

Abstract
More than 60 members of the Rab GTPase family exist in the human genome. However, our current understanding is only limited to the role of small Rab GTPases in membrane trafficking. Here we show that CRACR2A encodes a lymphocyte-specific “large Rab GTPase” containing multiple functional domains including EF-hand motifs, proline-rich and Rab GTPase domains with an unconventional prenylation site. We demonstrate its direct role in activation of the Ca2+ and the Jnk signaling pathways upon T cell receptor (TCR) stimulation using gene silencing and transgenic animal models. Mechanistically, vesicles containing this Rab GTPase translocate from the Golgi into the immunological synapse (IS) to activate these signaling pathways. The interaction between proline-rich domain of this Rab GTPase and a guanidine nucleotide exchange factor/scaffold protein Vav1 is essential for accumulation of these vesicles at the IS. Furthermore, we demonstrate that GTP binding and prenylation are closely linked to membrane association, stability, and thereby activation of downstream signaling by this large GTPase. Our findings reveal a novel function of a large Rab GTPase in TCR signaling pathways, which is potentially shared by other GTPases with similar domain architecture.
INTRODUCTION
Activation of T cells requires their direct contact with the antigen-presenting cells (APCs). The binding of TCRs to cognate peptide-major histocompatibility complexes (MHCs) induces clustering of the TCRs and recruitment of the kinases Lck (lymphocyte-specific protein tyrosine kinase) and ZAP70 (ζ-chain-associated protein kinase). These kinases phosphorylate a signaling adaptor Lat that forms a signalosome, which contains phospholipase C-γ1 (PLCy1) and Vav1 (1–3). PLCγ1 produces a second messenger inositol 1,4,5-triphosphate (InsP3) that binds to the InsP3 receptor on the endoplasmic reticulum (ER) and triggers depletion of the ER Ca2+ store. By sensing ER Ca2+ depletion, stromal interaction molecule 1 (STIM1) translocates to the plasma membrane (PM)-proximal regions, and activates Orai1, the pore subunit of the CRAC (Ca2+ release-activated Ca2+) channels (4–6). Vav1, a guanine nucleotide exchange factor (GEF) and adaptor molecule, accumulates at the immunological synapse (IS) and recruits small G proteins such as Rac1 and CDC42 (cell division control protein 42 homolog) to activate the c-Jun N-terminal kinase (Jnk) and p38 MAPK (mitogen-activated protein kinase) pathways (7). Activation of both the Ca2+ and MAPK signaling pathways are essential for the differentiation of helper T cells and dysregulation of these pathways result in various immune-related diseases in humans and mice (8–10).
In addition to its localization at the PM, Lat also exists in subsynaptic vesicles that translocate into the PM-proximal regions of the immunological synapse after TCR stimulation (11–13). Recruitment of this pool of Lat is important for its phosphorylation. Lat-containing vesicles utilize a SNARE (soluble N-ethylmaleimide-sensitive protein-attached protein receptor)-dependent trafficking mechanism for their recruitment. A v-SNARE protein VAMP7 guides these vesicles into the PM potentially by docking to the t-SNARE proteins, in a mechanism that does not involve actual membrane fusion (14). These results suggest that components or functional homologues of the molecular machinery utilized in trafficking of synaptic vesicles in the neuronal synapse such as SNAREs and small Rab GTPases play an important role in the trafficking of subsynaptic vesicles in T cells. However, the importance of these subsynaptic vesicles in TCR signaling has been uncovered only recently and the identity and functions of these subsynaptic vesicles in T cell activation needs further investigation.
More than 60 Rab GTPases exist to regulate vesicle trafficking between organelles in the human genome. Rab GTPases broadly control vesicle budding, uncoating, motility and fusion through recruitment of effector molecules including sorting adaptors, tethering factors and motors (15, 16). Functions of Rab GTPases (e.g. membrane association) are regulated by both GTP binding and prenylation (attachment of isoprenoid lipids) (17). GTP-bound Rab GTPases are retained at the donor membrane to initiate trafficking to the target organelles while GDP-bound forms (after GTP hydrolysis) detach from the membrane and move to the cytoplasm. GDP-bound Rab GTPases are recycled into the membranes by the exchange of GDP with GTP. C-terminal prenylation of Rab GTPases is also essential for membrane association. Depending on small GTPase families, different isoprenoid units are attached. Ras GTPases are farnesylated by farnesyl transferase while Rac and Rho GTPases are geranylgeranylated by type-I geranylgeranyl transferase (GGT). Rab GTPases are also geranylgeranylated, but by the type-II enzyme. Statin family drugs are useful tools to investigate protein prenylation because they are inhibitors of 3-hydroxyl-3-methyl-glutaryl-CoA (HMG-CoA) reductase, the key rate-liming enzyme in the cholesterol synthesis pathway. Statins suppress generation of farnesyl and geranygeranyl pyrophosphate, substrates of prenyl transferases, and thus inhibit prenylation of small G proteins (18). Statins including atorvastatin are widely prescribed for their cholesterol-lowering effects. Interestingly, statin treatment also decreases TCR signaling and these drugs are also used to suppress autoimmune diseases in clinics (19–21). These results emphasize the critical role of prenylation of small G proteins in intracellular signaling for T cell activation.
Although the role of Rab GTPases in membrane trafficking has been emphasized, surprisingly little is known about their direct involvement in intracellular signaling. Moreover, our current understanding of the Rab GTPase family is mostly limited to roles of small proteins of 20–25 kDa in size. Here we report a novel function of a unique “large Rab GTPase”, CRACR2A isoform a (CRACR2A-a) that contains multiple functional domains including the previously identified N-terminal CRAC channel-regulating domain (22), a proline-rich protein-interacting domain, and a C-terminal Rab GTPase domain. Our results show that various domains of this large Rab GTPase contribute to its translocation from the Golgi to the IS in subsynaptic vesicles that are clearly different from the known Lat-containing vesicles, to activate the Ca2+-NFAT and the Jnk signaling pathways. Mechanistically, the proline-rich domain was important for recruitment of these vesicles into the IS via Vav1 interaction and the GTPase domain regulated its membrane association by GTP binding and prenylation. These observations provide novel insights into the role of subsynaptic vesicles to mediate the signaling pathways for T cell activation.
RESULTS
A GTP-GDP switch regulates Golgi membrane association of a large Rab GTPase encoded by CRACR2A
Human genome database shows two transcriptional isoforms of CRACR2A, CRACR2A isoform a (NM_001144958.1, CRACR2A-a) and isoform c (NM_032680.3, CRACR2A-c). However, the current mouse genome database annotates only one isoform, CRACR2A-c (NM_001033464.3). In the previous work, we showed that CRACR2A-c, which was annotated in both the human and mouse databases, contains N-terminal EF hands and two coiled-coil domains that regulate CRAC channel function (22). In this study, we validated the existence of the long isoform, CRACR2A-a in both human and murine T cells, and examined its functions that are common and distinct from the short isoform, CRACR2A-c. CRACR2A-a was of particular interest because, based on the predicted amino acid sequence, it contains the Orai1 and STIM1-interacting domain (shared by CRACR2A-c) together with other potential functional domains (Fig. 1A). First, we validated the presence of CRACR2A proteins in human leukemic T cell line, Jurkat cells, by immunoblotting (CRACR2A-a, ~90 kDa; CRACR2A-c; ~45 kDa) (fig. S1A and see below for murine tissues). Next, to confirm the conserved function of Orai1 and STIM1-interacting domain in CRACR2A-a, we reconstituted its expression in Jurkat cells depleted of both CRACR2A isoforms. Consistent with previous observations, CRACR2A depletion showed a profound reduction in SOCE (fig. S1, B and C). Exogenous expression of either CRACR2A-a or CRACR2A-c isoforms rescued SOCE in CRACR2A-depleted Jurkat T cells, validating their conserved function in CRAC channel activation.
Figure 1. GTP binding regulates localization at the Golgi and activation of the Jnk MAPK pathway by CRACR2A-a after T cell receptor stimulation.
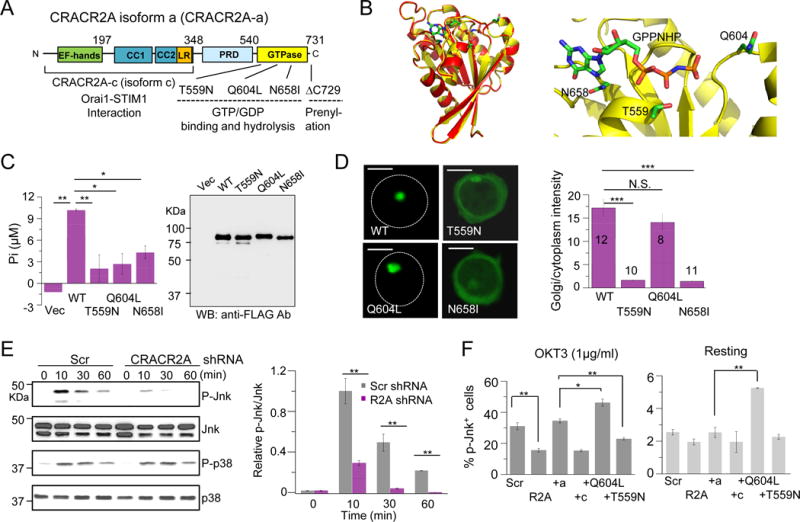
(A) Schematic showing the predicted domain structure of human CRACR2A-a. CRACR2A-a and CRACR2A-c share EF-hand motifs, coiled-coil domains (CC1 and CC2) and leucine-rich region (LR), which interact with the Orai1-STIM1 complex to regulate Ca2+ entry. CRACR2A-a contains additional proline-rich domain (PRD) and a predicted Rab GTPase domain with a putative prenylation site at the C terminus. The fragments and mutants of CRACR2A-a used in this study are indicated.
