

Abstract
Purpose
Later onset and progression of retinal dystrophy occur with some RPE65 missense mutations. We correlate the functional consequences of the novel P25L RPE65 mutation with its early childhood phenotype and compare it with other pathogenic missense mutations.
Methods
In addition to typical clinical tests, fundus autofluorescence (FAF), optical coherence tomography (OCT), and 2-color-threshold perimetry (2CTP) were measured. RPE65 mutations were screened by SSCP and direct sequencing. Isomerase activity of mutant RPE65 was assayed in 293F cells and quantified by HPLC analysis of retinoids.
Results
A very mild phenotype was detected in a now 7-y old boy homozygous for the P25L mutation in RPE65. Though abnormal dark adaptation was noticed early, best corrected visual acuity was 20/20 at age 5-y and 20/30 at age 7-y. Nystagmus was absent. Cone electroretinogram (ERG) was measurable, rod ERG severely reduced, and FAF very low. 2CTP detected mainly cone-mediated answers under scotopic conditions, light-adapted cone answers were about 1.5 log units below normal. High resolution spectral domain OCT revealed morphological changes. Isomerase activity in 293F cells transfected with RPE65/P25L was reduced to 7.7% of wildtype RPE65-transfected cells, while RPE65/L22P-transfected cells had 13.5%.
Conclusions
The mild clinical phenotype observed is consistent with the residual activity of a severely hypomorphic mutant RPE65. Reduction to < 10% of wildtype RPE65 activity by homozygous P25L correlates with almost complete rod function loss and cone amplitude reduction. We conclude that functional survival of cones is possible in patients with residual RPE65 isomerase activity. This patient should profit most from gene therapy.
Introduction
Human mutations in the gene for the highly preferentially expressed RPE protein RPE65 are associated with a spectrum of retinal dystrophies ranging from more severe early onset conditions variously described as Leber congenital amaurosis type 2/autosomal recessive childhood-onset severe retinal dystrophy or early onset severe retinal dystrophy (LCA2/arCSR, EOSRD) to later onset conditions described as autosomal recessive retinitis pigmentosa (arRP)1–7. Recently, RPE65 has been established as the isomerase central to the retinoid visual cycle,8–10. This cycle11 is crucial for supply of the chromophore 11-cis retinal for visual pigment regeneration.
Animal models have contributed greatly to our understanding of the role of RPE65 in the visual cycle, regeneration and retinal dystrophy. Rpe65 knockout mice display a biochemical phenotype consisting of extreme chromophore starvation (no rhodopsin) in the photoreceptors concurrent with overaccumulation of all-trans retinyl esters in the RPE10 and are extremely insensitive to light. This insensitivity to light protects Rpe65−/− mice from light damage, establishing rhodopsin as the mediator of light-induced retinal damage12. There is also a natural mutation in mouse Rpe65 called rd1213. Additionally, a mutation in the Briard dog RPE65 gene causes a severe retinal dystrophy in affected animals14,15. The utility of gene therapy was established by pre-clinical trials in these dogs16. These are all null mutations and represent one end of a spectrum of disease. Besides these, a hypomorphic animal model has been described: the L450M variant found in C57Bl/6 mice. Though it has been important in elucidating mechanisms in visual pigment regeneration, light damage, and retinal degeneration, etc.17–20, the M450 form of mouse RPE65 retains about 40% of wildtype (L450) activity21. This appears to be more than enough to maintain close to normal function.
In contrast, human RPE65 EOSRD displays a wide spectrum of severity, age of onset, and progression not seen in animal models1–7. Due to the wider range of mutational mechanisms operational (missense, nonsense, deletions or insertions, and splicing anomalies) and the various combinations of these mutations22, this diversity promises a wealth of potential clinical details to help understand the mechanisms underlying RPE65 retinal dystrophy. In practice, however, these details may be obscured by secondary degenerative changes and compounded by delay until a molecular genetics diagnosis is made. Consideration of such details may be required when assessing suitability of a patient for gene therapy.
In this paper we present the mild phenotypic consequences of a homozygous P25L missense mutation in a young patient and correlate these with the biochemical effect of this mutation on RPE65 activity. We show that even though the isomerase activity of the mutant RPE65 is quite impaired, the patient has near normal visual acuity. However, rod function is extremely impaired. Additionally, short wavelength cones appear more impaired than long wave cones, consistent with findings in other patients with RPE65 mutations that blue color vision is much more and earlier impaired than for red, opposite to the usual case for cone dystrophies. These findings are discussed in the context of emerging rationales for gene therapy of this disease.
Methods
Subject
The patient described was identified from a larger study investigating patients with known or suspected childhood onset retinal dystrophies from the genetics research facility of the Department of Paediatric Ophthalmology, Strabismology and Ophthalmogenetics of the Universitaetsklinikum Regensburg. The patient´s parents were informed about the objectives of the examination and volunteered to participate. Informed consent was taken according to the tenets of the Declaration of Helsinki. The study was approved by the Ethical Committees of the University of Regensburg and of the University of Giessen.
Clinical examination
Ophthalmic examination included best-corrected visual acuity BCVA, refraction, orthoptic evaluation, slit-lamp biomicroscopy and funduscopy, color vision testing using the Farnsworth-Munsell panel D 15 test, and fundus photography (RetCam120, Massie Lab, Pleasanton, CA, USA; TOPCON TRC 50X, Topcon Optical, Tokyo, Japan; Zeiss Funduscamera FF 450 plus, Carl Zeiss Meditec AG, Jena, Germany).
Perimetry
Kinetic Perimetry
Kinetic photopic visual fields were measured with a Goldmann perimeter (Haag-Streit, Bern, Switzerland) using 5 targets (V/4e, III/4e, I/3e, I/2e, I/1e).
Two-color threshold perimetry
Two-color threshold perimetry (2CT Perimetry) was performed under scotopic and photopic conditions to assess the spatial distribution of rod and cone mediated function. A modified Humphrey-Field-Analyzer (HFA, model 640 modified by F. Fitzke, Institute of Ophthalmology, London) was used as reported previously23. In the dark-adapted state, a neutral density filter (ND filter) was used for the stimulus generated with a 500 nm cut-off to remain in the effective range of the instrument. The lowest normal sensitivity for each tested locus was determined with the 10th percentile function.
To determine the photoreceptor system responsible for stimulus detection (mediation), the differences between the sensitivities with a 500 nm cut-off filter and a 600 nm cut-on filter were analyzed in the dark-adapted state. Photoreceptor mediation was defined as follows: (1) rod mediation of both long- and short-wavelength stimulus for differences of dark-adapted sensitivities above 4 decibels, (2) mixed mediation, e.g. rods detect the 500 nm cut-off filter generated stimulus and cones detect the 600 nm cut-on filter generated stimulus for differences between 3 and −11 decibel and (3) cone mediation for differences below −12 decibel. The negative values for mixed and cone mediation result from the fact that the sensitivities to the 500 nm cut-off filter generated stimulus are 20 dB lower than the real values due to the ND filter used. Dark adapted and light adapted photoreceptor sensitivity losses to 500 nm cut-off and 600 nm cut-on filter generated stimuli were calculated as the difference between the measured value and the 10th percentile of normals for each test locus.
