

Abstract
Xenografts are a popular model for the study of the action of new antitumor drugs. However, xenografts are highly heterogeneous structures, and therefore it is sometimes difficult to evaluate the effects of the compounds on tumor metabolism. In this context, imaging mass spectrometry (IMS) may yield the required information, due to its inherent characteristics of sensitivity and spatial resolution. To the best of our knowledge, there is still no clear analysis protocol to properly evaluate the changes between samples due to the treatment. Here we present a protocol for the evaluation of the effect of 2-hydroxyoleic acid (2-OHOA), an antitumor compound, on xenografts lipidome based on IMS. Direct treated/control comparison did not show conclusive results. As we will demonstrate, a more sophisticated protocol was required to evaluate these changes including the following: (1) identification of different areas in the xenograft, (2) classification of these areas (necrotic/viable) to compare similar types of tissues, (3) suppression of the effect of the variation of adduct formation between samples, and (4) normalization of the variables using the standard deviation to eliminate the excessive impact of the stronger peaks in the statistical analysis. In this way, the 36 lipid species that experienced the largest changes between treated and control were identified. Furthermore, incorporation of 2-hydroxyoleic acid to a sphinganine base was also confirmed by MS/MS. Comparison of the changes observed here with previous results obtained with different techniques demonstrates the validity of the protocol.
Graphical abstract
Xenografts are one of the most popular models to study tumor growth and their response to new treatments.1 They are generated in an immunodepressed mouse2 by injecting tumor cells either subcutaneously or onto a target organ. Although they are not as realistic as the studies with patients,3 they constitute a significantly better model than cultured cells, as the xenograft develops inside a living organism.4–7 However, there are certain aspects that should be taken into account when using this model to evaluate the effect of a new substance. First, the intrinsic heterogeneity of xenografts: cells at different metabolic stages, from viable to necrotic, coexist with no clear boundaries.8–10 Second, as in real tumors, irrigation may differ depending on the position of the cells inside the xenograft and on the local vascularization.9,11 Thus, drugs may not be able to reach equally all the tumor cells, and consequently, the response to the treatment may vary from one point to another within the xenograft.8,9 Therefore, a technique that offers location-dependent information is required to carry out such studies.
In this sense, matrix assisted laser desorption and ionization imaging mass spectrometry (MALDI-IMS) is particularly well fitted for this task, due to its inherent characteristics: it does not require previous labeling, and thanks to its high dynamic range, it allows one to record the distribution of hundreds of species in a single run and in very different concentrations.12–14 Furthermore, it offers very good resolution and sensitivity in the lipid region (m/z ~500–2500), especially if the correct matrix and preparation protocol are chosen,15–19 and allows for recording distributions with spatial resolutions from ~1 to 200 µm, depending on the sample and the abundance of the metabolite.12,20,21 On the other hand, lipids are good indicators of the metabolic stage of a cell, as they are involved in all major cellular processes: from cell growth to apoptosis.22–26 Consequently, their distribution inside the xenograft may give invaluable information about the viability of the cells27 and on the potential effect of a drug.
However, comparison of distribution maps between samples with very different geometry is not a simple task, as demonstrated by the number of publications covering IMS data analysis.28–35 This is particularly true in highly heterogeneous samples, such as xenografts, where very different regions may coexist inside each sample. Furthermore, the relative extension of these regions and their morphology vary substantially from sample to sample, even between sequential sections of the same xenograft. Some works on the distribution of metabolites along a series of samples may be found in refs 11, 36, and 37. Here we present a new methodology based on the use of IMS to assess the effect of the antitumor drug 2-hydroxyoleic acid (2-OHOA), on the lipid composition of A549 (human lung adenocarcinoma) xenografts.
Treatment of cultured tumor cells with 2-OHOA leads to profound changes on both phospholipid and fatty acid composition.38,39 The 2-OHOA mechanism of action involves the specific and sequential induction of cell cycle arrest,40 cell differentiation,41 and cell death in a wide range of human cancer cells.38,42 In cell culture, 2-OHOA is rapidly incorporated into the phospholipid fraction39 and significantly modifies cell lipid composition.38,43 Thus, the analysis by thin layer chromatography of cell lipid extracts shows that, in all tested tumor cell lines, sphingomyelin (SM) mass increased, while glycerophophatidylcholine (PC) and glycerophosphatidyl ethanolamine (PE) decreased.38 In addition, gas chromatography analysis shows that oleic acid content was severely compromised in both PC and PE fractions.43 Although 2-OHOA treatment has been efficient in reducing tumor size in xenograft models,41 the first attempts to assess the effect on their lipid composition by high performance thin layer chromatography (HPTLC) did not yield conclusive results (Table 1 and Supporting Information Figure S1). These analyses were done on the lipid extracts of whole xenografts, and consequently any local changes in composition may have been overlooked, due to the aforementioned reasons. In order to investigate this possibility, we obtained the lipid distribution in histological sections of xenografts obtained from 2-OHOA-treated (n = 10) and nontreated mice (control, n = 10) using MALDI-IMS.
Table 1.
Effect of 2-OHOA Treatment on Xenograft Phospholipid Composition Measured by HPTLCa
| % total phospholipid | ||||
|---|---|---|---|---|
| control | 2-OHOA-treated | |||
| phospholipid class | mean | SD | mean | SD |
| SM | 10.1 | 1.2 | 10.2 | 1.8 |
| PC | 47.9 | 6.6 | 53.2 | 7.9 |
| PS + PIb | 23.8 | 3.8 | 20.1 | 1.9 |
| PE | 18.2 | 3.1 | 16.6 | 8.4 |
Xenografts were analyzed as described in Material and Methods. Values are expressed as the percentage of total phospholipid and represent mean ± SD; n = 4–5.
PS = glycerophosphatidylserine; PI = glycerophosphatidylinositol.
A first statistical analysis of the images revealed the presence of at least three distinct regions inside each xenograft, corresponding to necrotic cells, viable cells, and an external region of conjunctive tissue usually developed by the host at the site of the xenograft formation to encapsulate it. As we will show, the changes in the lipidome induced by the treatment could not be unraveled by a simple comparison between average spectra. Indeed, a more sophisticated data analysis was required, which consisted of (1) identifying the number of different areas in the xenograft, (2) classifying these areas (necrotic/viable) to carry out the analysis using only the viable tissue, (3) suppressing the effect of the variation of adduct formation between samples, and (4) normalizing the variables using the standard deviation to eliminate the excessive impact of the stronger peaks. The procedure presented here could be easily adapted to other types of samples, and it may constitute a good starting point for a standardized protocol to analyze IMS results.
METHODS
The central subject of this work was the development of the protocol for data analysis. The detailed description of the experimental protocols may be found elsewhere.10 Therefore, for the sake of brevity, only the most relevant aspects of the experimental procedures will be highlighted here.