(B) Homology modeling of CRACR2A-a GTPase domain (yellow) with Rab3a (red). Sequence alignment between GTPase domain of CRACR2A-a and Rab3a gave a continuous alignment with sequence identity of 46% and similarity of 65% (Clustal Omega) (left). MODELLER (41) was used for homology modeling of CRACR2A-a GTPase domain to a high-resolution structure of a GPPNHP-bound Rab3a (PDB ID: 3RAB). A zoomed-in view of the GPPNHP binding site (right). GPPNHP and side-chains of residues important for GTP binding and hydrolysis, Thr559, Gln604 and Asn658 are shown in stick representation. A loop consisting of residues 561–570 was removed for clarity. Panels were generated using PyMOL (Version 1.5.0.4 Schrödinger, LLC).
(C) GTP hydrolysis activity of WT and mutant CRACR2A-a proteins. Intrinsic GTP hydrolysis activity was measured using immuno-purified FLAG-tagged indicated CRACR2A-a proteins expressed in HEK293 cells (left). Bar graph shows average ± SEM from one of three experiments, performed in triplicates. Similar protein levels in each sample were verified by immunoblotting with anti-FLAG antibody (right).
(D) Subcellular localization of WT and mutant CRACR2A-a proteins. Confocal microscopy images of GFP-fused WT and indicated mutant CRACR2A-a proteins expressed in Jurkat cells to examine their subcellular localization. White circles mark the periphery of the cells. Scale bar, 5 μm. Bar graph shows densitometry analysis (in indicated number of cells) of relative fluorescence intensity of GFP in the Golgi v/s the cytoplasm. Golgi was labeled by staining for endogenous Rab8a for quantification. Also see fig.S4.
(E) Activation of signaling pathways after TCR stimulation of control and CRACR2A-depleted Jurkat cells. Jurkat cells stably expressing control (Scr) or CRACR2A-depleting shRNAs were stimulated with anti-CD3 antibodies for indicated times and examined for phosphorylation of p38 and Jnk by immunoblotting. Blots are representative of three independent experiments. Right: Densitometry analysis of relative band intensities is shown as mean ± SEM of three independent experiments. Also see fig. S5A.
(F) Control (Scr), CRACR2A-depleted (R2A), and CRACR2A-depleted cells expressing shRNA-resistant WT CRACR2A-a (+a), CRACR2A-c (+c) or indicated mutant CRACR2A-a proteins were stimulated with anti-CD3 antibody for 10 mins (left) or kept under resting conditions (right) and examined for phosphorylated Jnk1 and Jnk2 by intracellular staining. Graphs show average ± SD from three independent experiments. N.S. Not Significant, * p<0.05, ** p<0.005, *** p<0.0005.
Compared to CRACR2A-c, CRACR2A-a contains additional proline-rich domain (PRD) and a Rab GTPase domain with a predicted prenylation site in its C terminus (Fig. 1A). The amino acid sequence of the GTPase domain showed high similarity to that of Rab GTPases and three-dimensional homology modeling to a high-resolution crystal structure of Rab3a (23) showed almost complete overlap (Fig. 1B). The predicted GTPase domain also closely resembles that of other small G proteins including Rac1 and CDC42 in the overall core fold (fig. S2) (23). The GTPase domain contains characteristic P-loop, switch I and II regions of small G proteins. P-loop and switch I region make contact with the γ-phosphate of GTP via Mg2+ ions while switch II region plays a major role in hydrolysis of GTP by interaction with phosphates and water molecules (23, 24). To confirm the role of these conserved residues, we created substitution of a Thr residue within the P-loop that is predicted to cause preferential GDP binding (T559N) (16, 24). The Gln in the switch II region which serves as a catalytic residue for intrinsic GTP hydrolysis was replaced with a Leu residue (Q604L) (24–26). Q604L mutant was predicted to be in a constitutively GTP-bound form. The conserved, predicted guanidine-binding G4 motif contained an Asn residue, which was replaced with Ile (N658I). This mutation was predicted to abolish binding of both GTP and GDP. Wild type (WT) CRACR2A-a hydrolysed GTP, which validates the presence of a functional GTPase domain (Fig. 1C). The mutants T559N, Q604L, and N658I showed a pronounced reduction in GTPase activity possibly due to defects in GTP binding (T559N and N658I) and hydrolysis (Q604L). These results suggest that the GTPase domain of CRACR2A-a, bearing a high similarity to those of small G proteins is functionally active.
Rab GTPases are known to be predominantly involved in vesicle trafficking and often localize in Golgi and secretory vesicles. Therefore, we determined the localization of fluorescently tagged CRACR2A-a by co-staining with various Golgi markers, including GM130, Golgin97, and Rab8a. Among these, CRACR2A-a showed significant colocalization with Rab8a, which marks TGN and associating vesicles (27) (fig. S3A). Endogenous CRACR2A-a also showed significant colocalization with Rab8a (fig. S3B). Therefore, we concluded that CRACR2A-a localizes primarily at the Golgi, more specifically with the membranes of the TGN and associating vesicles in resting T cells. CRACR2A-a was unique due to its predominant localization to Golgi membrane while other small GTPases such as Rac1 and CDC42 showed a broad distribution between Golgi membrane and the cytoplasm (fig. S3C). Next, we examined how GTP/GDP-binding influences the intracellular localization. GDP-bound T559N and guanidine-binding defective N658I mutants showed a predominantly cytoplasmic localization while GTP-bound Q604L mutant localized to the Golgi, similar to WT CRACR2A-a (Fig. 1D and fig. S4). Together, these data suggest that CRACR2A-a localizes to Golgi membrane in resting T cells, and GTP binding strictly regulates its localization.
CRACR2A-a plays a unique role in activation of the Jnk signaling pathway after TCR stimulation
To determine whether CRACR2A-a is important for intracellular signaling pathways other than Ca2+ signaling, we examined phosphorylation levels of various signaling molecules upon TCR stimulation from control and CRACR2A-depleted T cells. While CRACR2A depletion did not significantly affect phosphorylation of upstream signaling molecules such as PLCγ1 and Lat, or downstream signaling molecules including extracellular signal-related kinase (Erk) or p38, we observed a profound reduction in Jnk1/2 phosphorylation (Fig. 1E and fig. S5A). These results suggest that CRACR2A-a plays an important role in selective signaling pathways. To determine whether this function was common or unique to CRACR2A isoforms, we checked recovery of Jnk phosphorylation in presence of individual isoforms. Between the two isoforms, only CRACR2A-a expression rescued the defect in Jnk phosphorylation almost completely (Fig. 1F), identifying its unique role in modulation of the Jnk pathway.
To examine how GTP binding influences Jnk activation by CRACR2A-a, we expressed siRNA-resistant mutants of CRACR2A-a in Jurkat cells depleted of endogenous CRACR2A proteins. With TCR stimulation, GTP-bound Q604L mutant showed enhanced Jnk phosphorylation compared to WT while GDP-bound T559N did not show any rescue (Fig. 1F). These results suggested that GTP binding and Golgi localization are required for Jnk activation mediated by CRACR2A-a. Under resting conditions, only constitutively GTP-bound Q604L mutant significantly enhanced Jnk phosphorylation compared to WT (Fig. 1F). The Jnk pathway plays a broad role in T cell activation including production of cytokines (e.g. IL-2) and expression of T cell activation markers (e.g. CD69) via activation of the AP1 transcription factors (28). To examine the long-term outcomes of Jnk signaling, we measured CD69 expression in CRACR2A-depleted Jurkat T cells expressing wild type or mutant CRACR2A-a proteins. CRACR2A-depleted Jurkat T cells showed reduced CD69 expression that was rescued by WT CRACR2A-a, but not by CRACR2A-c and T559N, similar to data obtained for Jnk phosphorylation (fig. S5B). Interestingly, GTP-bound Q604L mutant that showed an enhanced rescue of Jnk phosphorylation after short-term stimulation, did not rescue CD69 expression in a long term, instead, it showed a dominant negative effect. This dominant negative effect by Q604L mutant may be caused by induction of a negative feedback on the AP1 signaling pathway due to excessive Jnk activation. In addition to the mutants involved in GTP binding, we also checked the effect of mutation within the Ca2+-binding EF-hand domain (97DAD99>AAA), which abolished Ca2+ binding in CRACR2A-c and influenced its interaction with Orai1-STIM1 complex, on CRACR2A-a localization and function (22). However, the EF-hand mutant of CRACR2A-a showed Golgi membrane localization similar to wildtype and could rescue Jnk phosphorylation when expressed in CRACR2A-depleted cells (fig. S5, C and D). Therefore, the Ca2+-sensing function of CRACR2A-a does not seem to play a role in subcellular localization or Jnk activation under these conditions. Together, these results not only validated the presence of CRACR2A-a in addition to CRACR2A-c in T cells, but also showed that CRACR2A-a is involved in regulation of both the Ca2+ and the Jnk signaling pathways while the role of CRACR2A-c is limited to Ca2+ signaling. Furthermore, these results indicate that CRACR2A-a-mediated phosphorylation of Jnk required its localization to the Golgi membranes, which is regulated by GTP binding.
