractions to analyze DDIT-3/CHOP expression at the transcriptional (semiquantitative RT-PCR) and protein levels, respectively. RT-PCR for DDIT-3/CHOP was carried out with forward (5′ TTGCCTTTCTCTTCGGACACT 3′) and reverse (5′ GCTAGCTGTGCCACTTTCC 3′) specific primers. Mouse monoclonal antibody to DDIT3/CHOP (Abcam) was used for western blots.
Acknowledgments
The laboratory of P.B. is supported by Kids Cancer Care Foundation of Alberta and Collaborative Research and Innovation Opportunities (CRIO). The authors want to thank Manfred Schwab (Deutches Krebsforschungszentrum, division B030 Tumor Genetics) for providing the TET-21N and TET-21C cells. The authors also want to thank Xiuling Wang at the Southern Alberta Cancer Research Institute Microarray and Genomics facility for her technical support and data interpretation, Douglas J. Mahoney for his critical review of the manuscript, Mana Alsheri for providing technical support, and Éva Nagy for allowing the performance of some experiments in her laboratory.
The authors declare no conflict of interest.
References
- Maris, JM, Hogarty, MD, Bagatell, R and Cohn, SL (2007). Neuroblastoma. Lancet 369: 2106–2120. [DOI] [PubMed] [Google Scholar]
- von Allmen, D, Grupp, S, Diller, L, Marcus, K, Ecklund, K, Meyer, J et al. (2005). Aggressive surgical therapy and radiotherapy for patients with high-risk neuroblastoma treated with rapid sequence tandem transplant. J Pediatr Surg 40: 936–41; discussion 941. [DOI] [PubMed] [Google Scholar]
- Grupp, SA, Stern, JW, Bunin, N, Nancarrow, C, Ross, AA, Mogul, M et al. (2000). Tandem high-dose therapy in rapid sequence for children with high-risk neuroblastoma. J Clin Oncol 18: 2567–2575. [DOI] [PubMed] [Google Scholar]
- Yu, AL, Gilman, AL, Ozkaynak, MF, London, WB, Kreissman, SG, Chen, HX et al. Children’s Oncology Group. (2010). Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 363: 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahller, YY, Williams, JP, Baird, WH, Mitton, B, Grossheim, J, Saeki, Y et al. (2009). Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PLoS One 4: e4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorence, RM, Reichard, KW, Katubig, BB, Reyes, HM, Phuangsab, A, Mitchell, BR et al. (1994). Complete regression of human neuroblastoma xenografts in athymic mice after local Newcastle disease virus therapy. J Natl Cancer Inst 86: 1228–1233. [DOI] [PubMed] [Google Scholar]
- Parato, KA, Senger, D, Forsyth, PA and Bell, JC (2005). Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer 5: 965–976. [DOI] [PubMed] [Google Scholar]
- Barber, GN (2005). VSV-tumor selective replication and protein translation. Oncogene 24: 7710–7719. [DOI] [PubMed] [Google Scholar]
- Fiola, C, Peeters, B, Fournier, P, Arnold, A, Bucur, M and Schirrmacher, V (2006). Tumor selective replication of Newcastle disease virus: association with defects of tumor cells in antiviral defence. Int J Cancer 119: 328–338. [DOI] [PubMed] [Google Scholar]
- Randall, RE and Goodbourn, S (2008). Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89(Pt 1): 1–47. [DOI] [PubMed] [Google Scholar]
- Diallo, JS, Vähä-Koskela, M, Le Boeuf, F and Bell, J (2012). Propagation, purification, and in vivo testing of oncolytic vesicular stomatitis virus strains. Methods Mol Biol 797: 127–140. [DOI] [PubMed] [Google Scholar]
- Paglino, JC and van den Pol, AN (2011). Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: rare resistance is overcome by blocking interferon pathways. J Virol 85: 9346–9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, L, Huang, TG, Meseck, M, Altomonte, J, Ebert, O, Shinozaki, K et al. (2008). rVSV(M Delta 51)-M3 is an effective and safe oncolytic virus for cancer therapy. Hum Gene Ther 19: 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl, DF, Lichty, BD, tenOever, BR, Paterson, JM, Power, AT, Knowles, S et al. (2003). VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4: 263–275. [DOI] [PubMed] [Google Scholar]
- Castle, VP, Heidelberger, KP, Bromberg, J, Ou, X, Dole, M and Nuñez, G (1993). Expression of the apoptosis-suppressing protein bcl-2, in neuroblastoma is associated with unfavorable histology and N-myc amplification. Am J Pathol 143: 1543–1550. [PMC free article] [PubMed] [Google Scholar]
