

Myeloproliferative neoplasms (MPN) are well-delineated diseases in adults but are rare, poorly understood conditions in children. Essential thrombocytosis (ET), one of the classical BCR-ABL-negative MPN, is overall a rare finding in the pediatric population but seems to be one of the more common MPN reported in children. It occurs less frequently and with more favorable outcomes in children than in adults. The proportions of pediatric patients with mutations in JAK2, MPL, and CALR (the genes most commonly mutated in adult patients) seem lower than in adults,1–4 raising the question of what alternative mutations may be contributing to disease in this population. Overall, the genetic landscape of pediatric ET has not been thoroughly evaluated.5 In an effort to understand the possible pathogenic lesions involved in pediatric ET better, we performed high-throughput sequencing using a comprehensive targeted gene panel on five pediatric patients with ET. We identified mutations in genes involved in transcriptional regulation and nuclear transport.
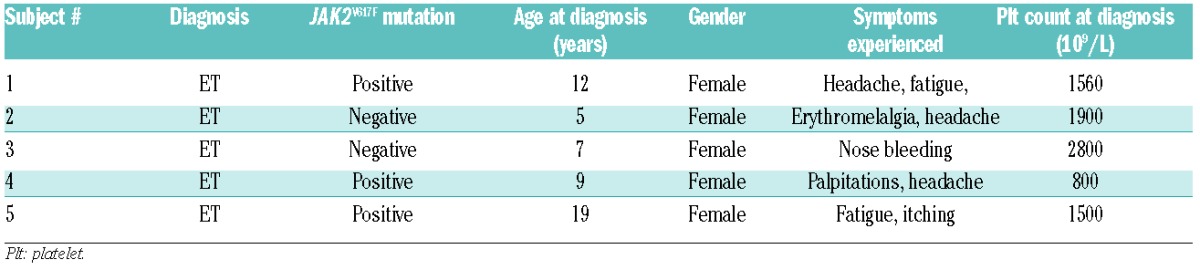
We examined five children in whom ET was diagnosed before 21 years of age at five different medical centers (Table 1). The patients were diagnosed according to World Health Organization 2008 criteria;6 bone marrow investigations were available for all but one patient who, based on JAK2V617F positivity and the findings of blood tests was given a diagnosis of ET. The patients’ family histories were assessed and were negative for MPN, myeloid disease and unprovoked thrombotic or bleeding events. Age at presentation ranged from 5 to 19 years old and presenting symptoms included bleeding, headache and fatigue. Most children had extreme thrombocytosis during their illness and platelet counts ranged as high as over 2000×109/L. Three of the children developed acquired von Willebrand disease with periods of extreme thrombocytosis. One patient developed pseudotumor cerebri (with no evidence of cerebral venous sinus thrombosis or stroke) but no other significant adverse events were reported. All subjects were treated with hydroxyurea at some point during their illness with improvements in symptoms and decreases in platelet count.
Table 1.
Clinical summary of enrolled subjects.
Commercial genetic testing was performed, and identified three of the five subjects as positive for JAK2V617F. No MPL or CALR mutations were identified. We performed high-throughput sequencing with a targeted deep sequencing assay of 585 genes (HemePACT). Tumor tissue (peripheral blood) was sequenced at an average coverage of 829x (with a standard deviation of 130) while germline tissue was sequenced at an average coverage of 220x (standard deviation of 150). We used Mutect to call single point variants, comparing our samples to a sample representing a pool of normal samples, and PINDEL to call short insertions and deletions, following previous recommendations.7 We then excluded all mutations either present at a high variant allele frequency in the matching germline samples (when available) or present in at least one database of known non-somatic variants (DBSNP and 1000 genomes) and absent from COSMIC. We used coverage information to look for copy number aberrations but did not find any.
Full panel sequencing was performed in both JAK-positive and JAK-negative patients. We confirmed the presence of the JAK2V617F mutation in the three patients in whom this variant had already been characterized (Figure 1). We did not find any additional mutations in CALR, MPL or JAK-family genes.