Electroretinography
Electroretinograms (ERGs) were recorded with DTL electrodes following white Ganzfeld (fullfield) stimulation using a Nicolet Spirit examination unit (Nicolet, Madison, WI, USA) according to the guidelines of the International Standard for Electroretinography24. Artefacts caused by nystagmus were rejected.
Fundus autofluorescence (FAF)
FAF was recorded using a standard confocal scanning laser ophthalmoscope and the most recent generation instrument (Heidelberg Retina Angiograph (HRA) and Spectralis HRA-OCT, Heidelberg Engineering, Heidelberg, Germany). A detailed description is given elsewhere25. In brief, to amplify the FAF signal generally 8 to 16 images were aligned using the integrated software and a mean of these images was calculated. With the Spectralis HRA-OCT, images are averaged automatically. The detector sensitivity of the laser scanning camera was adjusted to the amount of FAF.
Optical coherence tomography OCT
OCT scans were performed as previously described25. OCT images were recorded on a STRATUS OCT system (Carl Zeiss Meditec Inc., Dublin, CA) using 512 A-scans over 6 mm transverse scanning length for optimal sampling rate with 400 A-scans/sec. The best images were obtained using the hair cross scanning pattern. The fovea and areas of the posterior pole were scanned.
At the latest visit, high resolution spectral domain OCT (also known as Fourier OCT26; Spectralis HRA-OCT (Heidelberg Engineering, Heidelberg, Germany) using High Resolution Mode 40k-Hz scanning was applied. A series of 1536 A-scans around the fovea were acquired over a B-scan length of 4.3 mm with a series of 25 B-scans over 2.9 mm (volume scan).
Molecular genetic analysis
DNA was extracted from peripheral blood lymphocytes27 and subjected to PCR using oligonucleotide primers specific for RPE6528 and designed in our laboratory as described previously25. All coding exons of RPE65 including 162 bp of the 5’ UTR and 49 bp of the 3’ UTR were amplified. PCR products were analyzed using Single Strand Conformation Polymorphism (SSCP) analysis on PAGE gels (11 cm) using 10% polyacrylamide (19:1) with a 5 mm overlay of 6% polyacrylamide (19:1) in 1× TBE buffer. The gels were run for 12 – 14h at 140 – 180 V const. and 10 °C. Direct sequencing was applied to amplimers showing aberrant banding patterns in SSCP. Sequencing results were confirmed by a restriction endonuclease assay using Aci I (P25L).
Functional analysis
Transient transfection and cell culture
Cell culture methods and transient transfection protocols are as previously published10. For any given experiment, 3 ×107 293-F cells were transfected with 30 µg of pVitro2 plasmid (containing RPE65 and CRALBP open reading frames (ORFs)) and 30 µg of pVitro3 plasmid (containing LRAT and RDH5 ORFs) in the presence of 40 µl of 293fectin transfection reagent (Invitrogen, Carlsbad, CA), all in a total volume of 30 ml.
Site-directed Mutagenesis of RPE65
Site-directed mutagenesis of RPE65 ORF cloned in pVitro2 was done using a QuikChange XL Site-directed mutagenesis kit (Stratagene, La Jolla, CA). Oligonucleotide primer pairs used are listed in table 2. Mutants were verified by sequence analysis (Northwoods DNA, Solway, MN, USA) of DNA minipreps. Validated mutant and wildtype plasmids were grown up and purified using Qiagen purification kits (Maxi or Mega format, as appropriate; Qiagen, Valencia, CA).
Table 2.
Oligonucleotide primers used to prepare mutant clones.
| P25L | forward | 5’-GAA GAG CTG TCG TCG CTG CTC ACC GCC CAC GTG-3’ |
| reverse | 5’-CAC GTG GGC GGT GAG CAG CGA CGA CAG CTC TTC-3’ | |
| L22P | forward | 5’-GAA ACC GTG GAA GAG CCG TCG TCG CCG CTC ACC GCC-3’ |
| reverse | 5’-GGC GGT GAG CGG CGA CGA CGG CTC TTC CAC GGT TTC-3’ | |
| Y79H | forward | 5’-GAA GGA CAC GTC ACC CAT CAC AGA AGG TTC ATC CGC-3’ |
| reverse | 5’-GCG GAT GAA CCT TCT GTG ATG GGT GAC GTG TCC TTC-3’ | |
| E95Q | forward | 5’-GTC CGG GCA ATG ACC CAG AAA AGG ATC GTC ATA ACG-3’ |
| reverse | 5’-CGT TAT GAC GAT CCT TTT CTG GGT CAT TGC CCG GAC-3’. |
Retinoid extractions and HPLC
Culture fractions of 19 ml volumes of transfected 293-F cells were centrifuged and cells were harvested and retinoids extracted and saponified by the methods previously described10. Isomeric retinols were analyzed on 5 micron particle Lichrospher (Alltech, Deerfield, IL) normal phase columns (2 × 250 mm) on an isocratic HPLC system equipped with a diode-array UV-visible detector (Agilent 1100 series, Agilent Technologies, New Castle, DE), following the method of Landers and Olson29 as modified by us10. Data was analyzed on ChemStation32 software (Agilent).
Immunoblot analysis
Cell pellets (~ 2 ×106 cells) from 1 ml culture aliquots were lysed in 200 µl CytoBuster detergent (Novagen), incubated on ice for 10 min, centrifuged at 13,000 × g for 10 min, and the supernatant harvested for SDS-PAGE analysis. Denatured samples were separated on 12% BisTris NuPage (Invitrogen) gels and electrotransferred to nitrocellulose membranes. Blots were probed with antibodies by standard procedures and developed in color substrate. Primary antibodies used were: rabbit anti-bovine RPE65 antibody (1:4000) and rabbit anti-CRALBP antibody (1:20,000; gift of Dr. John Saari). Secondary antibody used was alkaline phosphatase-conjugated goat anti-rabbit IgG (1:10,000; Novagen). Expression levels of RPE65 were quantitated by fluorescent western blot. Gels and blots were prepared as above using Hybond-ECL nitrocellulose and ECL-plex Rainbow protein standard markers (GE Healthcare). Primary antibodies were rabbit anti-RPE65 (1:4000) and mouse monoclonal antibody anti-CRALBP (1:20,000; gift of Dr. John Saari). Secondary antibodies used were Cy5-conjugated goat anti-rabbit and Cy3-conjugated goat anti-mouse ECLplex fluorescent antibodies (both 1:2500). Processed blots, following manufacturer's protocols, were scanned in a Typhoon 9410 scanner (GE Healthcare) and quantitated using ImageQuant TL image analysis software. Wildtype and mutant RPE65 levels were normalized to co-expressed CRALBP levels and mutant levels were calculated relative to wildtype RPE65 expression (set at 100%).