- Seeger, RC, Brodeur, GM, Sather, H, Dalton, A, Siegel, SE, Wong, KY et al. (1985). Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med 313: 1111–1116. [DOI] [PubMed] [Google Scholar]
- Balachandran, S, Porosnicu, M and Barber, GN (2001). Oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras, or myc function and involves the induction of apoptosis. J Virol 75: 3474–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee, M, Hölzel, M, Bernard, S, Mailhammer, R, Schuhmacher, M, Reschke, J et al. (2007). C-myc activation impairs the NF-kappaB and the interferon response: implications for the pathogenesis of Burkitt’s lymphoma. Int J Cancer 120: 1387–1395. [DOI] [PubMed] [Google Scholar]
- Tulley, PN, Neale, M, Jackson, D, Chana, JS, Grover, R, Cree, I et al. (2004). The relation between c-myc expression and interferon sensitivity in uveal melanoma. Br J Ophthalmol 88: 1563–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, G, Barrett, JW, Stanford, M, Werden, SJ, Johnston, JB, Gao, X et al. (2006). Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc Natl Acad Sci USA 103: 4640–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn, DH and Thorne, SH (2009). Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer 9: 64–71. [DOI] [PubMed] [Google Scholar]
- Marcato, P, Shmulevitz, M and Lee, PW (2005). Connecting reovirus oncolysis and Ras signaling. Cell Cycle 4: 556–559. [DOI] [PubMed] [Google Scholar]
- Schwab, M, Westermann, F, Hero, B and Berthold, F (2003). Neuroblastoma: biology and molecular and chromosomal pathology. Lancet Oncol 4: 472–480. [DOI] [PubMed] [Google Scholar]
- Thiele, C (1998). Neuroblastoma. In: Masters J (ed.). Human Cell Culture. Kluwer Academic Publishers: Lancaster. [Google Scholar]
- Ciccarone, V, Spengler, BA, Meyers, MB, Biedler, JL and Ross, RA (1989). Phenotypic diversification in human neuroblastoma cells: expression of distinct neural crest lineages. Cancer Res 49: 219–225. [PubMed] [Google Scholar]
- Lutz, W, Stöhr, M, Schürmann, J, Wenzel, A, Löhr, A and Schwab, M (1996). Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene 13: 803–812. [PubMed] [Google Scholar]
- Furr, SR, Moerdyk-Schauwecker, M, Grdzelishvili, VZ and Marriott, I (2010). RIG-I mediates nonsegmented negative-sense RNA virus-induced inflammatory immune responses of primary human astrocytes. Glia 58: 1620–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miranda, J, Yaddanapudi, K, Hornig, M and Lipkin, WI (2009). Astrocytes recognize intracellular polyinosinic-polycytidylic acid via MDA-5. FASEB J 23: 1064–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi, SA, Salditt-Georgieff, M and Darnell, JE Jr (1995). Tyrosine-phosphorylated Stat1 and Stat2 plus a 48-kDa protein all contact DNA in forming interferon-stimulated-gene factor 3. Proc Natl Acad Sci USA 92: 3829–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang, JH, Chuang, HC, Huang, CC, Wu, CL, Du, YY, Kung, ML et al. (2011). Differential toll-like receptor 3 (TLR3) expression and apoptotic response to TLR3 agonist in human neuroblastoma cells. J Biomed Sci 18: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaben, M, Whitley, P, Bouwmeester, D, Setterquist, RA, Rottier, PJ and de Haan, CA (2008). Improved microarray gene expression profiling of virus-infected cells after removal of viral RNA. BMC Genomics 9: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinova, I, Forster, S, Yu, S, Kannan, A, Masse, M, Cumming, H et al. (2013). Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res 41(Database issue): D1040–D1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyce, RS, Taylor, K, Ciechonska, M, Collins, SE, Duncan, R and Mossman, KL (2011). Membrane perturbation elicits an IRF3-dependent, interferon-independent antiviral response. J Virol 85: 10926–10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinek, MA and Hassell, JA (1992). Reversion of middle T antigen-transformed Rat-2 cells by Krev-1: implications for the role of p21c-ras in polyomavirus-mediated transformation. Oncogene 7: 1687–1698. [PubMed] [Google Scholar]