CRACR2A deficiency causes a defect in the Ca2+-NFAT and Jnk signaling pathways in primary T cells
The current mouse genomic database annotates only one CRACR2A isoform, CRACR2A-c (NM_001033464.3). However, immunoblotting for detection of CRACR2A in murine tissues validated the presence of CRACR2A-a, which was expressed abundantly in the spleen and lymph nodes (Fig. 2A). To understand the gene structure of mouse CRACR2A-a, we cloned its cDNA using primers homologous to human cDNA. Amino acid sequence alignment between the human and mouse CRACR2A-a proteins (deduced from the cloned cDNA sequence) showed 80.1% sequence identity and 91.4% sequence similarity (Clustal Omega, fig. S6A). Using the cDNA sequence information, we generated an intron/exon map encoding murine CRACR2A-a (fig. S6B). Based on this map, we designed to delete exons 3 and 4 to abolish expression of both CRACR2A isoforms considering their redundant role in Ca2+-NFAT signaling (Fig. 2B and fig. S6C). CRACR2Afl/fl; CD4Cre mice were generated to ablate CRACR2A expression in the T cell lineage, which was validated by immunoblotting (Fig. 2B). In consistence with the role of CRACR2A proteins in Ca2+ signaling in Jurkat T cells, CRACR2A-deficient naïve T cells showed a significant decrease in Ca2+ entry after TCR stimulation and passive store depletion with thapsigargin (Fig. 2C). Expression of NFATc1, but not NFATc2 is known to be induced upon TCR stimulation in a Ca2+-NFAT pathway-dependent manner (29). Accordingly, we observed reduced auto-induction of NFATc1 in CRACR2A-deficient naïve T cells upon TCR stimulation while the abundance of NFATc2 remained unchanged, validating the role of CRACR2A in long-term regulation of the Ca2+-NFAT pathway (Fig. 2D). Furthermore, we observed a significant reduction in Jnk phosphorylation in CRACR2A-deficient naïve T cells upon TCR stimulation (Fig. 2E). Impaired activation of the Ca2+-NFAT and Jnk pathways led to reduced IL-2 production by CRACR2A-deficient cells in consistence with the results from CRACR2A-depleted Jurkat cells (fig. S7, A and B). These observations from the analyses of CRACR2A-deficient primary T cells affirm our prior data from CRACR2A-depleted Jurkat T cells demonstrating an important role of CRACR2A proteins in the Ca2+-NFAT and Jnk signaling pathways.
Figure 2. CRACR2A deficiency impairs activation of the Ca2+-NFAT and the Jnk MAPK signaling pathways in primary T cells.

(A) Detection of CRACR2A-a from indicated murine tissues by immunoblotting. β-actin – loading control.
(B) Schematic of targeting strategy to delete exons 3 and 4 of mouse CRACR2A gene (left). Also see fig. S6C. Expression of CRACR2A-a in control (WT, CRACR2Afl/fl; Cre−) and knockout (KO, CRACR2Afl/fl; CD4Cre) CD4+ T cells cultured under non-polarizing conditions for 4 days (right).
(C) Store-operated Ca2+ entry (SOCE) in WT and CRACR2A-deficient naïve T cells. Averaged (± SEM) SOCE responses from control (n = 81) and CRACR2A-deficient (n = 66) CD4+CD25− naïve T cells after stimulation with anti-CD3 antibody and subsequently ionomycin in the presence of external solution containing 2 mM Ca2+ (left). Bar graphs show average ± SEM from three independent experiments. Averaged (± SEM) SOCE responses from control (n = 73) and CRACR2A-deficient (n = 76) CD4+CD25− naïve T cells after passive store depletion with thapsigargin in the presence of external solution containing 0.5 or 2.0 mM Ca2+ as indicated (right). Bar graphs show average ± SEM from three independent experiments.
(D) NFATc1 and NFATc2 expression in whole cell lysates from control and CRACR2A-deficient naïve T cells stimulated with anti-CD3 and anti-CD28 antibodies for indicated times. β-actin –loading control. Right: Densitometry analysis of relative band intensities is shown as mean ± SEM of three independent experiments.
(E) Flow plots showing expression of phosphorylated Jnk in WT and CRACR2A-deficient T cells stimulated with anti-CD3 antibody for indicated times. The line graph shows average ± SDM from three independent experiments.
N.S. Not Significant, * p<0.05, ** p<0.005, *** p<0.0005.
CRACR2A-a translocates from the Golgi to the immunological synapse via subsynaptic vesicles distinct from Lat-containing vesicles
Next, we examined how the Golgi-resident CRACR2A-a plays an active role in TCR signaling. To examine if CRACR2A-a translocates from the Golgi into the IS where signaling molecules cluster to activate the Jnk pathway, we performed confocal microscopy analyses of T cells stimulated on anti-CD3 antibody-coated coverslips. Upon stimulation, endogenous CRACR2A-a translocated in vesicles from the Golgi to the central area of the cell-coverslip contact site (Fig. 3A and Movie S1). Furthermore, incubation of Jurkat T cells with superantigen-pulsed Raji B cells also showed translocation of CRACR2A-a-containing vesicles into the IS in a short time and the entire Golgi was polarized at longer time points (Fig. 3B). In addition, we identified an important role of the proline-rich domain of CRACR2A-a in translocation of these vesicles. We observed numerous CRACR2A-a-containing vesicles in ΔPRD-expressing Jurkat cells, however, these vesicles failed to accumulate at the IS (Fig. 3B). These data suggest that the PRD region may be important for protein interactions essential for the recruitment of CRACR2A-a-containing vesicles into the IS. The defect in translocation of the ΔPRD mutant also influenced downstream signaling. Compared to WT CRACR2A-a, expression of the ΔPRD mutant in CRACR2A-depleted T cells did not significantly rescue phosphorylation of Jnk or CD69 expression (Fig. 3C).
Figure 3. CRACR2A-a translocates from the Golgi to the immunological synapse via subsynaptic vesicles distinct from Lat-containing vesicles.

(A) CRACR2A-a translocates in vesicles from the Golgi to the contact site of T cells and anti-CD3 antibody-coated coverslips. Representative confocal images showing localization of endogenous CRACR2A-a in a Jurkat cell allowed to spread on stimulatory anti-CD3 antibody-coated coverslip for 10 mins. Golgi was labeled with WGA-594. Individual confocal sections at indicated depths from the bottom of the cell are shown. White boxes highlight the region magnified in the inset. Scale bar, 2 μm. Images are representative of 17 cells. Also see Movie S1.
(B) Localization of Vav1-GFP with mCherry-fused WT and ΔPRD mutant of CRACR2A-a at the immunological synapse formed between Jurkat cells and SEE-pulsed Raji B cells. Left most images show overlap of bright field and GFP images (APCs depicted by * in all the images). The top panel shows image of a cell incubated with an APC for a short time (10 mins), while the middle and bottom panels show images of cells incubated with APCs for 20 mins. White boxes highlight the immunological synapse, which is magnified in the inset. Scale bar, 5 μm, unless indicated. Images are representative of 18 (WT) or 12 (ΔPRD) cells.
(C) Control (Scr), CRACR2A-depleted (R2A), and CRACR2A-depleted Jurkat cells expressing shRNA-resistant WT or ΔPRD mutant CRACR2A-a were stimulated with anti-CD3 antibody for 10 mins to examine for phosphorylated Jnk1/2 by intracellular staining (left) and with anti-CD3 and anti-CD28 antibodies for 18 hours to examine surface expression of CD69 (right). Graphs show average ± SD from three independent experiments. ** p<0.005.
(D) CRACR2A-a-containing vesicles are distinct from those containing Lat. Jurkat cells expressing Lat-GFP and mCherry-CRACR2A-a were allowed to spread on anti-CD3 antibody-coated coverslip and their localization at the contact site (bottom) and 1.4 μm away from the contact site were determined by confocal microscopy. The Merge panel shows average Pearson’s correlation coefficient values (± SEM) from 13 cells. Scale bar, 5 μm unless indicated.
(E) Co-localization of Lat-GFP with mCherry-fused CRACR2A-a at the contact site between Jurkat cells and stimulatory coverslips. Live-cell TIRF images at indicated time points from a Jurkat cell coexpressing Lat-GFP and mCherry-CRACR2A-a dropped on anti-CD3 antibody-coated coverslip. The Merge panel shows average Pearson’s Correlation Coefficient values (± SEM) from 11 cells. Scale bar, 5 μm. Also see Movie S2.
It was generally thought that PM-resident TCR signaling molecules cluster at the IS to activate downstream signaling pathways. However, recently it has been found that subsynaptic vesicles also play an important role in recruitment of signaling molecules. The most well studied vesicles are the ones carrying Lat, translocation of which is regulated by the vesicle docking protein, VAMP7 (14). Therefore, we checked whether the CRACR2A-a-containing vesicles carry Lat as a cargo. However, we could not detect any significant co-localization between Lat and CRACR2A-a-containing vesicles in T cells stimulated on anti-CD3 antibody-coated coverslip (Fig. 3D). We also analyzed the accumulation of Lat and CRACR2A-a-containing vesicles at the PM-proximal regions using total internal reflection fluorescence (TIRF) microscopy. The TIRF images of Lat-GFP represented Lat molecules pre-localized at the PM as well as the translocated ones contained within the subsynaptic vesicles (Fig. 3E). However, we did not observe any significant co-localization between Lat and CRACR2A-a-containing vesicles (Movie S2). These results suggest that CRACR2A-a-containing vesicles are novel and distinct from the previously defined subsynaptic vesicles containing Lat.