Results
Genotypic analysis
The patient was born in 2001 to then 36 and 31 old consanguineous healthy parents. He had two older healthy siblings, a brother and a sister. The great-grandfathers of the parents were brothers. Molecular genetic analysis showed the patient to have a homozygous C to T mutation at nt 128 of RPE65, converting a proline at codon 25 to a leucine. This point mutation resulted in the loss of an Aci I restriction site which was used to confirm the sequencing results (data not shown). The parents were heterozygous for this mutation.
Phenotypic Analysis
The patient was first examined at age 3 years as the parents had noticed visual problems when the boy moved from daylight to dim light. His visual acuity was normal at age 3 and 4.5 y, and had slightly deteriorated at age 6. The clinical, imaging, electrophysiological and psychophysical data are summarized in table 1 and figures 1 – 4. The orthoptic exam was unremarkable with orthotropia at distance and a small exophoria of 3° at near, and good stereopsis (120”, TNO test). The anterior segments were normal. The fundus showed some uncharacteristic changes, i.e. a lightly pigmented appearance, increased visibility of choroidal vessels, reduced macular reflex, and some irregular pigmentation (fig. 1A, B). Fullfield ERGs were performed at ages 3, 5 and 6 years, and showed severely impaired rod responses, but relatively well preserved cone responses with amplitudes reduced to about 30 to 20%, and normal implicit times (fig. 4). Goldmann perimetry first performed at age 6 years showed some constriction to the I/4e isopter.
Table 1.
Clinical, electrophysiological and psychophysical data
| MV, born 2001 RPE65 mutation homozygous P25L |
At 3 years | At 4.5 years | At 6/7 years | ||
|---|---|---|---|---|---|
| BCVA OD / OS | 1.2 / 1.0 LEA 3m | 1.2 / 1.0 LEA 3m | 0.63 / 0.63 E | ||
| Refraction | OD +3.0 sph OS +3.0 sph |
OD +2.5 sph / −0.5 cyl A0° OS +2.75 sph |
OD +0.5 / −0.25 cyl A32° OS +0.75 / − 0.25 <A80° |
||
| Fundus | Relatively “blond” fundus (Fig. 1A) | “Blond” fundus and increased visibility of choroidal vessels |
Small macular reflex (Fig. 1B) | ||
| Goldmann Visual Field - OD + OS | nd | nd | V/4e, III/4e normal I/4e slightly constricted* |
||
| 2CT perimetry 30° OS | nd | nd | Dark adapted: mostly cone mediation Light adapted. CSL 1.5 log (Fig. 2) |
||
| Fullfield ERG | Fig. 5 | ||||
|
Scotopic Maximal Response |
Ampl |
OD OS |
Minimal residual Minimal residual |
16% / 21% 22% / 26% |
Minimal residual 25% / 15% |
| Impl |
OD OS |
√ √ |
√ | ||
|
Photopic 30 Hz-Flicker |
Ampl |
OD OS |
7% 12% |
50% 50% |
50% 50% |
| Impl |
OD OS |
√ √ |
√ √ |
√ √ |
|
|
Photopic Single Flash |
Ampl |
OD OS |
9% / 15% 16% / 23% |
32% / 34% 30% / 33% |
41% / 51% 15% / 26% |
| Impl |
OD OS |
√ √ |
√ √ |
√ √ |
|
| Lanthony Panel D15 OD + OS | nd | nd | Saturated normal Desaturated very few errors along blue axis |
||
| FAF OD + OS | Very low (Fig. 1C) | Very low | Very low (Fig. 1C, middle) | ||
| OCT Stratus | Layering grossly preserved (Fig. 2B) | Layering preserved | Layering grossly preserved (Fig 2C) | ||
| OCT Spectralis HRA-OCT | Altered layering outer retina + thinning (Figure 2D) |
||||
CSL, cone Sensitivity loss; nd, not done; sph, spheric; cyl, cylinder; LEA, Lea cards; E, E-signs; √, within normal range.
Figure 1.
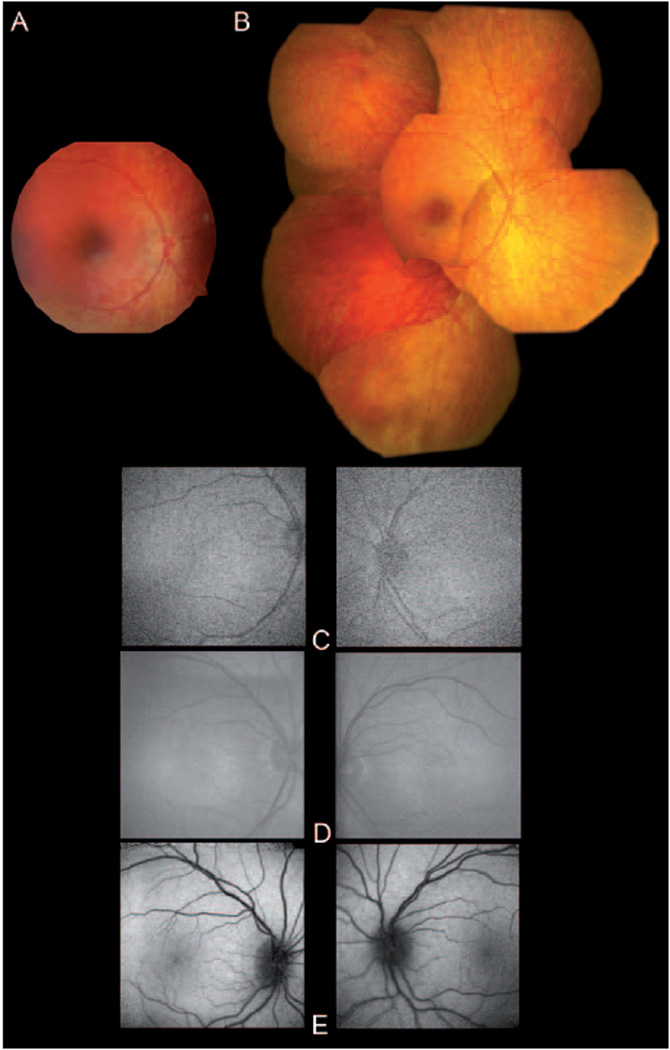
Fundus imaging of the patient. (A) Fundus photography of the right eye taken with a RetCam 120 system at age 3-y. (B) Fundus photomontage of the right eye taken with a Zeiss FF 450 plus at age 7-y. (C) Fundus autofluorescence images of both eyes of the patient at age 3-y (top), and 7-y (middle). Note the low resolution due to the high level of enhancement to record the minimal FAF. For comparison FAF of an 8-y old healthy individual is shown (bottom).
Figure 4.