Reagents
Ammonium acetate was purchased from Sigma–Aldrich (Steinheim, Germany). Water, methanol, acetonitrile, 2-propanol, and formic acid (Fisher Scientific, Fair Lawn, NJ, USA) were of Optima LC/MS grade. Leucine enkephalin acetate hydrate, ammonium acetate, and sodium hydroxide solution were purchased from Sigma–Aldrich Chemie (Steinheim, Germany). n-Hexane and chloroform were HPLC grade ACS ISO UV–vis purity (Scharlau, Barcelona, Spain); H3PO4 and CuSO4 were purchased from Sigma-Aldrich (Madrid, Spain).
Lipid standards lysophosphatidylcholine (LPC, synthetic), phosphatidylethanolamine (PE, bovine liver), and phosphatidylcholine (PC, bovine liver) were purchased from Avanti Polar-Lipids, Inc. (Alabaster, AL, USA). All stock standard solutions were prepared in chloroform at 1000 µg/mL and stored at −20 °C in darkness. For identification purposes, reference solutions of each lipid class were prepared in methanol–chloroform (2:1, v/v) at 100 µg/mL.
Chloroform and methanol used for xenograft lipid extraction for HPTLC analysis were HPLC grade ACS ISO UV–vis purity (Scharlau, Barcelona, Spain). In this case lipids were identified using commercially available standards from Larodan (Limhamm, Sweden).
Animals and Tumor Xenografts
Xenografts were generated as described in Supporting Information in ref 39. Animals were randomly divided into groups with a similar mean tumor volume, and they received daily oral treatments with the vehicle alone (water) or 2-OHOA (600 mg·kg−1, Avanti Polar Lipids) for 50 days. Then, tumors were removed and immediately frozen in liquid nitrogen before being stored at −80 °C until analysis. All experiments were carried out in accordance with the animal welfare guidelines of the European Union and the Institutional Committee for Animal Research of the University of the Balearic Islands. Sections of ~15 µm thicknesses were prepared using no embedding material with the aid of a cryostat (Leica CM3050S, Wetzlar, Germany) at −20 °C and placed on plain glass microscope slides. Sections were stored at −80 °C until analysis. For HPTLC analysis, frozen xenograft tissue samples were homogenized with a tissue blender (Ultra-Turrax, Janke and Kunkel). Lipids were extracted using n-hexane/2-propanol (3:2 (v/v), 1.8 mL/(100 mg of tissue)). Tissue extracts were centrifuged at 1000g to pellet debris. The lipid containing the organic phase was decanted and stored under nitrogen at −80 °C until analysis. Phospholipids were separated on Whatman silica gel-60 plates (10 × 10 cm2; Merck, Darmstadt, Germany) using chloroform/methanol/acetic acid/water 55:37.5:3:2 (by vol), which separates all major glycerophospholipids. Plates were air-dried after development, sprayed with 8% (w/v) H3PO4 containing 10% (w/v) CuSO4, and charred at 180 °C for 10 min. Then, lipids were quantified by photodensitometry (Quantity One software, Bio-Rad, Barcelona, Spain).
MALDI-IMS
The protocol employed here was adapted from ref 10. Ten sections of control xenografts and ten sections of treated xenografts were scanned in positive ion mode, at spatial resolutions between 100 and 150 µm/pixel, using the orbitrap analyzer of an LTQ-Orbitrap XL (ThermoFisher, San José, CA, USA), equipped with an N2 laser (100 µJ max power; elliptical spot of 150 µm diameter; 60 Hz repetition rate44). The sections were covered with a suitable matrix (2-mercaptobenzothiazole, MBT18) with the aid of a glass sublimator (Ace Glass 8023, Vineland, NJ, USA). The protocol was slightly modified from that described in ref 10, as it was demonstrated that leaving the vacuum system working during the sublimation process and using a sublimation temperature of 130 °C a deposition time of 10–12 min only was required, and matrix was deposited more uniformly.
Due to the amorphous nature of the sample, no anatomical structures were observed at any of the spatial resolutions tested, and therefore, after a first optimization stage, most of the samples were scanned at 150 µm/pixel. Mass resolutions of 60,000 and 100,000 were used to record the data in the m/z = 400–1000 region, using positive ion mode.
The spectra obtained were analyzed using dedicated software (MSIAnalyst, NorayBioinformatics S. L., Zamudio, Spain) and homemade algorithms built in Matlab (MathWorks, Natick, MA, USA). During parsing, the size of the data was reduced eliminating all peaks whose intensity was lower than the 0.5% of the strongest peak on the spectrum, and the spectra were normalized using two procedures: a total ion current algorithm45 and normalization to the intensity of the m/z = 760.5851 (PC 34:1+H+), although the results obtained with the second normalization procedure were more affected by experimental artifacts and were therefore discarded. The spectra were also aligned using the Xiong method46 and assuming a maximum misalignment of 0.02 amu.
Statistical analysis was carried out using principal component analysis (PCA)47 and k-means48 algorithms, depending on the information to be extracted. k-means performed better for the identification of areas in the tissues, while PCA was used for the classification process. Identification of the necrotic areas was confirmed by comparison with sequential sections dyed with acridine orange (data not shown).27
Lipid identification in MALDI-IMS was based on a direct comparison between the value of the m/z and the lipids in the software’s lipid database (>33,000 species plus their adducts) and with those in the lipid maps database (www.lipidmaps.org). Mass accuracy was always better than 8 ppm, and it was typically better than 3 ppm. In those cases where no univocal assignment was found, a comparison with the data from ultrahigh performance liquid chromatography (UHPLC) was performed. If any of the candidates was not detected by UHPLC-MS, and it was not detected as another adduct in the MALDI-IMS experiment, such a candidate was removed from the list.
UHPLC-MSE Analysis
Full description of the protocol may be found in ref 10. Briefly, UHPLC was carried out by using an ACQUITY UPLC system from Waters (Milford, MA, USA). Lipid extracts from xenografts were injected onto a column (Acquity UPLC HSS T3; 1.8 µm; 100 × 2.1 mm2; Waters) heated at 65 °C (see ref 10 for a more detailed description).
All UHPLC-MSE data were acquired on a SYNAPT G2 HDMS, with a quadrupole time-of-flight (Q-ToF) configuration, (Waters). Data acquisition took place over the mass range of 50–1200 amu in resolution mode (full width at half-maximum (fwhm) ~ 20,000) with a scan time of 0.5 s and an interscan delay of 0.024 s. The mass spectrometer was operated in the continuum MSE acquisition mode for both polarities.
