

The crystal structures of complexes of the SH3 domain of the Fyn tyrosine kinase with the high-affinity peptides VSL12 (class I) and APP12 (class II) have been solved at 1.6 and 2.1 Å resolution, respectively. These structures support an important role of water molecules in facilitating conformational changes to allow binding of the proline-rich motifs in the two available orientations in this family of SH3 domains.
Keywords: SH3 domain, X-ray crystal structure, Fyn tyrosine kinase, proline-rich motifs
Abstract
Interactions of proline-rich motifs with SH3 domains are present in signal transduction and other important cell processes. Analysis of structural and thermodynamic data suggest a relevant role of water molecules in these protein–protein interactions. To determine whether or not the SH3 domain of the Fyn tyrosine kinase shows the same behaviour, the crystal structures of its complexes with two high-affinity synthetic peptides, VSL12 and APP12, which are class I and II peptides, respectively, have been solved. In the class I complexes two water molecules were found at the binding interface that were not present in the class II complexes. The structures suggest a role of these water molecules in facilitating conformational changes in the SH3 domain to allow the binding of the class I or II peptides. In the third binding pocket these changes modify the cation–π and salt-bridge interactions that determine the affinity of the binding. Comparison of the water molecules involved in the binding of the peptides with previous reported hydration spots suggests a different pattern for the SH3 domains of the Src tyrosine kinase family.
1. Introduction
Protein–protein interactions are key in signal transduction and other important cell processes. Frequently, these interactions take place through the binding of proline-rich motifs (PRMs) to modular domains that recognize these sequences. At present, in spite of the large amount of structural, thermodynamic and kinetic information available, this molecular process is not fully understood (Martin-Garcia, Ruiz-Sanz et al., 2012 ▸). In the case of SH3 domains, binding studies show that the interaction of the consensus motif PpφP (where φ and p are frequently hydrophobic and proline residues, respectively) is usually weak, with affinity-constant values between 103 and 104 M −1. Besides, residues other than the prolines present in the flanking sequence are responsible for binding affinity and specificity (Viguera et al., 1994 ▸; Cussac et al., 1994 ▸; Lemmon et al., 1994 ▸) and drive the binding orientation of the PRMs: peptides with the motifs (K/R)xφPpφP and xPpφPx(K/R), classified as class I and class II motifs, respectively (Rickles et al., 1995 ▸; Feng et al., 1995 ▸; Lim et al., 1994 ▸).
Recently, a systematic study of the structures available to date of complexes of PRMs bound to SH3 domains revealed that solvent molecules might play a key role in the binding of these motifs (Palencia et al., 2010 ▸). The crystallographic structures of VSL12 and APP12 bound to c-Src SH3 have been solved at atomic resolution (Bacarizo & Camara-Artigas, 2013 ▸) and show some water molecules that might contribute to the energetics of the binding and also new features that were not revealed by the lower resolution NMR structures (Feng et al., 1994 ▸, 1995 ▸).
Determining the molecular basis of the binding affinity of PRMs to SH3 domains is fundamental to understand these transient interactions, and also for the design of high-affinity ligands for use as drugs. SH3 domains are characterized by their backbone dynamics, and an induced-fit mechanism has been proposed for their binding to PRMs (Horness et al., 2016 ▸). Transient interactions with solvent molecules might facilitate conformational states that are prone to bind PRMs in a specific orientation. Nevertheless, this information is not available in the NMR structures and only high-resolution crystal structures can provide this valuable information. Here, we present the crystallographic structures of the Fyn SH3 domain in complex with the high-affinity synthetic peptides VSL12 and APP12. These peptides also bind to Fyn SH3 with high affinity constants (∼106 M −1; Demers & Mittermaier, 2009 ▸). Comparison of these complex structures with that of the natural peptide derived from the nonstructural 5A protein from hepatitis virus (NS5A-2; A349PPIPPPRRKR359) shows some noticeable differences in the salt bridge that drives the binding orientation. Besides, the analysis of the water molecules at the binding site results in new hydration spots that have not been taken into account in previous analysis (Martin-Garcia, Ruiz-Sanz et al., 2012 ▸).