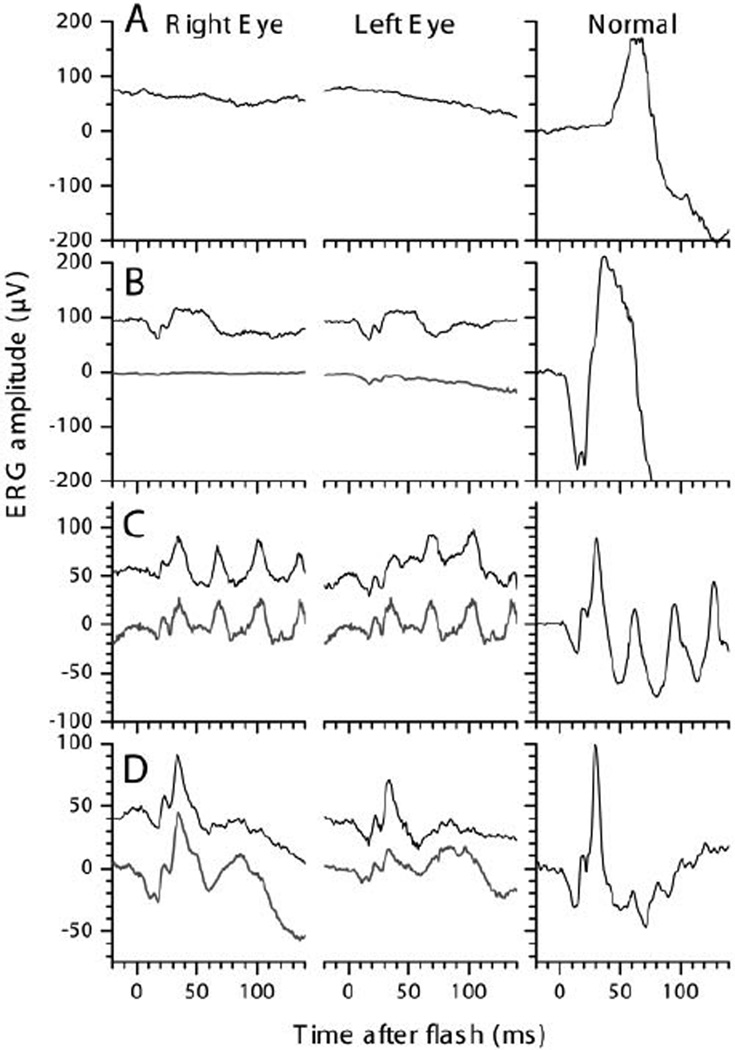
Ganzfeld-ERGs recordings of right and left eye of the patient at 5 (upper traces) and 6-y (lower traces). With a 2.75 log attenuated ISCEV Standard flash after 40 min of dark adaptation (A) hardly any rod response was obtained. Compared with a normal recording (right column), average rod-and-cone-responses to ISCEV Standard Flashes at 5 (B, upper trace) and 6-y (lower trace) were greatly reduced but not delayed. Responses to 30 Hz-Flicker after 10 min of light adaptation (C) were about half the normal size and within normal implicit time ranges. Light adapted responses to single Standard Flashes (D) were about 2/3 the normal size. Most responses obtained under general anaesthesia at age 3 years were even smaller (for more details see Table 1).
Fundus autofluorescence FAF was low but detectable, and did not change over the time of observation (fig. 1C).
Optical coherence tomography OCT (fig. 2) showed well preserved retinal layering at the initial exams, but at age 6 and 7-y the outer retina showed increased reflectivity and an increased transmission into the choroid. Spectralis OCT at age 7 detected retinal layer alterations and some thinning most pronounced in the outer retina (fig. 2D).
Figure 2.
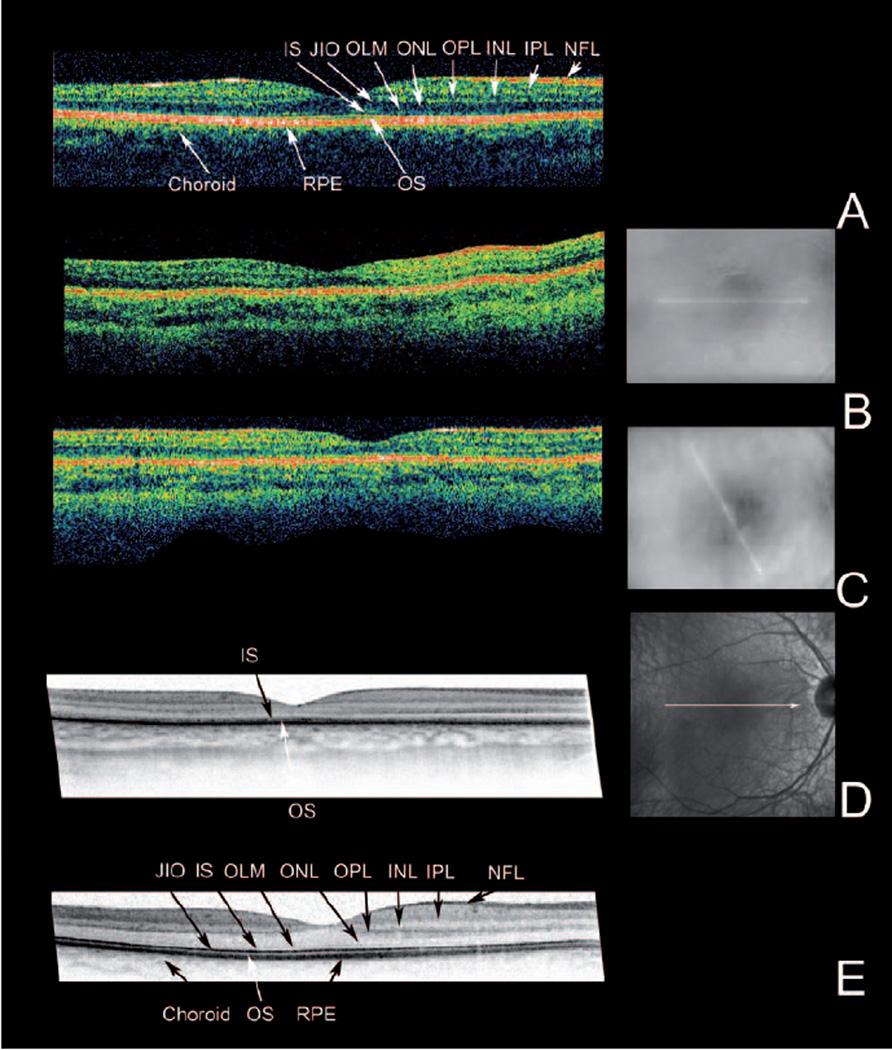
OCT imaging. For each eye the OCT image is accompanied by a fundus image showing the position of the scan. The findings were similar in both eyes. Only right eyes are shown.