- John, L, Thomas, S, Herchenröder, O, Pützer, BM and Schaefer, S (2011). Hepatitis E virus ORF2 protein activates the pro-apoptotic gene CHOP and anti-apoptotic heat shock proteins. PLoS One 6: e25378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y, Fung, TS, Huang, M, Fang, SG, Zhong, Y and Liu, DX (2013). Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J Virol 87: 8124–8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari, S and Mori, M (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11: 381–389. [DOI] [PubMed] [Google Scholar]
- Friedman, GK, Cassady, KA, Beierle, EA, Markert, JM and Gillespie, GY (2012). Targeting pediatric cancer stem cells with oncolytic virotherapy. Pediatr Res 71(4 Pt 2): 500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, SY, Hsieh, CL, Wu, D, Chung, LW and Johnstone, PA (2007). Tumor microenvironment promotes cancer progression, metastasis, and therapeutic resistance. Curr Probl Cancer 31: 36–100. [DOI] [PubMed] [Google Scholar]
- Ayala, G, Tuxhorn, JA, Wheeler, TM, Frolov, A, Scardino, PT, Ohori, M et al. (2003). Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin Cancer Res 9: 4792–4801. [PubMed] [Google Scholar]
- Liu, YP, Suksanpaisan, L, Steele, MB, Russell, SJ and Peng, KW (2013). Induction of antiviral genes by the tumor microenvironment confers resistance to virotherapy. Sci Rep 3: 2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moullan, N, Mouchiroud, L, Wang, X, Ryu, D, Williams, EG, Mottis, A et al. (2015). Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep 10: 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierle, EA, Ma, X, Stewart, J, Nyberg, C, Trujillo, A, Cance, WG et al. (2010). Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle 9: 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L, Iraci, N, Gherardi, S, Gamble, LD, Wood, KM, Perini, G et al. (2010). p53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer Res 70: 1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breit, S and Schwab, M (1989). Suppression of MYC by high expression of NMYC in human neuroblastoma cells. J Neurosci Res 24: 21–28. [DOI] [PubMed] [Google Scholar]
- Kuwahara, A, Hirabayashi, Y, Knoepfler, PS, Taketo, MM, Sakai, J, Kodama, T et al. (2010). Wnt signaling and its downstream target N-myc regulate basal progenitors in the developing neocortex. Development 137: 1035–1044. [DOI] [PubMed] [Google Scholar]
- Zemp, FJ, McKenzie, BA, Lun, X, Maxwell, L, Reilly, KM, McFadden, G et al. (2013). Resistance to oncolytic myxoma virus therapy in nf1(-/-)/trp53(-/-) syngeneic mouse glioma models is independent of anti-viral type-I interferon. PLoS One 8: e65801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devhare, PB, Chatterjee, SN, Arankalle, VA and Lole, KS (2013). Analysis of antiviral response in human epithelial cells infected with hepatitis E virus. PLoS One 8: e63793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, SY, Sanchez, DJ, Aliyari, R, Lu, S and Cheng, G (2012). Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci USA 109: 4239–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S, Weston, S, Kellam, P and Marsh, M (2014). IFITM proteins-cellular inhibitors of viral entry. Curr Opin Virol 4: 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins, JW, Wilson, SJ, Panis, M, Murphy, MY, Jones, CT, Bieniasz, P et al. (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472: 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbach, CJ, Paterson, JM, Lemay, CG, Falls, TJ, McGuire, A, Parato, KA et al. (2007). Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther 15: 1686–1693. [DOI] [PubMed] [Google Scholar]
- Morrow, MP, Grant, A, Mujawar, Z, Dubrovsky, L, Pushkarsky, T, Kiselyeva, Y et al. (2010). Stimulation of the liver X receptor pathway inhibits HIV-1 replication via induction of ATP-binding cassette transporter A1. Mol Pharmacol 78: 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed, M, Suzuki, R, Watanabe, N, Masaki, T, Tomonaga, M, Muhammad, A et al. (2011). Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J Biol Chem 286: 37264–37273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, A, Dinh, PX, Panda, D and Pattnaik, AK (2014). Interferon-inducible protein IFI35 negatively regulates RIG-I antiviral signaling and supports vesicular stomatitis virus replication. J Virol 88: 3103–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S, Juliana, C, Hong, S, Datta, P, Hwang, I, Fernandes-Alnemri, T et al. (2013). The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol 191: 4358–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J, Yang, B, Hu, Y, Zheng, Y, Zhou, H, Wang, Y et al. (2013). Negative regulation of Nmi on virus-triggered type I IFN production by targeting IRF7. J Immunol 191: 3393–3399. [DOI] [PubMed] [Google Scholar]