2. Materials and methods
2.1. Cloning, expression and purification of human Fyn SH3 domain
The plasmid pET-3d containing the Fyn SH3-domain gene was a generous gift from Dr L. Serrano (EMBL, Heidelberg; Noble et al., 1993 ▸). Plasmid-encoded SH3 domain was expressed in Escherichia coli strain BL21(DE3) (Novagen) using 1 mM IPTG as an inducing agent. Harvested cells were suspended in 300 mM sodium chloride, 50 mM sodium phosphate pH 8.0. After sonication, the cell lysate was centrifuged at 30 000g for 60 min at 4°C. The SH3 domain was precipitated from the supernatant with 75% saturated ammonium sulfate and was suspended in 500 mM sodium chloride, 50 mM sodium phosphate pH 6.5 (buffer A). The protein was further purified by size-exclusion chromatography on a Superdex 75 column (GE Healthcare Lifesciences) equilibrated and eluted with buffer A. Fractions containing the SH3 domain were pooled, concentrated and stored at −20°C. The Fyn domain is stable under these conditions for several months. The protein purity was checked by SDS–PAGE and the protein concentration was determined from the absorbance at 280 nm using an extinction coefficient of 16 800 M −1 cm−1. VSL12 (VSLARRPLPPLP) and APP12 (APPLPPRNRPRL) peptides were purchased from JPT Innovative Peptide Solutions (Germany).
2.2. Crystallization
Crystals of the complexes were obtained by the vapour-diffusion technique using a hanging-drop or sitting-drop setup at 25°C. Briefly, the protein complexes were prepared by mixing the Fyn SH3 domain (final concentration 10 mg ml−1 in 10 mM Tris buffer pH 8.0) with the peptides (3.4 mM in water) in a 1:2 molar ratio. Samples were kept overnight at 4°C and 4 µl droplets were then prepared by mixing 2 µl complex solution and 2 µl reservoir solution. The mixture was vapour-equilibrated against 1 ml reservoir solution. The best crystals of the Fyn SH3–VSL12 complex were obtained using 5.5 M sodium formate, 0.1 M MES pH 6.0. Crystals of the Fyn SH3–APP12 complex were obtained using 5.5 M sodium formate, 0.1 M HEPES pH 7.5.
2.3. Data collection and processing
For data collection, crystals were transferred into a cryoprotectant solution before flash-cooling in liquid nitrogen. Data sets were collected at 100 K on beamlines ID14-1 and ID23-1 at ESRF, Grenoble, France. Diffraction data were indexed and integrated with the autoPROC toolbox (Vonrhein et al., 2011 ▸) and the ISPyB information-management system (Delagenière et al., 2011 ▸). Data scaling was performed using AIMLESS (Evans & Murshudov, 2013 ▸) from the CCP4 suite (Winn et al., 2011 ▸). Data-collection statistics are collected in Table 1 ▸.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the outer shell.
| Fyn SH3–VSL12 | Fyn SH3–APP12 | |
|---|---|---|
| Diffraction source | ID14-1, ESRF | ID23-I, ESRF |
| Wavelength (Å) | 0.976 | 0.976 |
| Temperature (K) | 100 | 100 |
| Detector | ADSC Quantum Q315r | Dectris PILATUS 6M |
| Space group | P63 | P21 |
| a, b, c (Å) | 81.84, 81.84, 35.90 | 31.66, 76.66, 73.17 |
| α, β, γ (°) | 90.00, 90.00, 120.00 | 90.00, 94.74, 90.00 |
| Resolution range (Å) | 20–1.60 | 19.86–2.10 |
| Total No. of reflections | 192671 | 153807 |
| No. of unique reflections | 17446 (1318) | 20052 (1394) |
| Completeness (%) | 94.4 (72.5) | 98.2 (84.6) |
| Multiplicity | 11.0 (10.1) | 7.7 (3.7) |
| 〈I/σ(I)〉 | 71.1 (10) | 16.6 (3.0) |
| Wilson B factor (Å2) | 19.13 | 35.8 |
2.4. Structure solution and refinement
Solution and refinement of the structure were performed using the PHENIX suite (Adams et al., 2010 ▸). Molecular-replacement phasing using Phaser (Bunkóczi et al., 2013 ▸) was performed using the coordinates of the crystallographic structure of the monomeric structure of the Fyn SH3 domain (PDB code 3ua6, chain A; Martin-Garcia, Luque et al., 2012 ▸). Water molecules and residues of the n-Src loop were removed from the model. Manual model building was performed using Coot (Emsley & Cowtan, 2004 ▸; Emsley et al., 2010 ▸). Refinement was performed using phenix.refine in PHENIX (Afonine et al., 2012 ▸). The quality of the structures was checked using MolProbity (Chen et al., 2010 ▸) and PROCHECK (Laskowski, 2001 ▸). Structure-refinement statistics are collected in Table 2 ▸.