Interaction with Vav1 is crucial for recruitment of CRACR2A-a-containing subsynaptic vesicles into the immunological synapse
To elucidate the mechanism of the PRD in recruitment of CRACR2A-a-containing vesicles into the IS, we checked possible protein interactions between CRACR2A-a and SH3 domain-containing proximal TCR signaling molecules that have a high affinity for proline-rich sequences. These efforts lead into the identification of Vav1 as an interacting partner of the PRD region (Fig. 4A). Among the Ca2+-binding, coiled-coil, proline-rich, and GTPase domains, the PRD showed highest binding affinity for Vav1 in GST pulldown analysis. Conversely, we also determined the CRACR2A-a-interacting domain within Vav1. GST pulldown experiments with cellular lysates from cells expressing WT or ΔPRD mutant of CRACR2A-a upon incubation with GST-fused Vav1-C (C terminus of Vav1) fragment that contains SH3-SH2-SH3 domains showed a strong interaction between Vav1-C and CRACR2A-a, that was significantly reduced by truncation of the PRD (Fig. 4B). These results suggest that the PRD of CRACR2A-a is the primary binding site for Vav1. These results were further validated by immunoprecipitation experiments with endogenous proteins, which showed enhanced interaction of CRACR2A-a with Vav1 after TCR stimulation (Fig. 4C).
Figure 4. Vav1 interaction is important for accumulation of CRACR2A-a-containing vesicles at the immunological synapse.

(A) GST pulldown assay with functional domains of CRACR2A-a. CBD; Ca2+-binding domain; a.a. 1–197, CCD; coiled-coil domain; a.a. 198–348, PRD; proline-rich domain; a.a. 349–540, and GTPase; guanidine triphosphatase domain; a.a. 541–731. Purified recombinant GST-fusion proteins were incubated with lysates from Jurkat cells stimulated with anti-CD3 antibody and immunoblotted for endogenous Vav1. Lower panel - Ponceau S staining shows similar amounts of recombinant proteins.
(B) CRACR2A-a binds to the C terminus of Vav1. Purified recombinant GST-Vav1-C (C terminus of Vav1, a.a 637–870) was incubated with cellular lysates from Jurkat cells expressing GFP-fused WT or ΔPRD mutant of CRACR2A-a and analyzed by immunoblotting. Right panel - Ponceau S staining shows similar amounts of recombinant GST fusion proteins.
(C) CRACR2A-a co-immunoprecipitates with Vav1. Cell lysates from resting or anti-CD3 antibody-stimulated Jurkat cells were immunoprecipitated with anti-CRACR2A antibody, and analyzed by immunoblotting for Vav1 (top panel) and CRACR2A-a (lower panel). Bar graph shows densitometric analyses of Vav1 intensity normalized to that of CRACR2A-a (± SEM) from three independent experiments.
(D) Co-localization of Vav1-GFP with mCherry-fused WT or ΔPRD mutant of CRACR2A-a at the contact site between T cells and stimulatory coverslips. Live-cell TIRF images at indicated time points from CRACR2A-depleted Jurkat cells expressing mCherry-fused WT (top panel) or ΔPRD mutant (bottom panel) CRACR2A-a proteins dropped on anti-CD3 antibody-coated coverslips. Pearson’s correlation coefficient values (Rr) are shown in individual images. Scale bar, 5 μm. Scatter plot on the right shows average (± SEM) Pearson’s correlation coefficient values at different time points from indicated cell numbers (in parenthesis). The data were fitted with a double exponential waveform model and corresponding 95% confidence interval of the fit (shaded area) used to analyze the association and dissociation kinetics is shown. Also see Movies S3 and S4.
(E) Translocation of CRACR2A-a-containing vesicles into the contact site of a T cell with stimulatory coverslip is impaired in Vav1-deficient cells. WT (Control, left panels) and J.Vav1 (right panels) Jurkat cells expressing mCh-CRACR2A-a and ZAP70-GFP were dropped onto anti-CD3 antibody-coated coverslip and their translocation was monitored using live-cell TIRF microscopy. Scale bar, 5 μm.
(F) Quantification of translocation of CRACR2A-a-containing vesicles in Control and J.Vav1 cells stimulated on anti-CD3 antibody-coated coverslips. Line graph representing an average of normalized fluorescence intensity (± SEM) of mCherry-CRACR2A-a in Control and J.Vav1 Jurkat cells, respectively (left). Average number of mCherry-CRACR2A-a-containing vesicles per cell at the contact site at indicated time points (right). Numbers in parenthesis indicate total cell numbers used for these analyses. * p<0.05, ** p<0.005, ***p<0.0005.
Many TCR signaling molecules transiently associate and dissociate at the IS (30). To determine the association and dissociation kinetics of CRACR2A-a and Vav1 and to elucidate the role of Vav1 in recruitment of CRACR2A-a-containing vesicles, we performed real-time TIRF microscopy. In these experiments, we dropped Jurkat T cells expressing Vav1-GFP with mCherry-fused WT or ΔPRD mutant CRACR2A-a on anti-CD3 antibody-coated coverslips and monitored the recruitment of vesicles at the contact site. Association and dissociation phenomena were analyzed with a double-exponential waveform model (Fig. 4D) to extract the time-constants. WT CRACR2A-a-containing vesicles and Vav1 associated (t1/2 = 33 sec) at the central region of the contact site at very early time points (Fig. 4D, Movie S3). At later time points, Vav1 accumulated in the periphery of the cell whereas CRACR2A-a-containing vesicles localized near the central region of the contact site. Association of ΔPRD mutant-containing vesicles with Vav1 had similar kinetics to WT (t1/2 = 37 sec), but the strength of association was diminished as seen by much lower Pearson’s correlation coefficient, and its dissociation showed much faster kinetics compared to WT CRACR2A-a-containing vesicles (t1/2 = 144 sec for ΔPRD vs t1/2 = 312 sec for WT; Fig. 4D, Movie S4). In addition to ΔPRD mutant, we also checked whether Ca2+-binding defective 97DAD99>AAA mutant of CRACR2A affects its accumulation in vesicles. Upon stimulation on anti-CD3 antibody-coated coverslips, Jurkat cells expressing 97DAD mutant of CRACR2A-a accumulated at the contact sites similar to WT protein (fig. S8A). Together with previous results of rescue of Jnk phosphorylation (fig. S5D), these data suggest that a defect in Ca2+-sensing by CRACR2A-a does not alter its translocation in vesicles and Jnk activation under these conditions.
Next, we checked the effect of Vav1 on accumulation of CRACR2A-a-containing vesicles on anti-CD3 antibody-coated coverslips. Here, we used ZAP70 as a marker for the contact sites. In Vav1-deficient J.Vav1 cells, accumulation of CRACR2A-a-containing vesicles at the contact sites was profoundly reduced compared to that in control cells (Fig. 4E). Furthermore, Vav1 deficiency significantly decreased the rate of recruitment and number of CRACR2A-a-containing vesicles at the antibody-coverslip contact sites (Fig. 4F). Collectively, these data using the ΔPRD mutant and Vav1-deficient cells commonly demonstrated an essential role of Vav1 in recruitment of CRACR2A-a-containing subsynaptic vesicles. Vav1 acts as a GEF for Rac1 at the IS, however, via its C-terminal SH3 and SH2 domains, Vav1 can also assemble signaling complexes as a scaffold protein independent of its GEF activity (31). It is unlikely that Vav1 acts a GEF for CRACR2A-a because CRACR2A-a predominantly exists in a GTP-bound form at the Golgi. Therefore, the function of Vav1 as a signaling adaptor or scaffold protein may be important for recruitment of CRACR2A-a-containing vesicles from the Golgi into the IS.
In T cells, CRAC channel components, Orai1 and STIM1 co-cluster at the IS upon contact with APCs (32, 33). Because CRACR2A-a also translocated into the IS and can recover SOCE in CRACR2A-depleted Jurkat cells (fig. S1, B and C), we examined for its possible colocalization with CRAC channel components. TIRF imaging of Jurkat cells expressing STIM1-YFP and mCherry-CRACR2A-a after dropping on anti-CD3 antibody-coated coverslips showed a significant colocalization between the two proteins (fig. S8B). Interestingly, passive store depletion by thapsigargin also induced translocation of CRACR2A-a-containing vesicles in close proximity to STIM1 clusters, albeit to a lesser degree than TCR stimulation (fig. S8C). These experiments suggest that both passive and active store depletion can induce colocalization of CRACR2A-a-containing vesicles with STIM1, to mediate CRAC channel activation.