RESULTS AND DISCUSSION
Analysis of the results by this image technique was not straightforward. The simplest approach to evaluate differences between treated and control samples’ lipidomes consists of running statistical analysis using the average mass spectra of each xenograft as input data. Yet, this approach failed to correctly classify the samples into the treated/control categories, as it did with the HPTLC approach (Figure S1 of the Supporting Information) or even comparison of LC-MS data, pointing to the existence of additional variables that may have a stronger influence on the average spectrum than the treatment. Next, we describe the steps of the analysis required to surface the differences between treated and control samples.
Identification of Regions of Interest
The first step in the analysis deals with the intrinsic heterogeneity of each tissue section. The statistical analysis showed that at least four areas (regions of interest, ROIs; Figure 1) might be found in each xenograft. Statistics were carried out taking into account all of the m/z in the spectrum with intensities > 0.5%. In this case, we used k-means analysis, although it may work with PCA as well.
Figure 1.
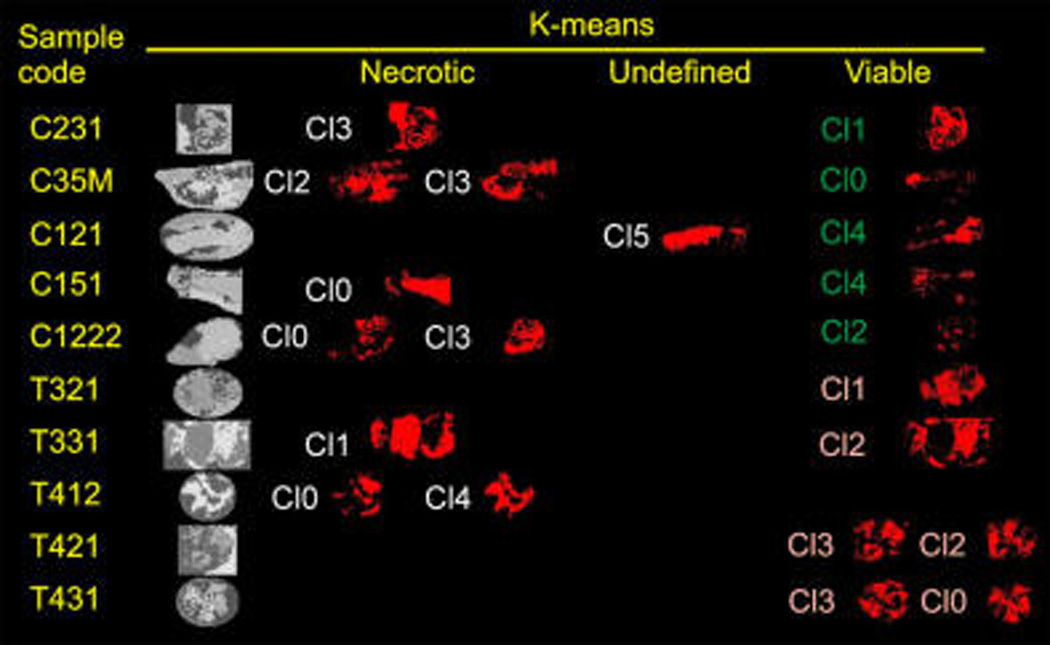
k-means analysis of eight sections of control (C) and treated (T) xenografts: gray scale images, ROIs identified by the algorithm in each section; necrotic, regions identified as containing mainly necrotic tissue; viable, regions identified as containing mainly viable tissue; undefined, sections in which necrotic or viable tissue was not clearly grouped.
Typically, four clusters were needed to account for all of the regions in the image, but some sections required up to eight clusters to correctly classify the different cellular areas in the xenograft. The k-means algorithm easily identified and grouped the spectra recorded outside the tissue in a single ROI that was excluded from the rest of the process, as the spectra contained only matrix peaks that would interfere with the analysis. There was an additional ROI present in all xenografts that was associated with the connective tissue developed by the host.10 The thickness of this tissue varied from one sample to another, but it was readily identified by its high content in neutral lipids (mainly triacylglycerols, TAG, species10), and it must also be discarded.
After this filtering stage, only those regions truly associated with tumor tissue should remain. In general, two ROIs were found in each section: one corresponding to necrotic tissue, while the other corresponded to the spectra recorded over viable tissue (see later text). In some sections, several additional regions were found, usually associated with viable areas. The latter could be due, for example, to the presence of cells at different metabolic stages within the xenograft or to differences in the blood supply.11
Classification of the ROIs into Necrotic and Viable Areas
Figure 2 shows the classification using the two first components of a PCA analysis of the ROIs that remained after the previous step. Still, it was necessary to eliminate from the spectra the peaks due to the matrix and the isotopic distributions. After this filtering process, only those species with intensities > 10% were taken into account for ROIs classification, as inclusion of less abundant species resulted in less clear separations. Although not all of the ROIs could be classified in this way (see ROIs at the center of the graph), there was a marked tendency of the analysis to establish two distinguishable groups. As it can be seen in the figure, the analysis did not segregate treated from control samples, but viable (left, blue) and necrotic tissue (right, red). Therefore, those ROIs lying at the center of the figure may correspond to areas of the tissue including a mixture of viable and necrotic cells. Developing a more sophisticated protocol for the separation at stage 1 could improve the whole analysis.
Figure 2.