Table 2. Structure solution and refinement.
Values in parentheses are for the outer shell.
| Fyn SH3–VSL12 | Fyn SH3–APP12 | |
|---|---|---|
| Resolution range (Å) | 19.658–1.600 (1.641–1.600) | 19.868–2.100 (2.152–2.100) |
| Completeness (%) | 94.4 (72.5) | 98.2 (84.6) |
| σ Cutoff | 0 | 2 |
| No. of reflections, working set | 17446 (884) | 20025 (1711) |
| No. of reflections, test set | 885 (43) | 1019 (70) |
| Final R cryst | 0.174 (0.245) | 0.180 (0.304) |
| Final R free | 0.195 (0.264) | 0.222 (0.418) |
| No. of non-H atoms | ||
| Protein | 555 | 2062 |
| Ligand | 1 | 1 |
| Water | 40 | 58 |
| Total | 596 | 2120 |
| R.m.s. deviations | ||
| Bonds (Å) | 0.018 | 0.010 |
| Angles (°) | 1.928 | 1.118 |
| Average B factors (Å2) | ||
| Protein | 23.7 | 50.4 |
| Water | 38.3 | 68.1 |
| Ramachandran plot | ||
| Most favoured (%) | 98 | 100 |
| Allowed (%) | 2 | 0 |
3. Results and discussion
3.1. Overall structure of the complexes of the Fyn SH3 domain with the high-affinity VSL12 and APP12
Crystals of the Fyn SH3–VSL12 complex belonged to space group P63 and contain only one molecule in the asymmetric unit, whereas crystals of the Fyn SH3–APP12 complex belonged to space group P21 and have four molecules in the asymmetric unit in a pseudo-tetragonal arrangement. Superposition of the backbone atoms of the four SH3 chains in the Fyn SH3–APP12 complex showed that all molecules in the asymmetric unit are practically identical, with r.m.s.d. values of ∼0.2 Å. Comparison of any of these chains with that present in the Fyn SH3–VSL12 complex showed an r.m.s.d. value of ∼0.4 Å (Fig. 1 ▸). One of the most important differences between the structures is a hydrogen bond formed by Asp100 and Tyr93. In Fyn SH3–APP12, the average distance between these residues is 2.6 Å, but in Fyn SH3–VSL12 a water molecule (w1) is located between these residues at a hydrogen-bonding distance from each side chain. This displacement seems to be correlated with different conformers of the residue Leu101 and the burial of a second water molecule (w2), which forms hydrogen bonds to the backbone atoms of Leu101 and Tyr132 (Fig. 1 ▸). A similar interaction pattern has been found in the crystallographic structures of these peptides with the c-Src SH3 domain (Bacarizo & Camara-Artigas, 2013 ▸).
Figure 1.

Superposition of the binding site of the VSL12 (blue) and APP12 (grey) peptides. (a) At the binding site of the VLS12 complex, residues Leu11-Pro12 and Leu8-Pro9 interact with the hydrophobic pockets formed by Tyr91 and Tyr137 and by Tyr93, Tyr137 and Trp119, respectively; in the APP12 complex the hydrophobic pockets formed by Tyr91 and Tyr137 and by Tyr93, Tyr137 and Trp119 are occupied by residues Ala1-Pro2 and Leu4-Pro5, respectively. (b) Detail of the third binding pocket. In both structures, the salt bridge between Arg6VSL12 (or Arg7APP12) and Asp100 shows a dihedral angle of near 0° between the planes of the guanidinium and carboxyl groups. The distance between the guanidinium group of Arg7APP12 and the aromatic ring of Trp119 is ∼3.5 Å, while the Arg6VSL12–Trp119 distance is >4 Å. Hydrogen bonds between nearby residues are shown as dashed lines (blue, VSL12; grey, APP12). Water molecules w1 and w2 (red) are shown as spheres.