Membrane dissociation by de-prenylation or GTP hydrolysis induces degradation of CRACR2A-a
Small Rab GTPases associate with the membranes of the Golgi and vesicles depending on their GTP bound/unbound state and C-terminal prenylation (17). PrePS sequence analysis tool predicts attachment of a 20-carbon geranylgeranyl moiety onto the C-terminal di-Cys motif of CRACR2A-a with high confidence score (E = 8.9 × 10−63; Fig. 5A). However, the C-terminal CCG residues of CRACR2A-a do not constitute a conventional prenylation motif for Rab GTPases (C×C or CC, where × is any amino acid). To determine whether this CCG motif is indeed geranylgeranylated and thus important for Golgi retention of CRACR2A-a, we truncated these three amino acids (ΔC729). Deletion of C-terminal CCG residues resulted in predominantly cytoplasmic localization, suggesting possible prenylation-induced Golgi membrane association of CRACR2A-a (Fig. 5A). Importantly, ΔC729 mutant could not rescue Jnk phosphorylation in CRACR2A-depleted Jurkat cells, suggesting an important role for prenylation in this function.
Figure 5. Degradation of CRACR2A-a by atorvastatin treatment, GTP hydrolysis and TCR stimulation.

(A) Prenylation is important for localization of CRACR2A-a. Sequence alignment of prenylation sites at the C termini of CDC42, Rac1, and CRACR2A-a. CRACR2A-a contains di-Cys residues that are potential targets of type II geranylgeranyl transferase. In addition, it also contains positively charged residues (++) before the prenylation site that can bind to phospholipids. Representative confocal images of Jurkat cells expressing GFP-fused WT and mutant CRACR2A-a proteins co-stained for endogenous Rab8a. White circles mark the cell periphery. Scale bar, 5 μm. Below: Bar graph (left) shows ratio of relative fluorescence intensity (of indicated cell numbers) of GFP in the Golgi v/s the cytoplasm. Right: Bar graph showing average (± SD) frequency of phosphorylated Jnk1/2 positive control (Scr), CRACR2A-depleted (R2A), and CRACR2A-depleted cells expressing shRNA-resistant WT or ΔC729 mutant CRACR2A-a after stimulation with anti-CD3 antibody for 10 mins.
(B) Confocal microscopy images of Jurkat cells expressing GFP-tagged Rac1 and CRACR2A-a treated with indicated concentrations of atorvastatin for 16 hours. Rac1, a known target of atorvastatin was used as a positive control. Bar graph below shows relative fluorescence intensities of GFP-Rac1 (left) or GFP-CRACR2A-a (right) in the Golgi v/s the surrounding area. Golgi was marked by WGA staining (also see fig. S9A). Scale bar, 5 μm.
(C) Atorvastatin induces robust degradation of CRACR2A-a. Jurkat T cells were treated with indicated concentrations of atorvastatin for 16 hours and examined for endogenous levels of indicated proteins by immunoblotting. β-actin, loading control. Below: Densitometry analysis of relative band intensities normalized to those of untreated cells is shown as mean ± SEM of three independent experiments.
(D) GTP/GDP-bound state of CRACR2A-a determines its stability. Lysates of HEK293T cells expressing N-terminally FLAG-tagged WT or mutant CRACR2A-a cDNAs and GFP from an IRES site were immunoblotted for detection of indicated proteins. All the lanes were derived from the same blot with identical exposure time. Bar graph shows densitometry analysis of relative amounts of various mutants normalized to that of WT CRACR2A-a (± SEM) from three independent experiments. The y-axis contains a break for clear presentation of data.
(E) Degradation of CRACR2A-a after TCR stimulation. Jurkat (left) or murine naïve CD4+ T (right) cells were stimulated with anti-CD3 antibody for indicated times and cellular lysates were analyzed for CRACR2A-a. Bar graph shows densitometric analysis of relative amounts of CRACR2A-a normalized to β-actin (± SEM) from three independent experiments.
We next examined how treatment with atorvastatin, an inhibitor of prenyl transferases affects the function of CRACR2A-a in comparison with a known target of statins. Treatment of Jurkat cells with atorvastatin significantly diminished Golgi membrane localization of Rac1, a positive control for atorvastatin treatment (Fig. 5B and fig. S9A). Under the same conditions, we observed a marked decrease in the Golgi membrane association of CRACR2A-a. Surprisingly, we noticed a profound decrease in fluorescence intensity of GFP-tagged CRACR2A-a in atorvastatin-treated cells. In consistence with this observation, protein levels of CRACR2A-a decreased dramatically after atorvastatin treatment while that of Rac1 was mildly influenced under the same conditions (Fig. 5C). These results suggested that lack of prenylation may induce robust degradation of CRACR2A-a.
Because de-prenylation by atorvastatin abolished Golgi membrane association and induced degradation of CRACR2A-a, we tested the hypothesis that GTP-GDP binding, which is also important for Golgi membrane association, may regulate degradation of CRACR2A-a. We expressed cDNA encoding FLAG-tagged WT or GTP-GDP binding-defective mutants of CRACR2A-a from a single mRNA with GFP, separated by an internal ribosomal entry site (IRES). We hypothesized that protein levels of WT or mutant CRACR2A-a, when normalized to those of GFP, would give an estimate of their stability because both proteins were translated from the same mRNA. Upon expression of WT or mutant CRACR2A-a proteins, we observed a substantially lower expression of GTP-binding defective T559N and N658I mutants while constitutively GTP-bound Q604L mutant showed pronounced increase in stability over WT (Fig. 5D). These results indicated that both prenylation and GTP binding are essential for the Golgi membrane association and stability of CRACR2A-a. To examine if this happened physiologically, we checked CRACR2A-a levels after TCR stimulation, which is expected to induce GTP hydrolysis. We observed robust degradation of CRACR2A-a after TCR stimulation in both human and mouse T cells (Fig. 5E). Together, these results identified a degradation mechanism of CRACR2A-a linked to its cytoplasmic localization caused by drug treatment-induced de-prenylation or more physiologically by TCR stimulation-induced GTP hydrolysis. Furthermore, these data also validate CRACR2A-a as a novel target of atorvastatin, degradation of which may contribute towards the anti-inflammatory effects observed by atorvastatin administration (19–21).
DISCUSSION
Human CRACR2A gene encodes two validated transcriptional isoforms, CRACR2A-a and CRACR2A-c. The short isoform CRACR2A-c was previously identified to facilitate CRAC channel function by enhancing Orai1-STIM1 interaction (22). Current mouse genome database shows presence of only CRACR2A-c. Our analyses validated presence of the long isoform CRACR2A-a in murine tissues as well, particularly, mouse lymphoid organs. Both these isoforms share conserved function in the Ca2+-NFAT pathway, but only CRACR2A-a has a unique role in activation of the Jnk MAPK pathway, emphasizing the importance of the PRD and GTPase domains in this distinctive function. Furthermore, our studies reveal a novel mechanism of a Rab GTPase in TCR signaling that includes recruitment in subsynaptic vesicles to the immunological synapse and inactivation by protein degradation. Activation and inactivation of CRACR2A-a consists of resting, effector, and termination steps controlled by GTP binding and protein degradation (fig. S9B). In resting T cells, to localize to the Golgi, CRACR2A-a needs to be GTP-bound (demonstrated by T559N, Q604L and N658I mutants) and prenylated (demonstrated by ΔC729 mutant and atorvastatin treatment). Unlike most small G proteins, CRACR2A-a predominantly exists in a GTP-bound form localized to the Golgi in resting T cells (resting state). After TCR stimulation, CRACR2A-a-containing vesicles are recruited into the IS via interaction with Vav1 to activate the downstream Jnk pathway (effector state). Interestingly, passive store depletion per se can also induce accumulation of CRACR2A-a-containing vesicles in close proximity to the ER-PM junctions to activate SOCE. The mechanism of translocation of these vesicles with TCR stimulation or store depletion needs further investigation. Upon TCR stimulation, CRACR2A-a seems to undergo posttranslational modification, as evidenced by detection of a higher molecular weight band by CRACR2A antibody from stimulated T cells (Fig. 4C and 5E). The identity and physiological relevance of this posttranslational modification of CRACR2A-a needs further investigation. Inactivation of CRACR2A-a occurs via GTP hydrolysis to induce its cytoplasmic localization and finally degradation (termination step). Currently, the composition and destiny of the CRACR2A-a-containing vesicles remain unknown due to a lack of additional marker molecules. Small GTPases bind to guanidine nucleotide dissociation inhibitors (GDIs) after GTP hydrolysis that allows their stabilization in the cytoplasm and eventually re-insertion into the membrane (16, 34). Contrary to small GTPases, CRACR2A-a is not re-cycled; rather it is consumed by entering a unidirectional pathway of protein degradation. Our studies also demonstrated that atorvastatin treatment influenced CRACR2A-a stability by inducing this unstable, cytoplasmic state through de-prenylation. High sensitivity of CRACR2A-a towards atorvastatin treatment could be due to lack of CRACR2A-a-specific GDI molecules.