Figure 1.
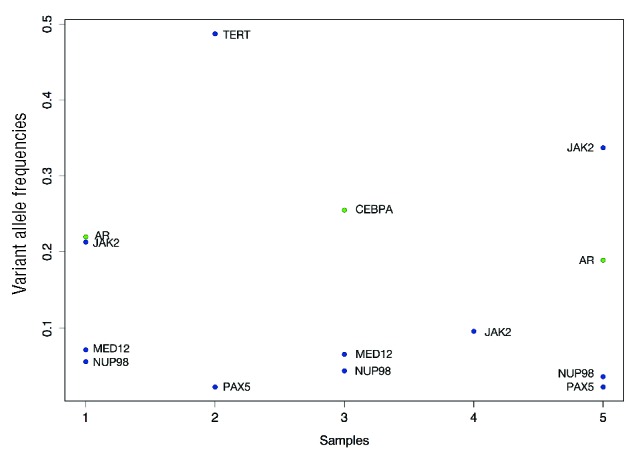
Genes mutated in pediatric ET patients. The figure illustrates the genes mutated for each of our samples with the corresponding variant allele frequencies. Insertions and deletions are colored green and single nucleotide variants are colored blue.
Mutations were identified in six additional cancer-associated genes: NUP98, MED12, PAX5, AR, CEBPA, and TERT.
NUP98 fusions have been reported in numerous patients with hematologic malignancies and, in our cohort three of the subjects expressed a missense mutation at NUP98H1636N. NUP98 is an important component of the nuclear pore complex and additional roles in transcriptional control and cell cycle progression have been suggested.8 This mutation was seen in both JAK2V617F –positive and -negative patients.
Two subjects expressed a missense mutation in MED12, the Mediator Complex Subunit 12 gene. MED12 encodes a subunit of the Mediator complex, which functions as a transciptional co-activator required for transcription of numerous genes. It interacts with CDK8, has been reported in both solid and lymphoid malignancies, and has been shown to play a role in preventing resistance to targeted therapeutics.9,10
PAX5 was also mutated in two subjects and has been associated with lymphoid malignancies but not previously with myeloid disease.
Mutations were also identified in AR, CEBPA, and TERT. These genes have enzymatic or transcriptional regulation functions and some have previously been identified to play a role in MPN and acute myeloid leukemia. AR encodes for the androgen receptor, a steroid hormone-activated transcription factor that is responsible for regulating steroid-responsive genes. There are numerous steroid-responsive genes and genes that interact with the androgen receptor, including CALR and STAT family members, implying that altered function of MPN-relevant genes may occur in AR-mutated patients.11–13
This is the first use of the IMPACT panel reported in pediatric patients with ET and is the broadest genetic examination to date in this population. We identified novel mutations in MPN that may play a role in disease pathogenesis. Common pathogenic mutations (besides JAK2V617F) seen more frequently in adult patients were not identified.
As no copy number events affect this dataset, variant allele frequency gives a good estimate of the cellular prevalence of each mutation. The group of mutations with the highest variant allele frequency gives us an indication of the tumor content of the sample and mutations with a significantly lower variant allele frequency are logically subclonal. As indicated by Figure 1, JAK2, when mutated, was always the mutation that yielded the highest or close to the highest frequency, from which we can infer that it was probably a clonal event. On the other hand, the MED12, NUP98 and PAX5 events were always present at a lower frequency than the estimated tumor content and were, therefore, probably subclonal events.
The World Health Organization 2008 diagnostic criteria for ET include a requirement for finding the presence of JAK2V617F, presence of another clonal marker if the patient is JAK2-negative, or absence of reactive thrombocytosis.6 Recent literature indicates that additional mutations may be relevant in pediatric disease14,15 and the impact of clonality needs further evaluation in these patients. It is not yet clear whether pediatric and adult disease represents the same or similar entities. Ultimately, revised criteria for pediatric diagnosis may be indicated as we learn more about these entities.
