
Figure 1. Workflow of the experimental strategy used in this study.
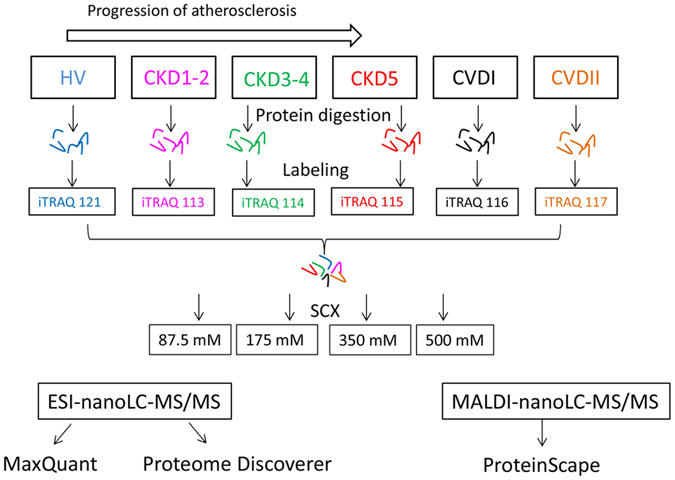
Plasma samples from six experimental groups were trypsin digested, labeled with isobaric tags, pooled and then purified and fractionated using SCX method. Quantitative proteomic analyses were simultaneously performed using ESI-nanoLC-MS/MS and MALDI-nanoLC-MS/MS and then obtained data were analyzed with three types of software: MaxQuant, ProteinScape and Proteome Discoverer. Only proteins identified by all software were found to have a differential accumulation level.