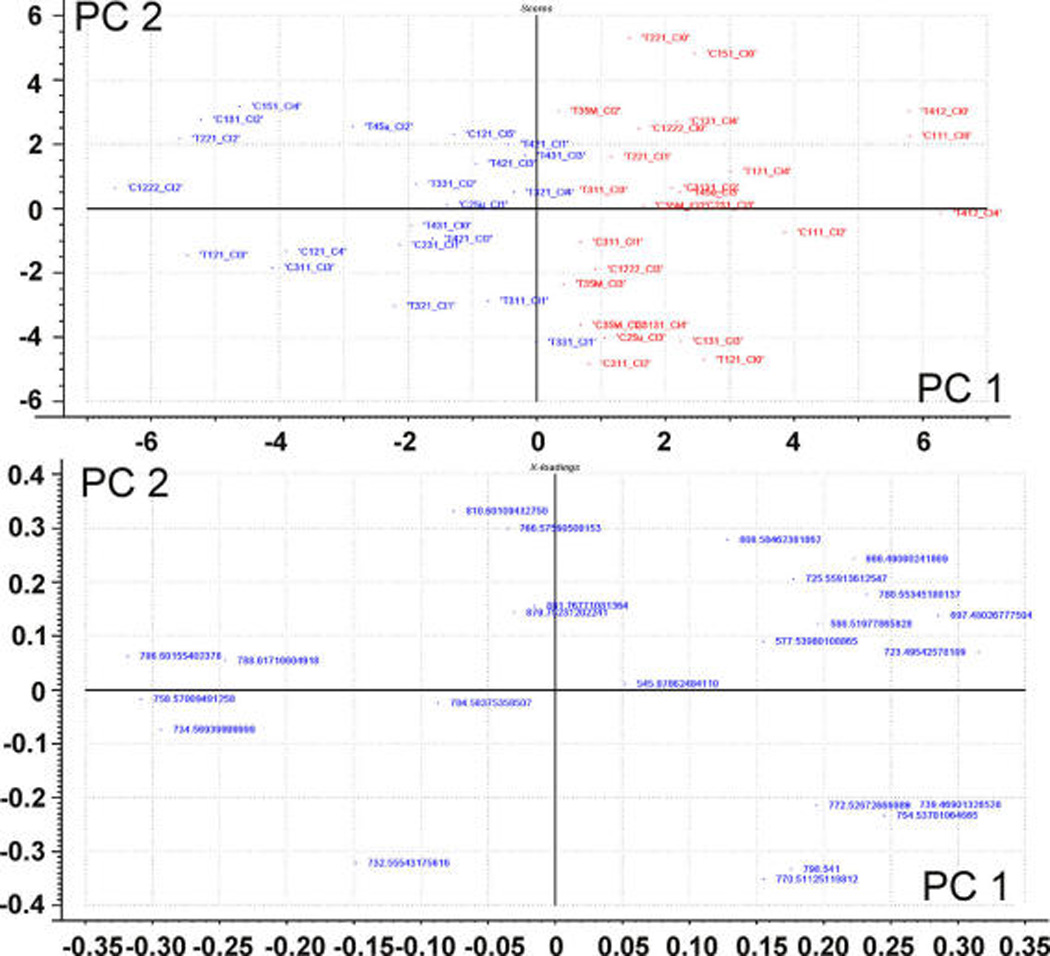
Scores plot (upper panel) and loadings (lower panel) of the ROIs selected in step 1. The algorithm was able to discriminate in the scores plot between necrotic (right, red) and viable (left, blue) regions.
The classification achieved in Figure 2 also means that, in this particular experiment, differences between necrotic and viable areas were statistically more significant than differences between treated and control groups. We have recently demonstrated27 that necrotic tissue is enriched in Na+ adducts. Consistently, a careful examination of the present results showed that the PC-Na+/PC-K+ adduct ratio was sufficient to classify the areas according to cell viability. Thus, the group on the right in Figure 2, presenting more intense Na+ adducts, would correspond to necrotic areas of the tissue, whereas the group on the left would contain the ROIs of viable areas. This assignment to necrotic/viable was further confirmed by comparison with sequential sections stained with acridine orange, a dye widely used to assess DNA damage in many cell types.27,49
According to our experience, the ROIs containing necrotic tissue must be removed from the analysis, as they do not present significant differences in composition between treated and control samples. Indeed, the analysis described later was performed in three different ways: using the average spectra of all of the samples, using the average spectra of only those ROIs corresponding to necrotic tissue, and, finally, using the average spectra of those ROIs containing viable tissue alone. Only in the latter case did differences between treated and control samples surface.
Suppression of Signal Intensity Variations Due to Changes in the Na+/K+ Ratio
Changes in adduct composition between treated and control were not as important as the changes between necrotic and viable tissue (Figure 3), but still this step proved to be critical, because the statistical noise introduced by the fluctuation in adduct composition will otherwise spoil the whole analysis. This step is probably the most difficult part of the protocol, as it involves identification of all lipid species that will be included in the analysis. However, if omitted, it is likely that only differences in adducts concentration would be observed after the comparison process. The most abundant lipid class detected in positive ion mode is PC, accounting for most of the intensity. Even for this abundant class, mass channels overlapping significantly complicates the analysis, and it is usually necessary to deconvolve the contribution of several species in each mass channel.
Figure 3.
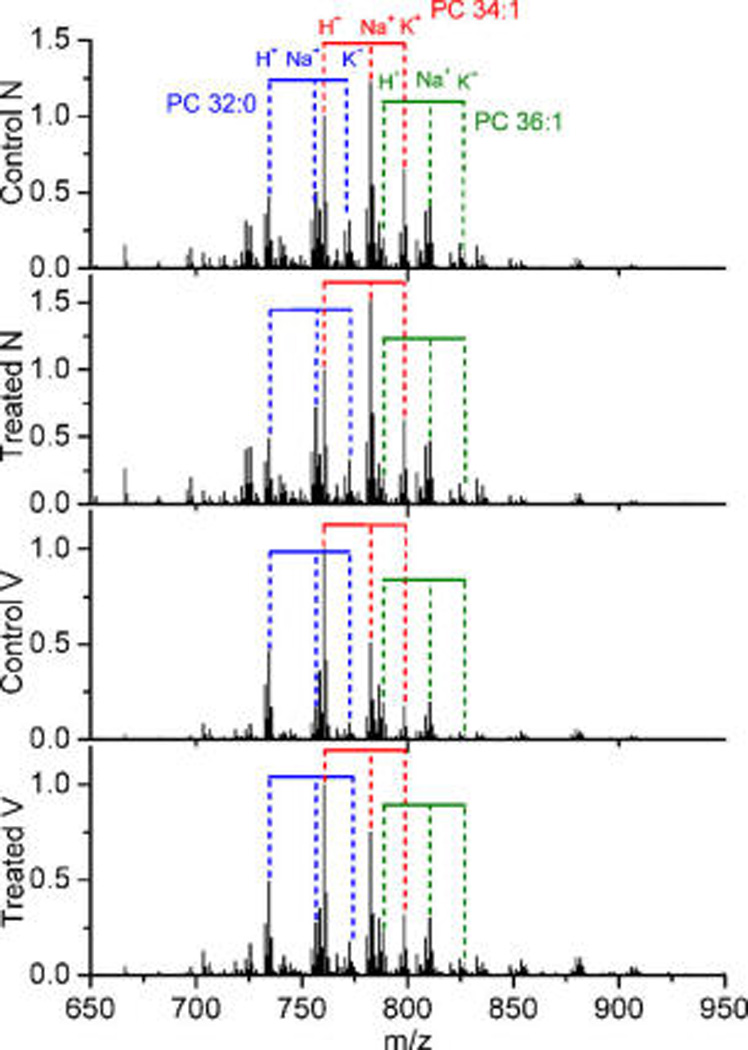
Average mass spectra over treated necrotic (treated N), control necrotic (control N), treated viable (treated V), and control viable (control V) ROIs. Important changes in the H+/Na+/K+ ratios were clearly visible.
The procedure followed in this section consisted of identifying the m/z channels by comparison with the values in our database and in lipid maps database (www.lipidmaps.org)50,51 and with the aid of the LC-MS data. Those species with univocal assignment in which all possible adducts were detected without interferences were used to determine the H+/Na+/K+ approximate average ratio for that lipid class. Then, this ratio was used to deconvolve the contribution from various species of the same m/z.