The use of targeted therapeutics in myeloid diseases has provided an exciting alternative to traditional therapies in many clinical situations. Given the alternative lesions that may play a role in pediatric MPN, it is possible that alternative pathways besides JAK/STAT may need to be targeted in children. This provides an opportunity for collaborative, multicenter clinical research trials in pediatric patients with MPN.
Further analysis of the transformative ability of potentially pathogenic mutations and samples from more patients are needed to determine the frequency of these mutations and identify additional lesions. Future analyses should also extend to other technologies, such as cytokine assays and RNA-seq which would allow us to profile expression and signaling as well as fusion events, and therefore enable us to distinguish potential subtypes of these diseases better and gain greater understanding of the symptomatology. Continued large-scale analyses, likely with whole-exome or whole-genome sequencing, as well as epigenomic exploration, should be done for children with MPN. There are unique ethical challenges raised by such broad evaluations in children and adolescents but these may be the necessary steps to develop a better understanding of the genotypic profile of these disorders. Ultimately, genotypic-phenotypic correlations could someday be made which would allow for better risk assessment and treatment guidelines. We are eager to continue this work and hope for multicenter collaboration as a means to improve care for these patients.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Teofili L, Giona F, Martini M, et al. The revised WHO diagnostic criteria for Ph-negative myeloproliferative diseases are not appropriate for the diagnostic screening of childhood polycythemia vera and essential thrombocythemia. Blood. 2007;110(9):3384–3386. [DOI] [PubMed] [Google Scholar]
- 2.Giona F, Teofili L, Moleti ML, et al. Thrombocythemia and polycythemia in patients younger than 20 years at diagnosis: clinical and biologic features, treatment, and long-term outcome. Blood. 2012; 119(10):2219–2227. [DOI] [PubMed] [Google Scholar]
- 3.Langabeer SE, Halsam K, McMahon C. CALR mutations are rare in childhood essential thrombocythemia. Pediatr Blood Cancer. 2014;61(8):1523. [DOI] [PubMed] [Google Scholar]
- 4.Kucine N, Chastain KM, Mahler MB, Bussel JB. Primary thrombocytosis in children. Haematologica. 2014;99(4):620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langabeer SE, Haslam K, McMahon C. The molecular landscape of childhood myeloproliferative neoplasms. Leuk Res. 2014;38(8):997–998. [DOI] [PubMed] [Google Scholar]
- 6.Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22(1):14–22. [DOI] [PubMed] [Google Scholar]
- 7.Cheng D, Mitchel TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSKIMPACT): a hydbridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gough SM, Slape CI, Aplan PD. NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood. 2011;118(24):6247–6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kämpjäarvi K, Jäarvinen TM, Heikkinen T, et al. Somatic MED12 mutations are associated with poor prognosis markers in chronic lymphocytic leukemia. Oncotarget. 2014;6(3):1884–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang S, Hölzel M, Knijnenburg T, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-β receptor signaling. Cell. 2012;151(5):937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fasan A, Haferlach C, Alpermann T, et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia. 2014;28(4):794–803. [DOI] [PubMed] [Google Scholar]
- 12.Tapper W, Jones AV, Kralovics R, et al. Genetic variation at MECOM, TERT, JAK2, and HSB1L-MYB predisposes to myeloproliferative neoplasms. Nat Commun. 2015;6:6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang M, Ma Y, Chen C, et al. Androgen-responsive gene database: integrated knowledge on androgen-responsive genes. Mol Endocrinol. 2009;23(11):1927–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karow A, Nienhold R, Lundberg P, et al. Mutational profile of childhood myeloproliferative neoplasms. Leukemia. 2015;29(12):2407–2409. [DOI] [PubMed] [Google Scholar]
- 15.Fu R, Liu D, Cao Z, et al. Distinct molecular abnormalities underlie unique clinical features of essential thrombocythemia in children. Leukemia. 2016;30(3):746–749. [DOI] [PMC free article] [PubMed] [Google Scholar]