(A) Stratus OCT recording of a 20-y old normal proband. (B) and (C) Stratus OCTs of the patient at age 3 and 7-y. (D) At the age of 7-y, a high resolution OCT was recorded with a Spectralis HRA-OCT. For comparison (E) a Spectralis OCT recording of the 20-y old normal proband is included. Note the overall reduced thickness of the retina in the patient (B, C, D). Spectralis imaging (D) shows that this loss of thickness is due to the loss of stratification in the inner (IS) and outer segment layers (OS) and thinning of the outer nuclear layer (ONL). NFL: Nerve fibre layer, IPL: inner plexiform layer, INL: inner nuclear layer, OPL: outer plexiform layer, ONL: outer nuclear layer, OLM: outer limiting membrane, IS: inner segment layer, JIO: junction between inner and outer segments, OS: outer segment layer: RPE: retinal pigment epithelium
2CT Perimetry (fig. 3) could be performed for the first time at age 6-y.
Figure 3.
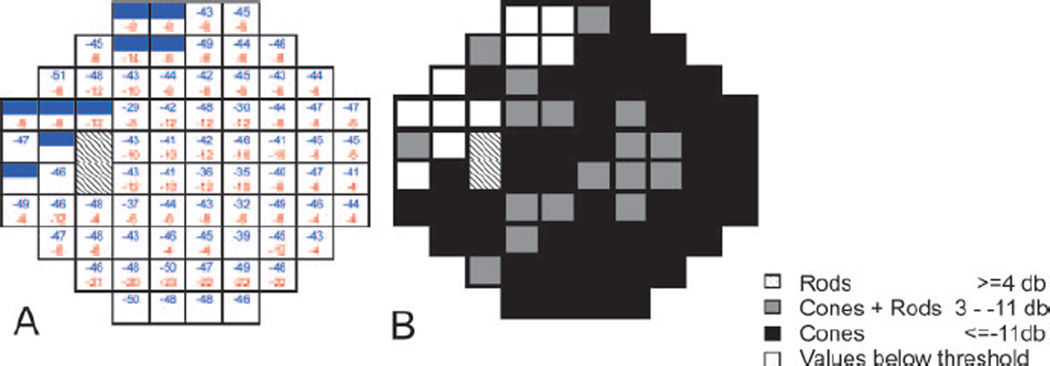
Two color threshold perimetry of a 30° visual field of the left eye of the patient at age 6 y. Hatched fields indicate the optic nerve head. (A) dark adapted sensitivity loss to 500 nm cut-off filter induced stimulus (blue) and light adapted sensitivity loss to 600 nm cut-on filter induced stimulus (red) for each tested target. Full color fields indicate sensitivity below threshold; empty fields indicate sensitivities within normal range. Shaded fields indicate sensitivity below 10th percentile of normal values. Numbers indicate sensitivity losses in dB. (B) Mediation; black = cone mediated answers at both wavelengths (pure cone mediation). Gray= rod mediation with a 500 nm cut-off filter and cone mediation with a 600 nm cut-on filter (mixed answer). Dark adapted responses were mostly cone mediated. Values close to pure cone mediation (12 loci 10 dB difference, 7 loci 8 dB difference).
In the dark adapted state, there was a severe sensitivity loss of 3 to 4 log units to the blue stimulus and of 1 to 1.4 log units to the red stimulus within the test field i.e. 30°. Most answers were cone mediated with only a few mixed answers i.e. rod-mediation for the blue and cone-mediation for the red stimulus. Cone sensitivity measured in the light adapted state showed a diffuse sensitivity loss of about 1.5 log units. Interestingly, cone sensitivity to red light (mediated by long wavelength cones) was higher than to blue light (mediated by short wavelength cones) which is in line with the clinical observation that most patients with RPE65 mutations report on difficulties with blue colors from early on.
Lowered isomerase activity of RPE65/P25L
We measured the isomerase activity of cells transfected with mutant constructs of RPE65. This activity is a combined phenotypic effect of the mutant of RPE65 on enzymatic activity and stability, comparable to an in vivo homozygous state for each allele. 293-F cells were transfected with pVitro3-LRAT+RDH5 and pVitro2-RPE65/P25L+CRALBP or pVitro2/wildtypeRPE65+CRALBP, grown for 24 hrs and then incubated with 2.5 µM all-trans retinol for 6 hours prior to harvest and analysis of isomeric retinols. Cells transfected with vector harboring the RPE65/P25L mutation produced only 7.75% of the 11-cis retinol made by cells transfected with pVitro2 containing wildtype RPE65 (Table 3). We compared the RPE65/P25L mutant isomerase activity with the activities of other missense mutants associated with cases of RPE65 EOSRD previously described as mild2,22, choosing to examine L22P, E95Q and Y79H. L22P was the least severely impaired at 13.5%, E95Q had an intermediate level of 6.1% of wildtype, while Y79H activity was almost abolished at 2.5% of wildtype activity (Table 3). Comparison with wildtype RPE65 of expression of the RPE65/P25L mutant was made by immunoblot (fig. 5) and quantitated (Table 4). We find that the level of P25L RPE65 protein is lowered to 25% of wildtype. The expression levels of each of the other mutants, like that of P25L, was reduced compared to wildtype. The level of expression of Y79H at 3% of wildtype (Fig 5 and Table 4) was the most adversely affected of all 4 mutants, concomitant with the low isomerase activity in cells transfected with its construct.
Table 3.
Effect of mutations on RPE65 isomerase activity
| Mutant | Mean Activity |
SD | n |
|---|---|---|---|
| P25L | 7.75 | 1.4 | 10 |
| L22P | 13.5 | 1.6 | 6 |
| Y79H | 2.5 | 0.6 | 7 |
| E95Q | 6.1 | 1.1 | 6 |
Activity is represented as % of wildtype RPE65 isomerase activity; with the SDS and number of replicates.
Figure 5.

Immunoblot analysis of RPE65 expression in 293-F cells transfected with wildtype, P25L, Y79H, E95Q and L22P mutants of RPE65 (top panel) and of CRALBP expression in the corresponding cells (bottom panel). It is seen that each of the mutants shows less expression than the wildtype construct. Lowest level of expression is seen for Y79H. Each lane was equivalently loaded with extract from 105 cells.
Table 4.
Effect of mutations on RPE65 protein expression
| Mutant | Mean Expression |
SD | n |
|---|---|---|---|
| P25L | 24.33 | 5.06 | 7 |
| L22P | 32.4 | 7.64 | 4 |
| Y79H | 3.31 | 0.38 | 4 |
| E95Q | 28.07 | 6.1 | 5 |
Mutant expression level was measured by fluorescence immunoblot and is normalized to and represented as % of wildtype RPE65 expression, with the SD and number of replicates.