It is well-known that tumors are usually in acidic environments that may result in variations in the H+/Na+/K+ ratio.52–54 Consequently, the correction described in this step may not only be required in treated/control comparisons but also in comparison between healthy and altered tissue, such as, for example, in the study of tumor biopsies.
An alternative and probably easier approach would be to use a salt to shift adduct formation toward a single species, for example, adding potassium salts. However, in that case, a different procedure would be required to identify the necrotic areas. In addition, matrix deposition could not be done using a sublimator.55,56
Elimination of Species Showing Meaningless Changes
There are lipid species whose concentrations experience large changes between experiments and/or samples due to the technique’s variability or to the interspecimen intrinsic differences, rather than due to the treatment by itself. Besides, relatively small changes in very abundant species may mislead the conclusion of the study, as their weight in the analysis is larger than those of less abundant species. Therefore, normalization of the changes in each species dividing its average in treated/control groups by the standard deviation of its m/z is required. Next, only the species experiencing statistically meaningful changes (i.e., standard deviation < average intensity) were conserved. In our case, only 50 species, with their adducts’ contribution added in a single m/z, passed this stage.
Statistical Analysis on the ROIs of Viable Tissue
At this stage, the number of m/z was reduced to only those whose changes were relevant to the study. Only now, a standard PCA analysis on the lipid species surviving stage 4 was able to classify them into two categories: one containing the viable ROIs of treated xenografts (Figure 4a, right, red) and one containing exclusively viable regions of control xenografts (left, blue).
Figure 4.
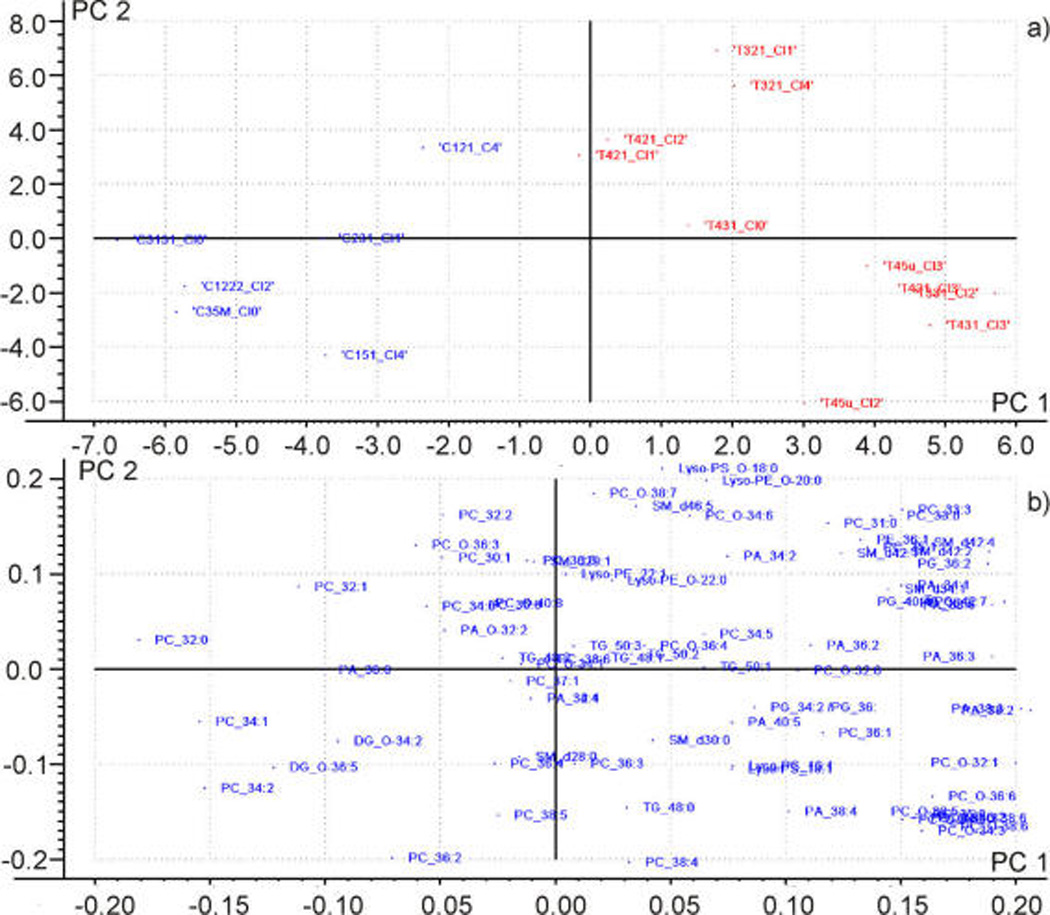
(a) Scores plot of the ROIs of viable tissue. Control samples (left) and treated samples (right) were well classified along the PC 1. (b) Loadings plot. The species contributing most to the separation are collected in Figure 5.
Figure 5 shows the average intensity of those species experiencing the largest changes in intensity after 2-OHOA treatment, i.e., the species at the far right and the far left in Figure 4b.
Figure 5.
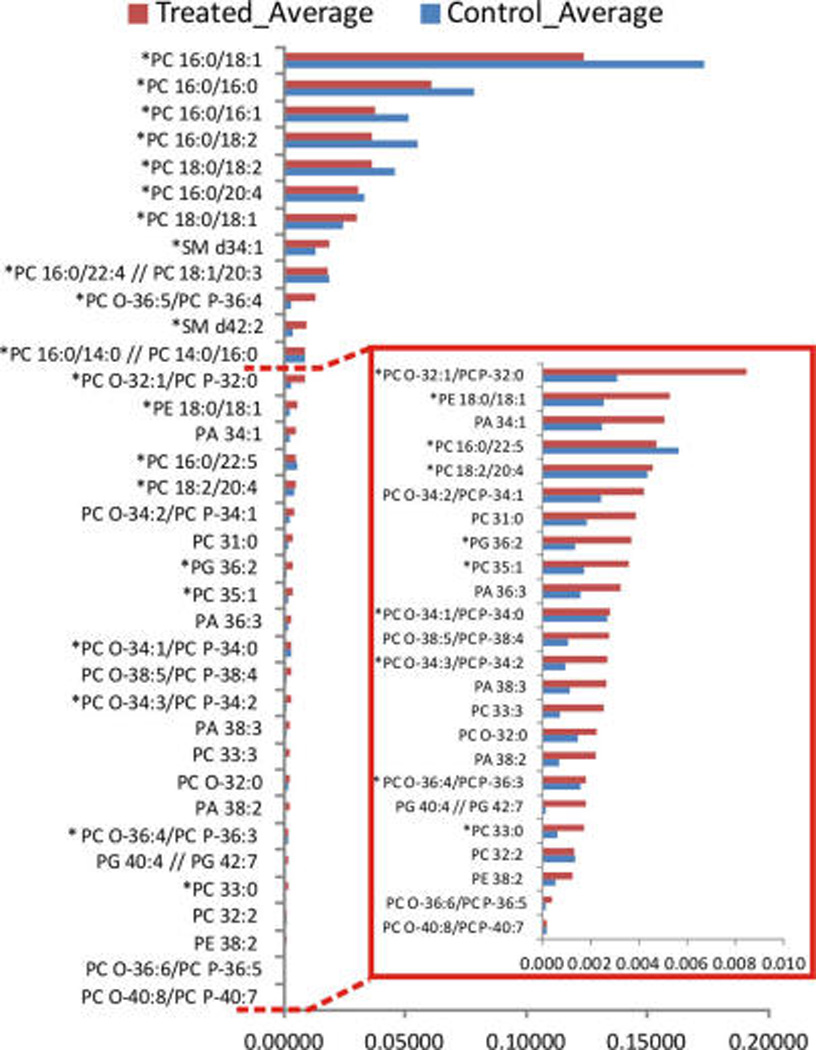
Lipid species experiencing the largest changes in intensity between treated and control samples, n = 10. Asterisks highlight those species whose presence was confirmed by LC/MS.