3.2. Comparison of the complex structures of the class II complexes Fyn SH3–APP12 and Fyn SH3–NS5A-2
The interaction between PRMs and SH3 domains typically shows low binding constants, as is expected for a transient interaction. However, some synthetic peptides show high binding constants, such as, for example, the APP12 and VSL12 peptides (Feng et al., 1995 ▸). The affinity of these peptides is even higher for the Fyn SH3 complexes (25°C, 20 mM phosphate buffer pH 7.0): the K d values for the Fyn SH3–VSL12 and Fyn SH3–APP12 complexes are 0.06 and 0.24 µM, respectively (unpublished results). Unexpectedly, the binding constant of the natural peptide NS5A-2 to the Fyn SH3 domain is also high, with a K d of 0.62 µM (Martin-Garcia et al., 2012 ▸). Fig. 2 ▸ shows a structural comparison of the two class II peptide complexes, Fyn SH3–NS5A-2 and Fyn SH3–APP12. The first and second binding pockets, which accommodate the canonical binding motif PpφP, do not show significant differences. In the third binding pocket, the most remarkable difference between these complexes is the geometry of the salt bridge that drives the orientation of the peptide: (i) Arg8NS5A-2 is placed between the aromatic residues Trp119 and Tyr132 and the cation–π interaction is in a T-shape form and (ii) the salt bridge between Arg8NS5A-2 and Asp100 shows a non-optimized geometry. The key role of this arginine-mediated salt bridge in the energetics of the binding of SH3 domains to PRMs is supported by the lower binding affinity of the methylated arginine in the formation of the Sam68 complexes (Bedford et al., 2000 ▸). The optimized geometry in the crystal structures of the high-affinity synthetic peptides but not in the complex of the natural peptide might account for the higher affinity of the VSL12 and APP12 peptides (Bacarizo et al., 2015 ▸). In the previous NMR structures of the c-Src SH3–VSL12 and c-Src SH3–APP12 complexes (PDB entries 1qwf and 1qwe, respectively) the salt bridge between Asp99c-Src SH3 and the arginine residue of the peptide has been modelled in a non-optimized geometry (Feng et al., 1994 ▸). In this way, molecular-dynamics calculations using these NMR structures will result in a poor estimation of the strength of this interaction.
Figure 2.

Superposition of the NS5A-2 (magenta) and APP12 (grey) peptides. The salt bridge between Asp100 and Arg7APP12 or Arg8NS5A-2 and the hydrogen bonds to nearby residues are shown as dashed lines (violet, NS5A-2; grey, APP12). The distances in Å from the guanidinium group of the arginine residue to the aromatic residues are indicated in the figure.
The conformation of aromatic residues at the binding site has been proposed to be important for the specificity of the peptide (Fernandez-Ballester et al., 2004 ▸). These authors reported that changes in the orientation of Trp119 were responsible for the binding of class I or II peptides. However, comparison of the Fyn SH3–VSL12 (class I) and Fyn SH3–APP12 (class II) structures shows the same orientation of this tryptophan residue. Nevertheless, we have identified some differences in this residue when comparing the Fyn SH3–APP12 and Fyn SH3–NS5A-2 structures, which are both class II peptides. The displacement of Trp119 in Fyn SH3–NS5A-2 allows a cation–π interaction in a T-shape form of the arginine residue in the flanking sequence (Arg8NS5A-2) with residues Trp119 and Tyr132. Even this cation–π interaction involves two aromatic residues; it is expected to be weaker than the cation–π interaction in parallel geometry found in the APP12 and VSL12 complexes (Gallivan & Dougherty, 1999 ▸).