Small G proteins including RhoA, Rac1 and CDC42 are important regulators of signaling pathways that control vital functions in T cells including thymocyte development, cytoskeleton dynamics, gene transcription and cell cycle progression (35). Small GTPases are present as GDP-bound inactive precursors, and are activated by GTP-GDP switch to mediate their downstream effects. CRACR2A-a is unique since it exists in a GTP and Golgi membrane-bound form in resting T cells and is degraded after GTP hydrolysis. CRACR2A-a resembles the atypical Rho GTPases that include RhoH, which has been shown to reside in a constitutively GTP-bound state (36). Hence, it appears that similar to RhoH, the activity of CRACR2A-a may be regulated by localization per se. At this stage, the detailed mechanism of how CRACR2A-a activates the downstream Jnk pathway is speculative. Rac1 and CDC42 are known to play an important role in stimulation of both p38 and Jnk. Activation of the Jnk pathway by Rac1 and CDC42 involves direct interaction with PAK1 or other MAPKKKs to phosphorylate Mkk4 and Mkk7 (10). It would be interesting to know whether CRACR2A-a acts as a signaling adaptor to directly activate the downstream Jnk pathway. Alternatively, it is also possible that CRACR2A-a is involved in trafficking of the vesicles carrying other signaling molecules involved in Jnk activation as cargo, supporting the conventional role of small Rab GTPases. Recently it was shown that a v-SNARE protein VAMP7 plays an important role in recruitment of Lat-containing subsynaptic vesicles into the TCR clusters and this recruitment is important for phosphorylation of Lat and formation of Lat signalosome (14). Further studies are required to know whether the same cellular machinery is used for the translocation of CRACR2A-a-containing vesicles. The identity and function of CRACR2A-a-containing vesicles is likely to be an exciting area for future investigation.
More than 60 members of the Rab GTPase family have been identified in humans. However, our knowledge of Rab proteins is limited within the scope of the role of small Rab GTPases in membrane trafficking. Here we elucidated a novel mechanism of a large Rab GTPase in intracellular signaling potentially mediated by vesicle trafficking in T cells. Unlike small G proteins, CRACR2A-a contains additional domains including N-terminal EF hands, a coiled coil domain and a proline-rich domain. Interestingly, we observed a strong interaction of CRACR2A-a with Vav1 via its proline-rich domain, which was important for the recruitment of CRACR2A-a-containing vesicles into the IS. These data suggest that additional domains of CRACR2A-a other than the GTPase domain are important to support its unique function. These results establish a foundation to understand the function of other large Rab GTPases including Rab44 and Rab45 that have a high similarity in their domain architecture with CRACR2A-a (fig. S10). Numerous gene linkage analyses of acute and chronic myeloid leukemia, and melanoma identified Rab45 (alternatively RASEF [RAS and EF-hand domain containing] or FLJ31614) as a potential tumor suppressor gene (37, 38). However, the molecular mechanism underlying its relationship with human diseases is not clearly understood. Therefore, our study of CRACR2A-a can help in uncovering possible roles of these large Rab GTPases in intracellular signaling and vesicle trafficking.
In summary, our study has identified CRACR2A proteins as an intracellular signaling module bridging two important proximal TCR signaling pathways, Ca2+-NFAT and Jnk, to affect T cell activation. While the short isoform CRACR2A-c was only involved in regulation of Ca2+-NFAT signaling, the long isoform and large Rab GTPase, CRACR2A-a was important for both the Ca2+-NFAT and the Jnk signaling pathways. An interesting aspect of CRACR2A-a is its presence in the newly identified subsynaptic vesicles that translocate from the Golgi into the IS after ligation of TCRs, which is important for activation of downstream signaling. Furthermore, CRACR2A-a function is uniquely regulated by membrane dissociation-induced degradation upon T cell stimulation. Therefore, this study will provide a conceptual framework to advance our understanding of the potential role of large Rab GTPases in intracellular signaling and their pathological mechanisms related to human diseases.
MATERIALS AND METHODS
Chemicals
Fura 2-AM and wheat germ agglutinin (WGA-594) were purchased from Invitrogen (Carlsbad, CA). Thapsigargin, PMA (Phorbol 12-myristate 13-acetate), and ionomycin were purchased from EMD Millipore. Brefeldin A was purchased from eBioscience. Atorvastatin was purchased from Sigma.
Plasmids and cells
Full-length cDNA of human CRACR2A-a (NCBI Reference Sequence: NM_001144958.1) was cloned from Jurkat cell cDNA library using primers described in Table S1 into pMSCV-CITE-eGFP-PGK-Puro vector with a N-terminal FLAG tag. The reverse primer was designed in the putative 3′ UTR region to sequence the endogenous STOP codon. Our clone contains an additional serine residue at amino acid position 425 within the proline-rich domain (codon 5′-AGT-3′). Various mutants of CRACR2A-a were generated by PCR amplification and site-directed mutagenesis using primers described in Table S1. CRACR2A-a plasmids were N-terminally tagged with a FLAG tag and subcloned into a lentiviral vector, FGllF (kind gift from Dr. Dong Sun An, UCLA). The cDNAs encoding WT and mutants of CRACR2A-a were also subcloned into pC1-mCherry and pEGFP-C1 vectors (Clontech) to generate N-terminally mCherry or GFP-fused proteins. HEK293 and Jurkat E6-1 T cell lines were obtained from American Type Culture Collection center (ATCC, Manassas, VA). Raji B cells were a kind gift from Dr. Sherie Morrison’s laboratory (UCLA). Vav1-GFP, ZAP70-GFP and GFP-Rac1 clones were purchased from Addgene. LAT1-CFP clone and J.Vav1 cells were a kind gift from Dr. Larry Samelson’s laboratory (NIH). LAT1 cDNA was excised out using EcoRI and BamH1 sites and subcloned into pEGFP-N1 vector.
Immunoblotting
For immunoblot analyses from tissues, wild-type mice were euthanized and perfused with PBS before harvesting organs. Tissues were snap frozen and lysed in RIPA buffer (10 mM Tris-Cl pH 7.5, 1% Triton X-100, 0.1% SDS, 140 mM NaCl, 1 mM EDTA, 0.1% sodium deoxycholate and protease inhibitor cocktail [Roche]), and centrifuged to remove debris. Samples were separated on 8–10% SDS-PAGE. Proteins were transferred to nitrocellulose membranes and subsequently analyzed by immunoblotting with relevant antibodies. For detection of NFAT, 5 × 106 Jurkat cells were stimulated with 10 μg/ml each of plate coated anti-CD3 and soluble anti-CD28 antibodies (OKT3, NCI clinical repository) for indicated times, harvested in PBS, lysed in RIPA buffer and centrifuged to remove debris. Same number of primary murine T cells were stimulated with 1 μg/ml each of plate coated anti-CD3 and soluble anti-CD28 antibodies for indicated times. Lysates were separated on 10% SDS-PAGE and immunoblotted for detection of NFATc1, NFATc2 and β-actin. For TCR signaling, 5 × 106 Jurkat or primary T cells were stimulated with 10 μg/ml of soluble anti-CD3 antibodies and cross-linked for indicated times and cell pellets were lysed in SDS loading dye. Lysates were separated on 10% SDS-PAGE and immunoblotted for indicated proteins. For atorvastatin treatment, 5 × 106 Jurkat T cells were treated with indicated concentrations of atorvastatin (in DMSO). Cell pellets were lysed in RIPA Buffer and centrifuged to remove debris. Lysates were separated on 12% SDS-PAGE and immunoblotted for detection of Rac1, CDC42, CRACR2A or β-actin. Antibodies used were anti-β-Actin (Santa Cruz, clone I-19), anti-FLAG (Sigma), anti-NFATc1 (BD Pharmingen, #556602), anti-NFATc2 (Santa Cruz, #7296), phospho-Erk (Cell Signaling, #4377), Erk (Cell Signaling, #9102), phospho-p38 (Cell Signaling, #4511), p38 (Cell Signaling, 9212), phospho-Jnk (Cell Signaling, #9255), Jnk (Cell Signaling, #9252), phospho-IkB (Cell Signaling, #9246), PLC-γ1 (Cell Signaling, # 2822), phospho-PLC-γ1 (Cell Signaling, # 2821), Rac1 (Cell Signaling, #2465), phosphor-Lat (Cell Signaling, #3581), Lat (Cell Signaling, #9166) and Vav1 (Cell Signaling, #2502). Polyclonal rabbit antibody for detection of CRACR2A was generated using purified human CRACR2A-c protein (Open Biosystems, Huntsville, AL) and used at 0.1 μg/ml concentration. Chemiluminescence images were acquired using an Image reader LAS-3000 LCD camera (FujiFilm).
shRNA-mediated depletion
pLKO.1 plasmids encoding shRNAs for depletion of CRACR2A were purchased from Open Biosystem (Thermofisher). The sequences of the shRNAs are described in Table S1. To generate lentiviruses for transduction, HEK293T cells were transfected with plasmid(s) encoding shRNA and packaging vectors (pMD2.G and psPAX2, purchased from addgene) using calcium phosphate transfection method. Culture supernatants were harvested at 48 and 72 hours post transfection and used for infection of Jurkat cells together with polybrene (8 μg/ml) using the spinfection method. Cells were selected with puromycin (1 μg/ml) 48 hours post infection.