The results show a clear impact of 2-OHOA treatment on PC species, decreasing the concentration of the most abundant ones, that is, PC 34:1, PC 32:0, PC 32:1, PC 34:2, and PC 36:2 which together account for ~75% of total PC. In addition, the two most abundant SM species detected by IMS in positive ion mode, SM d42:2 and SM d34:1, were significantly increased after the 2-OHOA treatment (24.1 vs 31.2% and 61.2 vs 73.0% of total SM, respectively). Both results, the decrease in PC and the increase in SM content, were in agreement with previous studies assessing the effect of 2-OHOA treatment on the lipid composition of cultured cells.38,39 Therefore, these results clearly support our initial hypothesis that the treatment with 2-OHOA induced local changes in lipid composition within the xenograft and that they are most probably vanished when the lipids of the entire sample are extracted. Furthermore, they emphasize the importance of IMS techniques as any technique based on the analysis of lipid extract previously used failed in detecting these differences.
It is worthy to note that the incorporation of 2-OHOA to a SM was also detected. Figure 6 shows the distribution of the m/z = 763.605 in representative sections of treated xenografts. This species was found only in treated tumors, and its molecular mass fits well with 2-OHOA bound to a sphinganine base. Furthermore, fragmentation of that peak showed fragments at m/z = 184 and 264, indicating that it is a SM, and a fragment at m/z = 297.2628, which is one proton smaller than the mass of 2-OHOA (298.2508). In fact, saturated 2-hydroxylated fatty acids are rather common in sphingolipids.55 So, it is possible that cells incorporated 2-OHOA into SM using enzymatic pathways similar to those used by the non-hydroxylated fatty acids. Interestingly, this lipid species accumulated preferentially in the necrotic areas of the xenografts, reinforcing the role of 2-OHOA in the size reduction observed in xenografts after treatment.41
Figure 6.
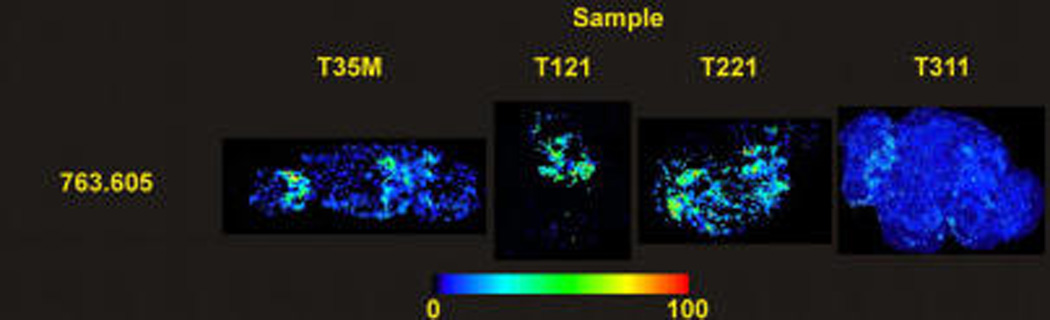
Distribution of m/z = 763.605 along representative sections of treated tumors, which seems to correspond to a SM that has incorporated 2-OHOA through an amide linkage.
CONCLUSIONS
We present here an optimized protocol for the analysis of xenograft samples treated with 2-OHOA. Several factors were taken into account to unravel the real changes induced by the treatment: tissue heterogeneity, changes in the adduct ratio due to factors other than the treatment, and normalization of the variables to eliminate statistical noise. The final procedure allowed us to detect that administration of the drug induced a reduction in the relative concentration of some PC species and an increase of SM species, in good agreement with previous studies in cultured cells.38,43 Furthermore, there is evidence that points to an incorporation of the drug, a 2-hydroxy fatty acid, into a sphingosine base. The fact that these changes were not detected during the analysis of lipid extracts suggests that the effect of the treatment was localized to certain areas, reinforcing the valuable information that IMS analysis may provide. The protocol described here may be a good starting point for a standardized protocol for the analysis of data from IMS experiments.
Supplementary Material
Acknowledgments
This study was partially supported by the Institute of Health Carlos III (Ministerio de Economía y Competitividad) and the EC (European Regional Development Fund, ERDF; Grant CP12/03338), BIO2010-21132, (Ministerio de Ciencia e Innovación, Spain) by UPV/EHU (UFI 11/23) and by the Basque Government (consolidated groups). G.B.-C.’s contract was supported by the Miguel Servet program of the Institute of Health Carlos III. J.G. thanks the UPV/EHU for a predoctoral fellowship. S.T. holds a contract from the INSERM (Institut National de la santé de la recherche médical), Délégation regional de Bordeaux. F.G.-S. holds a contract from the Asociación Española Contra el Cáncer (AECC). Technical and human support provided by the Servicio de Lipidómica of the SGIKER (UPV/EHU, MICINN, GV/EJ, ESF) is gratefully acknowledged.