Discussion
Herein we correlate the phenotypic effects of a homozygous P25L mutation in RPE65 on visual function in a young patient with in vitro measurement of the isomerase activity of RPE65/P25L mutant protein. We show that the mild clinical phenotype not yet complicated by long-term degeneration, is consistent with residual activity of a severely hypomorphic mutant enzyme. When tested in vitro, P25L RPE65 has only about 8% of wildtype activity. Yet, in vivo, this residual activity can maintain a physiologically relevant level of regeneration in psychophysical testing. These findings have relevance to understanding the natural history of RPE65-EOSRD and in providing rational indicators for the application of therapies.
The most important primary effect of RPE65 deficiency is its impact on visual pigment regeneration30,31. Various combinations of null and missense mutations can give a spectrum of disease from mild to severe. How RPE65 activity governs the dynamic range of visual pigment regeneration will become clearer as less severe late onset forms of RPE65-related retinal dystrophy disease are described. To put the biochemical data in context we compared the RPE65/P25L mutant isomerase activity with RPE65 EOSRD missense mutations previously described as mild, including L22P, E95Q and Y79H. Unlike the P25L patient, however, early clinical data is almost completely lacking. L22P was a compound heterozygote with H68Y2 and the patient had “a milder phenotype in childhood and a slower progression of the disease”, but visual function was severely affected at the time of molecular diagnosis (at 40-y)2. We have previously shown that H68Y essentially abolishes RPE65 isomerase activity (<2%;10). We estimate that the averaged level of RPE65 isomerase activity ((13.5%+2%)/2=7.75%) of the L22P/H68Y patient to be similar to the P25L patient (7.75%). In the Y79H/E95Q22 patient there was also a later time of onset (night blindness at 3 years, RP at age 20, and residual vision at age 5822). The averaged level of RPE65 isomerase activity ((6.1%+2.5%)/2=4.3%) of Y79H/E95Q is estimated to be less than P25L/P25L. Overall, however, these residual levels (<10%) of RPE65 activity may be enough to delay time of onset and delay, but not prevent slow cumulative progression/degeneration.
Efficient regeneration is required under conditions of high ambient light when photoisomerizations are at peak level31. Insufficient supply of 11-cis retinal causes night blindness due to insufficient formation of rhodopsin. Ligand-free opsin constitutively activates the phototransduction cascade and causes photoreceptor degeneration by mimicking a constant light-adapted state32. Thus, any activity of RPE65 supports survival of photoreceptors by formation of rhodopsin and control of its state of activity. While faster regeneration is beneficial under conditions of higher ambient light it confers a higher susceptibility to light damage33 due to greater turnover of activated phototransduction species and greater A2E accumulation18,34, both of which are linked to retinal degeneration in animal models35,36.
FAF detects lipofuscin accumulation in the RPE and lipofuscin /A2E accumulation is correlated with RPE65 activity: Rpe65−/− mice accumulate less than 10% of lipofuscin fluorophore as wildtype mice34. Similarly, FAF, as a clinically accessible surrogate for A2E/lipofuscin accumulation in the RPE, measures RPE65 activity. This patient’s reduced FAF correlates with the lower RPE65 activity of P25L mutant RPE65. Evaluation of FAF plays an important role in the differential diagnosis of RPE65 EOSRD/LCA. Of paramount importance is the association of normal fundus with low or no autofluorescence25. In severe RPE65 mutations, FAF is non detectable from early on. Lowered FAF also occurs with severe retinal changes on ophthalmoscopy, and normal FAF occurs with abnormal fundus, but neither of these situations occurs in RPE65 EOSRD/LCA. Interestingly, in this regard, Simonelli et al.37 present a lower than normal FAF image for an RPE65 patient, in association with a normal fundus, thus supporting our interpretation. In their own interpretation of this data, more weight is given to the actual existence of FAF than to its relative diminishment in the RPE65 patients, underestimating its importance in RPE65 EOSRD diagnosis.
Cone vision in many non-null cases of human RPE65-EOSRD is relatively spared compared to rod vision (night-blindness and lack of rod ERG), in contrast to the knockout mouse model, which is by definition null. In the Rpe65−/− mouse cones degenerate much more rapidly than rods, and this raises concern as to timing of rescue38. Is the situation observed in mice directly relevant to humans? Certainly, both cone and rod visual functions are severely impacted in many patients described7,22, but the chronology of rod vs. cone demise is still not known. Greater preservation of foveal structure concomitant with relatively spared cone-mediated function is seen in patients with a variety of RPE65 mutations39. Thus, the low chromophore level in the P25L patient, and possibly in other patients, may spare cone function. Conversely, how is cone survival affected in mice with a knock-in missense mutation? Recently, the phenotype of an RPE65 R91W missense knockin mouse has been published40. This mouse makes 5% of wildtype level of 11-cis retinal and has less severe morphological and physiological changes, including better cone function, than the Rpe65 knockout. Another strikingly consistent finding is that blue cone vision is more markedly affected in RPE65-EOSRD patients than is red cone vision7. Why is this so? It may be due to a combination of the biochemistry of cone pigments in general, combined with the abundance/distribution of blue cones in particular. Cone opsins make a reversible Schiff’s base with 11-cis retinal, one not requiring photoisomerization to break it41,42, while in rod opsin the bond is essentially irreversible without photoisomerization. Thus cone pigments more easily release chromophore, which if captured by rod pigment is not relinquished. The second consideration is the abundance and distribution of blue cones, comprising only about 10% of all cones. In the fovea, where blue cones are excluded from the central fovea43, when chromophore is limiting blue cones may be out-competed by the abundance of red/green cones. In the peripheral retina, where rod cells are overwhelming, the biochemical properties of rod opsin will ensure that blue cones are out-competed when chromophore is limiting. However, as RPE65 is more abundant centrally than peripherally39, this may further reduce peripheral rod and blue cone access to chromophore compared to central red/green cones and may help explain the serious rod deficit as well as the blue cone deficit.
In conclusion, this study represents the earliest time point yet comprehensively analyzed in the natural history of RPE65-associated severe retinal dystrophy in human. The residual activity of the hypomorphic P25L RPE65 can maintain some, but not all, features of normal vision. Of note, despite relative normal vision, morphological changes are already detected at that age by high resolution spectral domain OCT. In the Briard dog, Rpe65 knockout and rd12 mice morphological changes are also already evident at early ages. Nevertheless this does not prevent rescue by gene therapy44,45. The combination of clinical measurements with functional biochemical data in this patient strongly reinforces the concept that functional survival of cones is possible in patients with residual RPE65 activity and is a practical target for therapy. Though the future outcome of this patient is not clear, it is likely to be slowly progressing. The relative sparing of cone function might make this patient an ideal candidate for RPE65 somatic gene therapy, trials for which are currently in progress46,47.
Acknowledgments
Grant information:
DFG LO 457/5, Pro Retina Deutschland e.V., and by the Intramural Research Program of the National Eye Institute, NIH (T.M.R. and E.P.).
We thank our little patient and his parents for excellent cooperation. We thank Birgit Langer, Ursula Biendl, Ulrike Brauer, Dagmar Glatz, Karin Heinfling, Günter Schuch, and Susan Gentleman for excellent technical help.