The differences found in the comparison between the Fyn SH3–VSL12, Fyn SH3–APP12 and Fyn SH3–NS5A-2 complexes are also present in the c-Src SH3–VSL12, c-Src SH3–APP12 (Bacarizo & Camara-Artigas, 2013 ▸) and c-Src SH3–NS5A-2 complexes (Bacarizo et al., 2015 ▸): the salt bridge is optimized in the crystal structures of the VSL12 and APP12 complexes, but not in the case of the NS5A-2 complex. These crystal structures support the importance of the salt-bridge geometry in the binding of PRMs, but also indicate a relevant role of the cation–π interaction between the guanidinium group of arginine and the aromatic residues in the third binding pocket. Another important difference between the structures of the high-affinity complexes and those of the natural peptide NS5A-2 is the position of the arginine residue that forms the salt bridge. In the high-affinity peptides, the arginine is placed in the n + 2 position from the canonical motif PpφP (+xPpφP or PpφPx+, where + represents the positive charge of the arginine residue). In the NS5A-2 peptide the arginine residue is placed in the n + 3 position from the canonical motif (PpφPxx+) and no residues beyond Arg8 have been modelled in the NS5A-2 peptide. The lack of electron density in the difference maps to model residues 9–11 of the NS5A-2 peptide indicates the high flexibility of these residues as a consequence of the absence of steady interactions. However, in the Fyn SH3–APP12 complex the Asn9 side chain interacts with the backbone atoms of Trp119 and Asp118, but also with a water molecule that is also linked to the backbone atoms of Pro6, Asn8 and Asp118 (water molecule 3; w3). In the Fyn SH3–VSL12 complex the Ala4 backbone O atom is linked to the Trp119 backbone N atom, but no water molecules are found in this case.
3.3. Comparison of the hydration spots present in the Fyn SH3 complexes
Martin-Garcia, Ruiz-Sanz et al. (2012 ▸) performed a thorough analysis of all of the SH3-domain crystallographic structures available to date to determine the presence of some water molecules that are buried upon binding in the SH3 domain–PRM interface. The goal of this work was to explain the unexpected thermodynamic signature of the binding. Five hydration spots were identified based on the results obtained from analysis of the crystallographic structures of the Abl SH3 domain, as the wild type and several mutants, both free and in complex with the high-affinity synthetic peptide P41 (Palencia et al., 2010 ▸; Fig. 3 ▸). In this analysis, only water molecules mediating interactions between the protein and the ligand at the binding interface were investigated. Taking into account both thermodynamic and structural data, a dual binding mechanism was proposed in which the hydrophobic interactions of the proline residues with the aromatic residues on the surface of the SH3 domains was complemented by the formation of hydrogen bonds between water molecules and the protein/peptide residues. Unfortunately, at this time no crystallographic structures of c-Src and Fyn SH3 domains in complex with PRMs were available, with the exception of the structure of the Fyn SH3 domain in complex with the synthetic peptide 3BP-2 (PPAYPPPPPVP; PDB entry 1fyn; Musacchio et al., 1994 ▸). Nevertheless, this structure must be analysed with care as the peptide molecule is placed in a special position and two molecules of the peptide with opposite orientations are overlaid.
Figure 3.
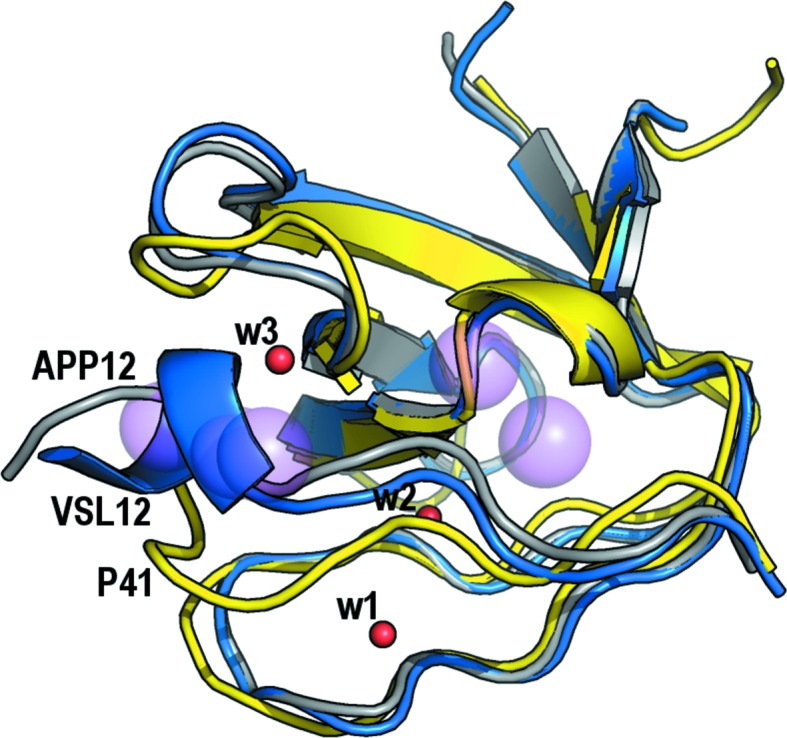
Comparison of the hydration spots present in the Fyn SH3–VSL12 (blue) and Fyn SH3–APP12 (grey) complexes with those described in the Abl SH3–P41 complex (yellow). Water molecule w3 (red sphere) mediates hydrogen bonds between the backbone atoms of the APP12 peptide and residues in the n-Src loop of the Fyn SH3 domain. The three water molecules found in the Fyn SH3 complexes are located at any of the hydration spots previously reported in the Abl SH3–PRM complexes (pink spheres).