Footnotes
ASSOCIATED CONTENT
Supporting Information
- HPTLC of lipid extracts from treated and control xenografts, examples of average spectra over sections of xenografts from treated and control animals, and residual variance for the final PCA analysis (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Batubara A, Carolan VA, Loadman PM, Sutton C, Shnyder SD, Clench MR. Rapid Commun. Mass Spectrom. 2015;29:1288–1296. doi: 10.1002/rcm.7223. [DOI] [PubMed] [Google Scholar]
- 2.Morton CL, Houghton PJ. Nat. Protoc. 2007;2:247–250. doi: 10.1038/nprot.2007.25. [DOI] [PubMed] [Google Scholar]
- 3.Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti JM, Schepartz S, Kalyandrug S, Christian M, Arbuck S, Hollingshead M, Sausville EA. Br. J. Cancer. 2001;84:1424–1431. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mischek D, Steinborn R, Petznek H, Bichler C, Zatloukal K, Sturzl M, Gunzburg WH, Hohenadl C. J. Biomed. Biotechnol. 2009;2009:437284. doi: 10.1155/2009/437284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fiebig HH, Maier A, Burger AM. Eur. J. Cancer. 2004;40:802–820. doi: 10.1016/j.ejca.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Junttila MR, de Sauvage FJ. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 7.Berns AP, Pandolfi PP. EMBO Rep. 2014;15:458–459. doi: 10.1002/embr.201438794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chughtai K, Heeren RMA. Chem. Rev. 2010;110:3237–3277. doi: 10.1021/cr100012c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huber K, Feuchtinger A, Borgmann DM, Li Z, Aichler M, Hauck SM, Zitzelsberger H, Schwaiger M, Keller U, Walch A. Anal. Chem. 2014;86:10568–10575. doi: 10.1021/ac502177y. [DOI] [PubMed] [Google Scholar]
- 10.Fernández R, Lage S, Abad-García B, Barceló-Coblijn G, Terés S, López DH, Guardiola-Serrano F, Martín ML, Escribá PV, Fernández JA. J. Am. Soc. Mass Spectrom. 2014;25:1237–1246. doi: 10.1007/s13361-014-0882-3. [DOI] [PubMed] [Google Scholar]
- 11.Jiang L, Chughtai K, Purvine SO, Bhujwalla ZM, Raman V, Pasa-Tolic L, Heeren RMA, Glunde K. Anal. Chem. 2015;87:5947–5956. doi: 10.1021/ac504503x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellis S, Bruinen A, Heeren RA. Anal. Bioanal. Chem. 2014;406:1275–1289. doi: 10.1007/s00216-013-7478-9. [DOI] [PubMed] [Google Scholar]
- 13.Hanrieder J, Phan NTN, Kurczy ME, Ewing AG. ACS Chem. Neurosci. 2013;4:666–679. doi: 10.1021/cn400053c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eriksson C, Masaki N, Yao I, Hayasaka T, Setou M. Mass Spectrom. 2013;2:S0022–S0022. doi: 10.5702/massspectrometry.S0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson SN, Wang H-YJ, Woods AS, Ugarov M, Egan T, Schultz JA. J. Am. Soc. Mass Spectrom. 2005;16:133–138. doi: 10.1016/j.jasms.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Jackson SN, Wang HYJ, Woods AS. Anal. Chem. 2005;77:4523–4527. doi: 10.1021/ac050276v. [DOI] [PubMed] [Google Scholar]
- 17.Tempez A, Ugarov M, Egan T, Schultz JA, Novikov A, Della-Negra S, Lebeyec Y, Pautrat M, Caroff M, Smentkowski VS, Wang HJ, Jackson SN, Woods AS. J. Proteome Res. 2005;4:540–545. doi: 10.1021/pr0497879. [DOI] [PubMed] [Google Scholar]
- 18.Astigarraga E, Barreda-Gomez G, Lombardero L, Fresnedo O, Castaño F, Giralt MT, Ochoa B, Rodriguez-Puertas R, Fernández JA. Anal. Chem. 2008;80:9105–9114. doi: 10.1021/ac801662n. [DOI] [PubMed] [Google Scholar]
- 19.Barceló-Coblijn G, Fernández JA. Front. Physiol. 2015;6:3. doi: 10.3389/fphys.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernández JA, Ochoa B, Fresnedo O, Giralt MT, Rodriguez-Puertas R. Anal. Bioanal. Chem. 2011;401:29–51. doi: 10.1007/s00216-011-4696-x. [DOI] [PubMed] [Google Scholar]
- 21.Angel PM, Caprioli RM. Biochemistry. 2013;52:3818–3828. doi: 10.1021/bi301519p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Meer G. EMBO J. 2005;24:3159–3165. doi: 10.1038/sj.emboj.7600798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang F, Du G. World J. Biol. Chem. 2012;3:167–174. doi: 10.4331/wjbc.v3.i8.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown HA, Marnett LJ. Chem. Rev. 2011;111:5817–5820. doi: 10.1021/cr200363s. [DOI] [PubMed] [Google Scholar]
- 25.Shen H, Kuo CC, Chou J, Delvolve A, Jackson SN, Post J, Woods AS, Hoffer BJ, Wang Y, Harvey BK. FASEB J. 2009;23:1958–1968. doi: 10.1096/fj.08-123281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woods AS, Colsch B, Jackson SN, Post J, Baldwin K, Roux A, Hoffer B, Cox BM, Hoffer M, Rubovitch V, Pick CG, Schultz JA, Balaban C. ACS Chem. Neurosci. 2013;4:594–600. doi: 10.1021/cn300216h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernández R, Garate J, Lage S, Terés S, Higuera M, Bestard-Escalas J, López D, Guardiola-Serrano F, Escribá P, Barceló-Coblijn G, Fernández JA. J. Am. Soc. Mass Spectrom. 2015;26:1–11. doi: 10.1007/s13361-015-1268-x. [DOI] [PubMed] [Google Scholar]
- 28.Trim PJ, Henson CM, Avery JL, McEwen A, Snel MF, Claude E, Marshall PS, West A, Princivalle AP, Clench MR. Anal. Chem. 2008;80:8628–8634. doi: 10.1021/ac8015467. [DOI] [PubMed] [Google Scholar]
- 29.Veselkov KA, Mirnezami R, Strittmatter N, Goldin RD, Kinross J, Speller AVM, Abramov T, Jones EA, Darzi A, Holmes E, Nicholson JK, Takats Z. Proc. Natl. Acad. Sci. U. S. A. 2014;111:1216–1221. doi: 10.1073/pnas.1310524111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexandrov T, Chernyavsky I, Becker M, von Eggeling F, Nikolenko S. Anal. Chem. 2013;85:11189–11195. doi: 10.1021/ac401420z. [DOI] [PubMed] [Google Scholar]