Single-cell Ca2+ imaging, TIRF and confocal microscopy
Jurkat or primary T Cells were loaded at 1 × 106 cells/ml with 1 μM Fura 2-AM for 30 min at 25°C and attached to poly-L-lysine–coated coverslips. Intracellular [Ca2+]i measurements were performed using essentially the same methods as previously described (39). For TIRF analysis, coverslip bottom dishes were coated with 10 μg/ml of anti-CD3 antibody (OKT3) at 37 °C for 1 hr, washed with PBS and used for experiments. Cells were resuspended in Ringer’s solution containing (in mM): 155 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 10 D-glucose, and 5 Na-HEPES (pH 7.4) and dropped onto anti-CD3 antibody-coated coverslips and used either for time course imaging or fixed after stimulation for 10 mins with 2.5% PFA at room temperature and used for confocal analysis. TIRF microscopy was performed using an Olympus IX2 illumination system mounted on an Olympus IX51 inverted microscope using recently described methods (22). Acquisition and image analysis including measurement of Pearson’s correlation coefficient was performed using Slidebook (Intelligent Imaging Innovations, Inc.) software and graphs were plotted using OriginPro8.5 (Originlab). For quantification of TIRF intensity across different cells, individual regions of interest were selected and data were analyzed as the ratio of fluorescence intensity at each time-point (F) to that at the start of the experiment (F0). Vesicle number quantification was performed using automated counting options in Image J. For confocal analysis eGFP and mCherry were excited sequentially on a Zeiss LSM inverted microscope (Axiovert 100LSM, Carl Zeiss) fitted with a 63X water immersion objective lens (C-Apochromat 63/1.2 W corr; Carl Zeiss). Images were acquired and processed for enhancement of brightness/contrast using Pascal5 software (Carl Zeiss).
GTPase activity assay
HEK293T cells were transfected with empty vector or plasmids encoding WT or mutants of CRACR2A-a. 48 hours after transfection, cells were harvested, lysed in lysis buffer (20 mM Tris-Cl [pH 7.5], 2 mM EDTA, 100 mM NaCl, 10% glycerol, 1.0 % Igepal CA-630, protease inhibitor cocktail [Roche]) and centrifuged to remove the debris. The lysate was then precleared with protein A-Sepharose and immunoprecipitated overnight with anti-FLAG antibody-conjugated agarose (Sigma). Immunoprecipitates were washed in lysis buffer three times and two more times in lysis buffer without any EDTA and Igepal CA-630. Immunoprecipitates were directly used for analysis of GTPase activity using a colorimetric GTPase assay kit (Novus Biologicals) and the absorbance was read on a POLARstar Omega plate reader (BMG Labtech).
Measurement of Jnk phosphorylation by intracellular staining
Exponentially growing Jurkat cells (5 × 106) were transfected with empty vector or plasmids encoding CRACR2A-c and wild type or mutants of CRACR2A-a by electroporation at 200V using ECM 830 electroporator (Harvard Instruments). At 48 hours post transfection, cells were left untreated or stimulated with 10 μg/ml anti-CD3 antibody (OKT3) for 10 mins. Naïve CD4+ T cells isolated from control and CRACR2Afl/fl-CD4Cre animals were stimulated with 1 μg/ml anti-CD3 antibody (1452C11) for indicated times. Cells were fixed with 4% PFA, permeabilized with ice-cold methanol and stained with phospho-SAPK/Jnk mAb (Cell Signaling, #9257). Cells were washed twice with PBS and analyzed with a FACSCalibur flow cytometer (Becton Dickinson) and FlowJo software.
Immunological synapse and Immunofluorescence analysis
For immunological synapse analysis, Jurkat cells were electroporated with indicated plasmids and used 24 hours later. To visualize Jurkat cells interacting with superantigen pulsed B-cells, CMAC blue dye (Invitrogen)-loaded Raji B cells were pulsed with 1 μg/ml staphylococcal enterotoxin E (SEE) toxin (Toxin Technology, Sarasota, FL) for 45 min in complete medium. After rinsing, Raji B cells were mixed with Jurkat cells at a ratio of 1:1 for 10 or 20 mins on poly-D-lysine-coated coverslips at 37°C, fixed with 2.5% PFA and rinsed with PBS. Immune conjugates were examined on an inverted Zeiss LSM (Axiovert 100 LSM) confocal microscope as described above. For immunofluorescence staining for endogenous CRACR2A Jurkat cells fixed with 2.5% PFA were washed 3 times with PBS, permeabilised with 0.5% Igepal CA-630, blocked with 2.5% FBS and stained with purified anti-CRACR2A antibody for 1 hr at room temperature. Cells were washed with PBS and incubated with FITC labeled anti-rabbit antibody for 30 mins, mounted and examined on Zeiss LSM confocal microscope. Rab8a antibody (clone 3G1) was purchased from Abnova, GM130 antibody (D6B1) was purchased from Cell Signaling Technology, Golgin antibody (# A21270) was purchased from Invitrogen.
GST pulldown analysis and Immunoprecipitation
2 × 107 Jurkat cells were harvested in PBS and lysed in lysis buffer (20 mM Tris-Cl, 2 mM EDTA, 100 mM NaCl, 10% glycerol, 1.0 % Igepal CA-630, protease inhibitor cocktail [Roche]) and centrifuged to remove debris before preclearing with glutathione sepharose 4B beads. Lysates were incubated with 20 μg of GST or GST-tagged CRACR2A fragments of Ca2+-binding domain (CBD, a.a. 1–197), coiled-coil domain (CCD, a.a. 198–348), proline-rich domain (PRD, a.a. 349–540), and GTPase domain (a.a. 541–731) for 18 hours in binding buffer (20 mM Tris-HCl, 100 mM NaCl, 2 mM EDTA, 1.0% Igepal CA-630, protease inhibitors, 10% glycerol). Pulldown samples were washed five times with lysis buffer and analyzed by immunoblotting for indicated proteins. For GST pulldown with Vav1 C-terminus, 5 × 106 HEK293T cells expressing GFP-tagged wild type or ΔPRD mutant of CRACR2A were lysed as described above and incubated with 20 μg of GST or GST-tagged C-terminal fragment of Vav1 (a.a. 637–870) overnight using binding conditions described above. For immunoprecipitation experiments, 2 × 107 Jurkat T cells were lysed in lysis buffer (20 mM Tris-Cl, 2 mM EDTA, 100 mM NaCl, 10% glycerol, 1.0 % Igepal CA-630, protease inhibitor cocktail [Roche]), centrifuged to remove debris and precleared with Protein A sepharose. Precleared lysates were incubated with 1.0 μg of Rabbit IgG or anti-CRACR2A antibody for 18 hours; immunoprecipitates were washed five times in lysis buffer and analyzed by immunoblotting for detection of Vav1 and CRACR2A.
Generation of CRACR2A knockout animals
Targeting of CRACR2A gene was performed by flanking exons 3 and 4 with LoxP sites by homologous recombination in AB2.2 (129SvEv) embryonic stem (ES) cells. Aberrant splicing of exon 2 to exon 5 causes a shift in reading frame resulting in truncation of the protein. Deletion of exons 3 and 4 would result in loss of expression of both the isoforms of CRACR2A. G418 resistant clones were PCR screened for homologous recombination at both the homology arms. Chimeric mice with floxed CRACR2A alleles were generated by blastocyst injection of heterozygous CRACR2Afl/+ ES cell clones. Founder CRACR2Afl/+ chimeric mice were bred with Flp-deleter mice (Jackson Laboratories) to remove neomycin resistance gene cassette. CRACR2A mice were backcrossed to C57BL/6 mice for at least 10 generations and then bred with CD4Cre mice (Jackson Laboratories) to generate T cell-specific deletion of CRACR2A. All animals were maintained in pathogen-free barrier facilities and used in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of California, Los Angeles.
T cell purification
T cell purification, activation and transduction were carried out as previously described (40). CD4+ T cells were purified by magnetic sorting from single-cell suspensions generated by mechanical disruption of spleens and lymph nodes of adult mice according to the manufacturer’s instructions (Invitrogen). For effector T cell differentiation, cells were stimulated with 1 μg/ml of anti-CD3 antibody (1452C11, Bio X Cell) and 1 μg/ml of anti-CD28 antibody (Pharmingen) for 48 hours on a plate coated with 0.3 mg/ml of goat anti-hamster antibody (MP Biomedicals).
Statistical analysis
Statistical comparisons were performed using two-tailed Student’s t-test. Normal distribution of data was evaluated using the Shapiro-Wilk test and equal variance was tested using the F-test. For all the tests, a P value of <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Drs. Valarie Barr and Lawrence Samelson (National Institute of Health) for sharing Lat plasmid, protocols for anti-CD3 antibody and APC stimulation of Jurkat T cells, and J.Vav1 cell line. We also thank Drs. Masatsugu Oh-hora (Kyushu University, Japan) and Bernard Ribalet for helpful suggestions.
Funding: This work was supported by National Institute of Health grant AI083432 (Y.G.) and the American Heart Association grant 12SDG12040188 from (S.S). Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by the JCCC grant P30 CA016042, and the CFAR grant 5P30 AI028697.
Footnotes
Author Contributions: S.S. and Y.G. designed research; S.S. cloned and generated CRACR2Afl/fl animals and performed all the Ca2+ imaging, microscopy and statin treatment experiments; K.D.K performed the in vitro T cell analysis and JNK experiments with help from J.S.W.; Y.Y.G. performed biochemical experiments of CRACR2A and Vav1 interaction and GTPase assay; S.G performed immunoblots for expression of CRACR2A and NFAT; G.C helped with statistical analyses; A.P. and J.A. built the structural model; M.J. helped with generation of CRACR2Afl/fl animals; S.S and Y.G wrote the manuscript with input from other authors.
Competing Financial Interests: The authors do not have any competing financial interests.