The binding of PRMs to the Fyn SH3 domain is also characterized by the same unexpected thermodynamic signature as found in other SH3 domains: favourable enthalpy and unfavourable entropy changes (Demers & Mittermaier, 2009 ▸; Martin-Garcia, Luque et al., 2012 ▸). However, the water molecules previously reported to explain this unexpected thermodynamic signature in the Abl SH3–P41 complex are not present in the complexes of the Fyn and c-Src SH3 domains (Fig. 3 ▸). Besides, the water molecules found in the Fyn SH3–VSL12 complex, w1 and w2, are not present in the APP12 complex, while w3 appears in the Fyn SH3–APP12 complex but not in the Fyn SH3–VSL12 complex. The contribution of water molecules to enthalpy and entropy might come from both implicit water molecules, such as those discussed in this work, and also explicit solvent water molecules. In addition, other contributions must be taken into account: salt-bridge formation is expected to make an unfavourable contribution to the enthalpy because of the increase in hydration enthalpy when two charged species are separated; cation–π interactions are expected to make energetic contributions of the same order of magnitude as hydrogen bonds. Although electrostatic interactions are long-distance interactions, in our case, where an arginine residue is responsible for the interactions, the geometry is also a factor to take into account.
Computer modelling using reliable structural information is able to dissect the particular contribution of each interaction. However, these calculations must also bear in mind the flexibility of the ligand and the protein and its contribution to the conformational entropy. In this way, computer modelling of the energetic contribution of each interaction present in the structures of the complexes must bear in mind the solvent effect and its role in the conformational changes observed upon binding. Water molecules found in the two complex structures, class I and II, as well as conformational changes in the SH3 domain are also present in the crystallographic structures of the unbound SH3 domain. Thus, the Fyn SH3-domain crystal structure contains two molecules in the asymmetric unit with different conformations, the same as the crystal structure of the c-Src SH3 domain. Each molecule has a different conformation: chain B contains the same water molecules w1 and w2 as found in the Fyn SH3–VSL12 complex, while chain A does not (Bacarizo et al., 2014 ▸). Additionally, Leu101 (Leu100 in c-Src SH3) shows different conformers in each chain and we have proposed a relevant role of this residue in the conformational changes that take place to accommodate the binding of PRMs in the two available orientations (Bacarizo et al., 2015 ▸).
The crystallographic structures reported in this work show some important conformational changes in the SH3 domain upon the binding of PRMs and suggest a role of the protein dynamics, as has been addressed using spectroscopic techniques (Horness et al., 2016 ▸).