References
- 1.Gu S, Thompson DA, Srisailapathy Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat. Genet. 1997;17(2):194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 2.Marlhens F, Griffoin JM, Bareil C, Arnaud B, Claustres M, Hamel CP. Autosomal recessive retinal dystrophy associated with two novel mutations in the RPE65 gene. Eur. J Hum. Genet. 1998;6(5):527–531. doi: 10.1038/sj.ejhg.5200205. [DOI] [PubMed] [Google Scholar]
- 3.Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or Leber congenital amaurosis. Proceedings of the National Academy of Science USA. 1998;95(6):3088–3093. doi: 10.1073/pnas.95.6.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenz B, Gyürüs P, Preising M, Bremser D, Gu S, Andrassi M, Gerth C, Gal A. Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Investigative Ophthalmology and Visual Science. 2000;41(9):2735–2742. [PubMed] [Google Scholar]
- 5.Hamel CP, Griffoin JM, Lasquellec L, Bazalgette C, Arnaud B. Retinal dystrophies caused by mutations in RPE65: assessment of visual functions. British Journal of Ophthalmology. 2001;85(4):424–427. doi: 10.1136/bjo.85.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yzer S, van den Born LI, Schuil J, Kroes HY, van Genderen MM, Boonstra FN, van den HB, Brunner HG, Koenekoop RK, Cremers FP. A Tyr368His RPE65 founder mutation is associated with variable expression and progression of early onset retinal dystrophy in 10 families of a genetically isolated population. Journal of Medical Genetics. 2003;40(9):709–713. doi: 10.1136/jmg.40.9.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paunescu K, Wabbels B, Preising MN, Lorenz B. Longitudinal and cross-sectional study of patients with early-onset severe retinal dystrophy associated with RPE65 mutations. Graefes Arch. Clin. Exp. Ophthalmol. 2005;243(5):417–426. doi: 10.1007/s00417-004-1020-x. [DOI] [PubMed] [Google Scholar]
- 8.Jin M, Li S, Moghrabi WN, Sun H, Travis GH. Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell. 2005;122(3):449–459. doi: 10.1016/j.cell.2005.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moiseyev G, Takahashi Y, Chen Y, Gentleman S, Redmond TM, Crouch RK, Ma JX. RPE65 is an iron(II)-dependent isomerohydrolase in the retinoid visual cycle. J Biol. Chem. 2006;281(5):2835–2840. doi: 10.1074/jbc.M508903200. [DOI] [PubMed] [Google Scholar]
- 10.Redmond TM, Poliakov E, Yu S, Tsai JY, Lu Z, Gentleman S. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proceedings of the National Academy of Science USA. 2005;102(38):13658–13663. doi: 10.1073/pnas.0504167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamb TD, Pugh EN., Jr Dark adaptation and the retinoid cycle of vision. Prog. Retin. Eye Res. 2004;23(3):307–380. doi: 10.1016/j.preteyeres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Grimm C, Wenzel A, Hafezi F, Yu S, Redmond TM, Reme CE. Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nat. Genet. 2000;25(1):63–66. doi: 10.1038/75614. [DOI] [PubMed] [Google Scholar]
- 13.Pang JJ, Chang B, Hawes NL, Hurd RE, Davisson MT, Li J, Noorwez SM, Malhotra R, McDowell JH, Kaushal S, Hauswirth WW, Nusinowitz S, Thompson DA, Heckenlively JR. Retinal degeneration 12 (rd12): a new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA) Molecular Vision. 2005;11:152–162. 152-162. [PubMed] [Google Scholar]
- 14.Veske A, Nilsson SE, Narfström K, Gal A. Retinal dystrophy of Swedish Briard/Briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics. 1999;57(1):57–61. doi: 10.1006/geno.1999.5754. [DOI] [PubMed] [Google Scholar]
- 15.Aguirre GD, Baldwin V, Pearce Kelling S, Narfström K, Ray K, Acland GM. Congenital stationary night blindness in the dog: common mutation in the RPE65 gene indicates founder effect. Molecular Vision. 1998;4:23. [PubMed] [Google Scholar]
- 16.Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, Jacobson SG, Hauswirth WW, Bennett J. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001;28(1):92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- 17.Wenzel A, Reme CE, Williams TP, Hafezi F, Grimm C. The Rpe65 Leu450Met Variation Increases Retinal Resistance Against Light-Induced Degeneration by Slowing Rhodopsin Regeneration. J. Neurosci. 2001;21(1):53–58. doi: 10.1523/JNEUROSCI.21-01-00053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim SR, Fishkin N, Kong J, Nakanishi K, Allikmets R, Sparrow JR. Rpe65 Leu450Met variant is associated with reduced levels of the retinal pigment epithelium lipofuscin fluorophores A2E and iso-A2E. Proc. Natl. Acad Sci U. S A. 2004;101(32):11668–11672. doi: 10.1073/pnas.0403499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyubarsky AL, Savchenko AB, Morocco SB, Daniele LL, Redmond TM, Pugh EN., Jr Mole quantity of RPE65 and its productivity in the generation of 11-cis-retinal from retinyl esters in the living mouse eye. Biochemistry. 2005;44(29):9880–9888. doi: 10.1021/bi0505363. [DOI] [PubMed] [Google Scholar]
- 20.Samardzija M, Wenzel A, Naash M, Reme CE, Grimm C. Rpe65 as a modifier gene for inherited retinal degeneration. Eur. J Neurosci. 2006;23(4):1028–1034. doi: 10.1111/j.1460-9568.2006.04639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Redmond TM, Weber CH, Poliakov E, Yu S, Gentleman S. Effect of Leu/Met variation at residue 450 on isomerase activity and protein expression of RPE65 and its modulation by variation at other residues. Molecular Vision. 2007;13:1813–1821. 1813-1821. [PubMed] [Google Scholar]
- 22.Thompson DA, Gyürüs P, Fleischer LL, Bingham EL, McHenry CL, Apfelstedt-Sylla E, Zrenner E, Lorenz B, Richards JE, Jacobson SG, Sieving PA, Gal A. Genetics and Phenotypes of RPE65 Mutations in Inherited Retinal Degeneration. Investigative Ophthalmology and Visual Science. 2000;41(13):4293–4299. [PubMed] [Google Scholar]
- 23.Lorenz B, Andrassi M, Kretschmann U. Phenotype in two families with RP3 associated with RPGR mutations. Ophthalmic Genetics. 2003;24(2):89–101. doi: 10.1076/opge.24.2.89.14001. [DOI] [PubMed] [Google Scholar]
- 24.Marmor MF, Holder GE, Seeliger MW, Yamamoto S. Standard for clinical electroretinography (2004 update) Doc. Ophthalmol. 2004;108(2):107–114. doi: 10.1023/b:doop.0000036793.44912.45. [DOI] [PubMed] [Google Scholar]