References and Notes
- 1.Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol. 2013;13:257–269. doi: 10.1038/nri3403. [DOI] [PubMed] [Google Scholar]
- 2.Villalba M, Hernandez J, Deckert M, Tanaka Y, Altman A. Vav modulation of the Ras/MEK/ERK signaling pathway plays a role in NFAT activation and CD69 up-regulation. Eur J Immunol. 2000;30:1587–1596. doi: 10.1002/1521-4141(200006)30:6<1587::AID-IMMU1587>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 3.Wang H, Kadlecek TA, Au-Yeung BB, Goodfellow HE, Hsu LY, Freedman TS, Weiss A. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb Perspect Biol. 2010;2:a002279. doi: 10.1101/cshperspect.a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cahalan MD. STIMulating store-operated Ca(2+) entry. Nat Cell Biol. 2009;11:669–677. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrasco S, Meyer T. STIM Proteins and the Endoplasmic Reticulum-Plasma Membrane Junctions. Annu Rev Biochem. 2010 doi: 10.1146/annurev-biochem-061609-165311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis RS. Store-operated calcium channels: new perspectives on mechanism and function. Cold Spring Harb Perspect Biol. 2011;3:a003970. doi: 10.1101/cshperspect.a003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tybulewicz VL. Vav-family proteins in T-cell signalling. Curr Opin Immunol. 2005;17:267–274. doi: 10.1016/j.coi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol. 1997;15:297–322. doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- 9.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 11.Purbhoo MA, Liu H, Oddos S, Owen DM, Neil MA, Pageon SV, French PM, Rudd CE, Davis DM. Dynamics of subsynaptic vesicles and surface microclusters at the immunological synapse. Sci Signal. 2010;3:ra36. doi: 10.1126/scisignal.2000645. [DOI] [PubMed] [Google Scholar]
- 12.Williamson DJ, Owen DM, Rossy J, Magenau A, Wehrmann M, Gooding JJ, Gaus K. Pre-existing clusters of the adaptor Lat do not participate in early T cell signaling events. Nat Immunol. 2011;12:655–662. doi: 10.1038/ni.2049. [DOI] [PubMed] [Google Scholar]
- 13.Balagopalan L, Barr VA, Kortum RL, Park AK, Samelson LE. Cutting edge: cell surface linker for activation of T cells is recruited to microclusters and is active in signaling. J Immunol. 2013;190:3849–3853. doi: 10.4049/jimmunol.1202760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larghi P, Williamson DJ, Carpier JM, Dogniaux S, Chemin K, Bohineust A, Danglot L, Gaus K, Galli T, Hivroz C. VAMP7 controls T cell activation by regulating the recruitment and phosphorylation of vesicular Lat at TCR-activation sites. Nat Immunol. 2013;14:723–731. doi: 10.1038/ni.2609. [DOI] [PubMed] [Google Scholar]
- 15.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 16.Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91:119–149. doi: 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pfeffer S, Aivazian D. Targeting Rab GTPases to distinct membrane compartments. Nat Rev Mol Cell Biol. 2004;5:886–896. doi: 10.1038/nrm1500. [DOI] [PubMed] [Google Scholar]
- 18.Greenwood J, Steinman L, Zamvil SS. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat Rev Immunol. 2006;6:358–370. doi: 10.1038/nri1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L, Zamvil SS. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 20.Weber MS, Youssef S, Dunn SE, Prod’homme T, Neuhaus O, Stuve O, Greenwood J, Steinman L, Zamvil SS. Statins in the treatment of central nervous system autoimmune disease. J Neuroimmunol. 2006;178:140–148. doi: 10.1016/j.jneuroim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 21.Dunn SE, Youssef S, Goldstein MJ, Prod’homme T, Weber MS, Zamvil SS, Steinman L. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J Exp Med. 2006;203:401–412. doi: 10.1084/jem.20051129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srikanth S, Jung HJ, Kim KD, Souda P, Whitelegge J, Gwack Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat Cell Biol. 2010;12:436–446. doi: 10.1038/ncb2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dumas JJ, Zhu Z, Connolly JL, Lambright DG. Structural basis of activation and GTP hydrolysis in Rab proteins. Structure. 1999;7:413–423. doi: 10.1016/s0969-2126(99)80054-9. [DOI] [PubMed] [Google Scholar]
- 24.Lee MT, Mishra A, Lambright DG. Structural mechanisms for regulation of membrane traffic by rab GTPases. Traffic. 2009;10:1377–1389. doi: 10.1111/j.1600-0854.2009.00942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pereira-Leal JB, Seabra MC. The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J Mol Biol. 2000;301:1077–1087. doi: 10.1006/jmbi.2000.4010. [DOI] [PubMed] [Google Scholar]
- 26.Stroupe C, Brunger AT. Crystal structures of a Rab protein in its inactive and active conformations. J Mol Biol. 2000;304:585–598. doi: 10.1006/jmbi.2000.4236. [DOI] [PubMed] [Google Scholar]
- 27.Banton MC, Inder KL, Valk E, Rudd CE, Schneider H. Rab8 binding to immune cell-specific adaptor LAX facilitates formation of trans-Golgi network-proximal CTLA-4 vesicles for surface expression. Mol Cell Biol. 2014;34:1486–1499. doi: 10.1128/MCB.01331-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castellanos MC, Munoz C, Montoya MC, Lara-Pezzi E, Lopez-Cabrera M, de Landazuri MO. Expression of the leukocyte early activation antigen CD69 is regulated by the transcription factor AP-1. J Immunol. 1997;159:5463–5473. [PubMed] [Google Scholar]
- 29.Chuvpilo S, Jankevics E, Tyrsin D, Akimzhanov A, Moroz D, Jha MK, Schulze-Luehrmann J, Santner-Nanan B, Feoktistova E, Konig T, Avots A, Schmitt E, Berberich-Siebelt F, Schimpl A, Serfling E. Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity. 2002;16:881–895. doi: 10.1016/s1074-7613(02)00329-1. [DOI] [PubMed] [Google Scholar]
- 30.Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair DA, Waite J, Sacristan C, Victora GD, Zanin-Zhorov A, Dustin ML. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol. 2010;28:79–105. doi: 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cantrell DA. GTPases and T cell activation. Immunol Rev. 2003;192:122–130. doi: 10.1034/j.1600-065x.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 32.Barr VA, Bernot KM, Srikanth S, Gwack Y, Balagopalan L, Regan CK, Helman DJ, Sommers CL, Oh-Hora M, Rao A, Samelson LE. Dynamic Movement of the Calcium Sensor STIM1 and the Calcium Channel Orai1 in Activated T-Cells: Puncta and Distal Caps. Mol Biol Cell. 2008;19:2802–2817. doi: 10.1091/mbc.E08-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lioudyno MI, Kozak JA, Penna A, Safrina O, Zhang SL, Sen D, Roos J, Stauderman KA, Cahalan MD. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc Natl Acad Sci U S A. 2008;105:2011–2016. doi: 10.1073/pnas.0706122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Bliek AM. A sixth sense for Rab5. Nat Cell Biol. 2005;7:548–550. doi: 10.1038/ncb0605-548. [DOI] [PubMed] [Google Scholar]
- 35.Rougerie P, Delon J. Rho GTPases: masters of T lymphocyte migration and activation. Immunol Lett. 2012;142:1–13. doi: 10.1016/j.imlet.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Aspenstrom P, Ruusala A, Pacholsky D. Taking Rho GTPases to the next level: the cellular functions of atypical Rho GTPases. Exp Cell Res. 2007;313:3673–3679. doi: 10.1016/j.yexcr.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 37.Kaplon J, Homig-Holzel C, Gao L, Meissl K, Verdegaal EM, van der Burg SH, van Doorn R, Peeper DS. Near-genomewide RNAi screening for regulators of BRAF(V600E) - induced senescence identifies RASEF, a gene epigenetically silenced in melanoma. Pigment Cell Melanoma Res. 2014;27:640–652. doi: 10.1111/pcmr.12248. [DOI] [PubMed] [Google Scholar]
- 38.Sweetser DA, Peniket AJ, Haaland C, Blomberg AA, Zhang Y, Zaidi ST, Dayyani F, Zhao Z, Heerema NA, Boultwood J, Dewald GW, Paietta E, Slovak ML, Willman CL, Wainscoat JS, Bernstein ID, Daly SB. Delineation of the minimal commonly deleted segment and identification of candidate tumor-suppressor genes in del(9q) acute myeloid leukemia. Genes Chromosomes Cancer. 2005;44:279–291. doi: 10.1002/gcc.20236. [DOI] [PubMed] [Google Scholar]
- 39.Srikanth S, Gwack Y. Measurement of intracellular Ca2+ concentration in single cells using ratiometric calcium dyes. Methods Mol Biol. 2013;963:3–14. doi: 10.1007/978-1-62703-230-8_1. [DOI] [PubMed] [Google Scholar]
- 40.Kim KD, Srikanth S, Tan YV, Yee MK, Jew M, Damoiseaux R, Jung ME, Shimizu S, An DS, Ribalet B, Waschek JA, Gwack Y. Calcium Signaling via Orai1 Is Essential for Induction of the Nuclear Orphan Receptor Pathway To Drive Th17 Differentiation. J Immunol. 2014;192:110–122. doi: 10.4049/jimmunol.1302586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morreale A, Venkatesan M, Mott HR, Owen D, Nietlispach D, Lowe PN, Laue ED. Structure of Cdc42 bound to the GTPase binding domain of PAK. Nat Struct Biol. 2000;7:384–388. doi: 10.1038/75158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.