Supplementary Material
PDB reference: Fyn SH3 domain, complex with the high-affinity peptide VSL12, 4eik
PDB reference: complex with the high-affinity peptide APP12, 4znx
Acknowledgments
This research was funded by the Spanish Ministry of Science and Innovation and Ministry of Economy and Competitiveness and FEDER (EU) (BIO2009-13261-C02-01/02 and BIO2012-39922-C02-01/02) and by the Andalusian Regional Government and FEDER (EU) (P09-CVI-5063). Data collection was supported by the European Synchrotron Radiation Facility (ESRF), Grenoble, France (BAG proposals MX-1406, MX-1541 and MX-1629) and ALBA, Barcelona, Spain (BAG 2012010072 and 2012100378).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Bacarizo, J. & Camara-Artigas, A. (2013). Acta Cryst. D69, 756–766. [DOI] [PubMed]
- Bacarizo, J., Martínez-Rodríguez, S. & Cámara-Artigas, A. (2015). J. Struct. Biol. 189, 67–72. [DOI] [PubMed]
- Bacarizo, J., Martinez-Rodriguez, S., Martin-Garcia, J. M., Andujar-Sanchez, M., Ortiz-Salmeron, E., Neira, J. L. & Camara-Artigas, A. (2014). PLoS One, 9, e113224. [DOI] [PMC free article] [PubMed]
- Bedford, M. T., Frankel, A., Yaffe, M. B., Clarke, S., Leder, P. & Richard, S. (2000). J. Biol. Chem. 275, 16030–16036. [DOI] [PubMed]
- Bunkóczi, G., Echols, N., McCoy, A. J., Oeffner, R. D., Adams, P. D. & Read, R. J. (2013). Acta Cryst. D69, 2276–2286. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cussac, D., Frech, M. & Chardin, P. (1994). EMBO J. 13, 4011–4021. [DOI] [PMC free article] [PubMed]
- Delagenière, S. et al. (2011). Bioinformatics, 27, 3186–3192. [DOI] [PubMed]
- Demers, J.-P. & Mittermaier, A. (2009). J. Am. Chem. Soc. 131, 4355–4367. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Feng, S., Chen, J. K., Yu, H., Simon, J. A. & Schreiber, S. L. (1994). Science, 266, 1241–1247. [DOI] [PubMed]
- Feng, S., Kasahara, C., Rickles, R. J. & Schreiber, S. L. (1995). Proc. Natl Acad. Sci. USA, 92, 12408–12415. [DOI] [PMC free article] [PubMed]
- Fernandez-Ballester, G., Blanes-Mira, C. & Serrano, L. (2004). J. Mol. Biol. 335, 619–629. [DOI] [PubMed]
- Gallivan, J. P. & Dougherty, D. A. (1999). Proc. Natl Acad. Sci. USA, 96, 9459–9464. [DOI] [PMC free article] [PubMed]
- Horness, R. E., Basom, E. J., Mayer, J. P. & Thielges, M. C. (2016). J. Am. Chem. Soc. 138, 1130–1133. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A. (2001). Nucleic Acids Res. 29, 221–222. [DOI] [PMC free article] [PubMed]
- Lemmon, M. A., Ladbury, J. E., Mandiyan, V., Zhou, M. & Schlessinger, J. (1994). J. Biol. Chem. 269, 31653–31658. [PubMed]
- Lim, W. A., Richards, F. M. & Fox, R. O. (1994). Nature (London), 372, 375–379. [DOI] [PubMed]
- Martin-Garcia, J. M., Luque, I., Ruiz-Sanz, J. & Camara-Artigas, A. (2012). Acta Cryst. D68, 1030–1040. [DOI] [PubMed]
- Martin-Garcia, J. M., Ruiz-Sanz, J. & Luque, I. (2012). Biochem. J. 442, 443–451. [DOI] [PubMed]
- Musacchio, A., Saraste, M. & Wilmanns, M. (1994). Nature Struct. Biol. 1, 546–551. [DOI] [PubMed]
- Noble, M. E. M., Musacchio, A., Saraste, M., Courtneidge, S. A. & Wierenga, R. K. (1993). EMBO J. 12, 2617–2624. [DOI] [PMC free article] [PubMed]
- Palencia, A., Camara-Artigas, A., Pisabarro, M. T., Martinez, J. C. & Luque, I. (2010). J. Biol. Chem. 285, 2823–2833. [DOI] [PMC free article] [PubMed]
- Rickles, R. J., Botfield, M. C., Zhou, X.-M., Henry, P. A., Brugge, J. S. & Zoller, M. J. (1995). Proc. Natl Acad. Sci. USA, 92, 10909–10913. [DOI] [PMC free article] [PubMed]
- Viguera, A. R., Arrondo, J. L., Musacchio, A., Saraste, M. & Serrano, L. (1994). Biochemistry, 33, 10925–10933. [DOI] [PubMed]
- Vonrhein, C., Flensburg, C., Keller, P., Sharff, A., Smart, O., Paciorek, W., Womack, T. & Bricogne, G. (2011). Acta Cryst. D67, 293–302. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Fyn SH3 domain, complex with the high-affinity peptide VSL12, 4eik
PDB reference: complex with the high-affinity peptide APP12, 4znx