- 25.Lorenz B, Wabbels B, Wegscheider E, Hamel CP, Drexler W, Preising MN. Lack of fundus autofluorescence to 488 nanometers from childhood on in patients with early-onset severe retinal dystrophy associated with mutations in RPE65. Ophthalmology. 2004;111(8):1585–1594. doi: 10.1016/j.ophtha.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 26.Drexler W, Fujimoto JG. State-of-the-art retinal optical coherence tomography. Prog. Retin. Eye Res. 2008;27(1):45–88. doi: 10.1016/j.preteyeres.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 27.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, Eliaou C, Liu SY, Harris E, Redmond TM, Arnaud B, Claustres M, Hamel CP. Mutations in RPE65 cause Leber's congenital amaurosis. Nat. Genet. 1997;17(2):139–141. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- 29.Landers GM, Olson JA. Rapid, simultaneous determination of isomers of retinal, retinal oxime and retinol by high-performance liquid chromatography. J Chromatogr. 1988;438(2):383–392. doi: 10.1016/s0021-9673(00)90269-3. [DOI] [PubMed] [Google Scholar]
- 30.Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet. 1998;20(4):344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- 31.Lamb LE, Simon JD. A2E: a component of ocular lipofuscin. Photochem. Photobiol. 2004;79(2):127–136. doi: 10.1562/0031-8655(2004)079<0127:aacool>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 32.Fan J, Rohrer B, Moiseyev G, Ma JX, Crouch RK. Isorhodopsin rather than rhodopsin mediates rod function in RPE65 knock-out mice. Proc. Natl. Acad Sci U. S A. 2003;100(23):13662–13667. doi: 10.1073/pnas.2234461100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wenzel A, Grimm C, Marti A, Kueng-Hitz N, Hafezi F, Niemeyer G, Reme CE. c-fos controls the "private pathway" of light-induced apoptosis of retinal photoreceptors. J. Neurosci. 2000;20(1):81–88. doi: 10.1523/JNEUROSCI.20-01-00081.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katz ML, Redmond TM. Effect of rpe65 knockout on accumulation of lipofuscin fluorophores in the retinal pigment epithelium. Investigative Ophthalmology and Visual Science. 2001;42(12):3023–3030. [PubMed] [Google Scholar]
- 35.Sparrow JR, Zhou J, Cai B. DNA is a target of the photodynamic effects elicited in A2E-laden RPE by blue-light illumination. Investigative Ophthalmology and Visual Science. 2003;44(5):2245–2251. doi: 10.1167/iovs.02-0746. [DOI] [PubMed] [Google Scholar]
- 36.Reme CE. The dark side of light: rhodopsin and the silent death of vision the proctor lecture. Invest Ophthalmol. Vis. Sci. 2005;46(8):2672–2682. doi: 10.1167/iovs.04-1095. [DOI] [PubMed] [Google Scholar]
- 37.Simonelli F, Ziviello C, Testa F, Rossi S, Fazzi E, Bianchi PE, Fossarello M, Signorini S, Bertone C, Galantuomo S, Brancati F, Valente EM, Ciccodicola A, Rinaldi E, Auricchio A, Banfi S. Clinical and molecular genetics of Leber's congenital amaurosis: a multicenter study of Italian patients. Invest Ophthalmol. Vis. Sci. 2007;48(9):4284–4290. doi: 10.1167/iovs.07-0068. [DOI] [PubMed] [Google Scholar]
- 38.Rohrer B, Lohr HR, Humphries P, Redmond TM, Seeliger MW, Crouch RK. Cone Opsin Mislocalization in Rpe65−/− Mice: A Defect That Can Be Corrected by 11-cis Retinal. Invest Ophthalmol. Vis. Sci. 2005;46(10):3876–3882. doi: 10.1167/iovs.05-0533. [DOI] [PubMed] [Google Scholar]
- 39.Jacobson SG, Aleman TS, Cideciyan AV, Sumaroka A, Schwartz SB, Windsor EA, Traboulsi EI, Heon E, Pittler SJ, Milam AH, Maguire AM, Palczewski K, Stone EM, Bennett J. Identifying photoreceptors in blind eyes caused by RPE65 mutations: Prerequisite for human gene therapy success. Proc. Natl. Acad. Sci. U. S. A. 2005;102(17):6177–6182. doi: 10.1073/pnas.0500646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samardzija M, von LJ, Tanimoto N, Oberhauser V, Thiersch M, Reme CE, Seeliger M, Grimm C, Wenzel A. R91W mutation in Rpe65 leads to milder early-onset retinal dystrophy due to the generation of low levels of 11-cis-retinal. Human Molecular Genetics. 2008;17(2):281–292. doi: 10.1093/hmg/ddm304. [DOI] [PubMed] [Google Scholar]
- 41.Matsumoto H, Tokunaga F, Yoshizawa T. Accessibility of the iodopsin chromophore. Biochim. Biophys. Acta. 1975;404(2):300–308. doi: 10.1016/0304-4165(75)90337-2. [DOI] [PubMed] [Google Scholar]
- 42.Kefalov VJ, Estevez ME, Kono M, Goletz PW, Crouch RK, Cornwall MC, Yau KW. Breaking the covalent bond--a pigment property that contributes to desensitization in cones. Neuron. 2005;46(6):879–890. doi: 10.1016/j.neuron.2005.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeMonasterio FM, Schein SJ, McCrane EP. Staining of blue-sensitive cones of the macaque retina by a fluorescent dye. Science. 1981;213(4513):1278–1281. doi: 10.1126/science.7268439. [DOI] [PubMed] [Google Scholar]
- 44.Narfstrom K, Katz ML, Bragadottir R, Seeliger M, Boulanger A, Redmond TM, Caro L, Lai CM, Rakoczy PE. Functional and structural recovery of the retina after gene therapy in the RPE65 null mutation dog. Invest Ophthalmol. Vis. Sci. 2003;44(4):1663–1672. doi: 10.1167/iovs.02-0595. [DOI] [PubMed] [Google Scholar]
- 45.Pang JJ, Chang B, Kumar A, Nusinowitz S, Noorwez SM, Li J, Rani A, Foster TC, Chiodo VA, Doyle T, Li H, Malhotra R, Teusner JT, McDowell JH, Min SH, Li Q, Kaushal S, Hauswirth WW. Gene therapy restores vision-dependent behavior as well as retinal structure and function in a mouse model of RPE65 Leber congenital amaurosis. Mol. Ther. 2006;13(3):565–572. doi: 10.1016/j.ymthe.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 46.Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR. Effect of Gene Therapy on Visual Function in Leber's Congenital Amaurosis. New England Journal of Medicine. 2008 doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 47.Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell'osso L, Hertle R, Ma JX, Redmond TM, Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ, McDonnell JW, Auricchio A, High KA, Bennett J. Safety and Efficacy of Gene Transfer for Leber's Congenital Amaurosis. New England Journal of Medicine. 2008 doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]