- Wang, J, Wang, Y, Liu, J, Ding, L, Zhang, Q, Li, X et al. (2012). A critical role of N-myc and STAT interactor (Nmi) in foot-and-mouth disease virus (FMDV) 2C-induced apoptosis. Virus Res 170: 59–65. [DOI] [PubMed] [Google Scholar]
- Müller, U, Steinhoff, U, Reis, LF, Hemmi, S, Pavlovic, J, Zinkernagel, RM et al. (1994). Functional role of type I and type II interferons in antiviral defense. Science 264: 1918–1921. [DOI] [PubMed] [Google Scholar]
- Ben-Israel, H, Sharf, R, Rechavi, G and Kleinberger, T (2008). Adenovirus E4orf4 protein downregulates MYC expression through interaction with the PP2A-B55 subunit. J Virol 82: 9381–9388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domachowske, JB, Bonville, CA and Rosenberg, HF (2000). Cytokeratin 17 is expressed in cells infected with respiratory syncytial virus via NF-kappaB activation and is associated with the formation of cytopathic syncytia. J Infect Dis 182: 1022–1028. [DOI] [PubMed] [Google Scholar]
- Harris, D and Engelman, A (2000). Both the structure and DNA binding function of the barrier-to-autointegration factor contribute to reconstitution of HIV type 1 integration in vitro. J Biol Chem 275: 39671–39677. [DOI] [PubMed] [Google Scholar]
- Shinohara, ML, Lu, L, Bu, J, Werneck, MB, Kobayashi, KS, Glimcher, LH et al. (2006). Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat Immunol 7: 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager, R, Penin, F, Lohmann, V and André, P (2011). Assembly of infectious hepatitis C virus particles. Trends Microbiol 19: 95–103. [DOI] [PubMed] [Google Scholar]
- Goodwin, EC, Lipovsky, A, Inoue, T, Magaldi, TG, Edwards, AP, Van Goor, KE et al. (2011). BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. MBio 2: e00101–e00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, WB, Zhu, J, Hart, B, Sui, B, Weng, K, Chang, S et al. (2009). Identification of PTCH1 requirement for influenza virus using random homozygous gene perturbation. Am J Transl Res 1: 259–266. [PMC free article] [PubMed] [Google Scholar]
- Moorman, NJ and Shenk, T (2010). Rapamycin-resistant mTORC1 kinase activity is required for herpesvirus replication. J Virol 84: 5260–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrada, L, Liang, XH, Zheng, W, Johnston, C and Levine, B (2002). Age-dependent resistance to lethal alphavirus encephalitis in mice: analysis of gene expression in the central nervous system and identification of a novel interferon-inducible protective gene, mouse ISG12. J Virol 76: 11688–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap, MW and Stoye, JP (2012). TRIM proteins and the innate immune response to viruses. Adv Exp Med Biol 770: 93–104. [DOI] [PubMed] [Google Scholar]
- Clarke, P, Meintzer, SM, Gibson, S, Widmann, C, Garrington, TP, Johnson, GL et al. (2000). Reovirus-induced apoptosis is mediated by TRAIL. J Virol 74: 8135–8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, B and Cheng, G (2007). Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J Biol Chem 282: 11817–11826. [DOI] [PubMed] [Google Scholar]
- Mullan, PB, Quinn, JE and Harkin, DP (2006). The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene 25: 5854–5863. [DOI] [PubMed] [Google Scholar]
- Tang, WF, Yang, SY, Wu, BW, Jheng, JR, Chen, YL, Shih, CH et al. (2007). Reticulon 3 binds the 2C protein of enterovirus 71 and is required for viral replication. J Biol Chem 282: 5888–5898. [DOI] [PubMed] [Google Scholar]
- Tabah, AA, Tardif, K and Mansky, LM (2014). Anti-HIV-1 activity of Trim 37. J Gen Virol 95(Pt 4): 960–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan, K, Waggoner, L, Pham, ST, Hendricks, GL, Waggoner, SN, Conlon, J et al. (2014). TRIM13 is a negative regulator of MDA5-mediated type I interferon production. J Virol 88: 10748–10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda, D, Das, A, Dinh, PX, Subramaniam, S, Nayak, D, Barrows, NJ et al. (2011). RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc Natl Acad Sci USA 108: 19036–19041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.