- 31.Verbeeck N, Yang J, De Moor B, Caprioli RM, Waelkens E, Van de Plas R. Anal. Chem. 2014;86:8974–8982. doi: 10.1021/ac502838t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdelmoula WM, Škrášková K, Balluff B, Carreira RJ, Tolner EA, Lelieveldt BPF, van der Maaten L, Morreau H, van den Maagdenberg AMJM, Heeren RMA, McDonnell LA, Dijkstra J. Anal. Chem. 2014;86:9204–9211. doi: 10.1021/ac502170f. [DOI] [PubMed] [Google Scholar]
- 33.Fonville JM, Carter CL, Pizarro L, Steven RT, Palmer AD, Griffiths RL, Lalor PF, Lindon JC, Nicholson JK, Holmes E, Bunch J. Anal. Chem. 2013;85:1415–1423. doi: 10.1021/ac302330a. [DOI] [PubMed] [Google Scholar]
- 34.Dill AL, Eberlin LS, Costa AB, Zheng C, Ifa DR, Cheng L, Masterson TA, Koch MO, Vitek O, Cooks RG. Chem. - Eur. J. 2011;17:2897–2902. doi: 10.1002/chem.201001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdelmoula WM, Carreira RJ, Shyti R, Balluff B, van Zeijl RJM, Tolner EA, Lelieveldt BFP, van den Maagdenberg AMJM, McDonnell LA, Dijkstra J. Anal. Chem. 2014;86:3947–3954. doi: 10.1021/ac500148a. [DOI] [PubMed] [Google Scholar]
- 36.Hanselmann M, Kothe U, Kirchner M, Renard BY, Amstalden ER, Glunde K, Heeren RMA, Hamprecht FA. J. Proteom. Res. 2009;8:3558–3567. doi: 10.1021/pr900253y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eberlin LS, Norton I, Dill AL, Golby AJ, Ligon KL, Santagata S, Cooks RG, Agar NYR. Cancer Res. 2012;72:645–654. doi: 10.1158/0008-5472.CAN-11-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barcelo-Coblijn G, Martin ML, de Almeida RF, Noguera-Salva MA, Marcilla-Etxenike A, Guardiola-Serrano F, Luth A, Kleuser B, Halver JE, Escriba PV. Proc. Natl. Acad. Sci. U. S. A. 2011;108:19569–19574. doi: 10.1073/pnas.1115484108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin ML, Barceló-Coblijn G, de Almeida RFM, Noguera-Salvá MA, Terés S, Higuera M, Liebisch G, Schmitz G, Busquets X, Escribá PV. Biochim. Biophys. Acta, Biomembr. 2013;1828:1405–1413. doi: 10.1016/j.bbamem.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 40.Martinez J, Gutierrez A, Casas J, Llado V, Lopez-Bellan A, Besalduch J, Dopazo A, Escriba PV. J. Pharmacol. Exp. Ther. 2005;315:466–474. doi: 10.1124/jpet.105.088716. [DOI] [PubMed] [Google Scholar]
- 41.Teres S, Llado V, Higuera M, Barcelo-Coblijn G, Martin ML, Noguera-Salva MA, Marcilla-Etxenike A, Garcia-Verdugo JM, Soriano-Navarro M, Saus C, Gomez-Pinedo U, Busquets X, Escriba PV. Proc. Natl. Acad. Sci. U. S. A. 2012;109:8489–8494. doi: 10.1073/pnas.1118349109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Llado V, Teres S, Higuera M, Alvarez R, Noguera-Salva MA, Halver JE, Escriba PV, Busquets X. Proc. Natl. Acad. Sci. U. S. A. 2009;106:13754–13758. doi: 10.1073/pnas.0907300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin ML, Liebisch G, Lehneis S, Schmitz G, Alonso-Sande M, Bestard-Escalas J, Lopez DH, Garcia-Verdugo JM, Soriano-Navarro M, Busquets X, Escriba PV, Barcelo-Coblijn G. J. Lipid Res. 2013;54:1457–1465. doi: 10.1194/jlr.M036749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garate J, Fernandez R, Lage S, Bestard-Escalas J, Lopez DH, Reigada R, Khorrami S, Ginard D, Reyes J, Amengual I, Barcelo-Coblijn G, Fernandez JA. Anal. Bioanal. Chem. 2015;407:4697–4708. doi: 10.1007/s00216-015-8673-7. [DOI] [PubMed] [Google Scholar]
- 45.Hankin JA, Barkley RM, Murphy RC. J. Am. Soc. Mass Spectrom. 2007;18:1646–1652. doi: 10.1016/j.jasms.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiong XC, Xu W, Eberlin LS, Wiseman JM, Fang X, Jiang Y, Huang ZJ, Zhang YK, Cooks RG, Ouyang Z. J. Am. Soc. Mass Spectrom. 2012;23:1147–1156. doi: 10.1007/s13361-012-0361-7. [DOI] [PubMed] [Google Scholar]
- 47.Deininger SO, Cornett DS, Paape R, Becker M, Pineau C, Rauser S, Walch A, Wolski E. Anal. Bioanal. Chem. 2011;401:167–181. doi: 10.1007/s00216-011-4929-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arthur D, Vassilvitskii S. Proceedings of the Eighteenth Annual ACM-SIAM Symposium on Discrete Algorithms; New Orleans, LA, USA. Jan. 1–9, 2007; Philadelphia, PA, USA: Society of Industrial and Applied Mathematics (SIAM); 2007. pp. 1027–1035. [Google Scholar]
- 49.Söderström K-O, Parvinen L, Parvinen M. Experientia. 1977;33:265–266. doi: 10.1007/BF02124103. [DOI] [PubMed] [Google Scholar]
- 50.Schmelzer K, Fahy E, Subramaniam S, Dennis EA. Methods Enzymol. 2007;432:171–183. doi: 10.1016/S0076-6879(07)32007-7. [DOI] [PubMed] [Google Scholar]
- 51.Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CRH, Shimizu T, Spener F, van Meer G, Wakelam MJO, Dennis EA. J. Lipid Res. 2009;50:S9–S14. doi: 10.1194/jlr.R800095-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griffiths JR. Br. J. Cancer. 1991;64:425–427. doi: 10.1038/bjc.1991.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kato Y, Ozawa S, Miyamoto C, Maehata Y, Suzuki A, Maeda T, Baba Y. Cancer Cell Int. 2013;13:89. doi: 10.1186/1475-2867-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kato Y, Ozawa S, Tsukuda M, Kubota E, Miyazaki K, St-Pierre Y, Hata R. FEBS J. 2007;274:3171–3183. doi: 10.1111/j.1742-4658.2007.05848.x. [DOI] [PubMed] [Google Scholar]
- 55.Sugiura Y, Setou M. Rapid Commun. Mass Spectrom. 2009;23:3269–3278. doi: 10.1002/rcm.4242. [DOI] [PubMed] [Google Scholar]
- 56.He Q, Takizawa Y, Hayasaka T, Masaki N, Kusama Y, Su J, Mineta H, Setou M. Anal. Bioanal. Chem. 2014;406:5815–5825. doi: 10.1007/s00216-014-7890-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Merrill AH, Wang MD, Park M, Sullards MC. Trends Biochem. Sci. 2007;32:457–468. doi: 10.1016/j.tibs.2007.09.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.