
Figure 1. MiSeq®-based amplicon sequencing has an intrinsic mononucleotide run indel error rate.
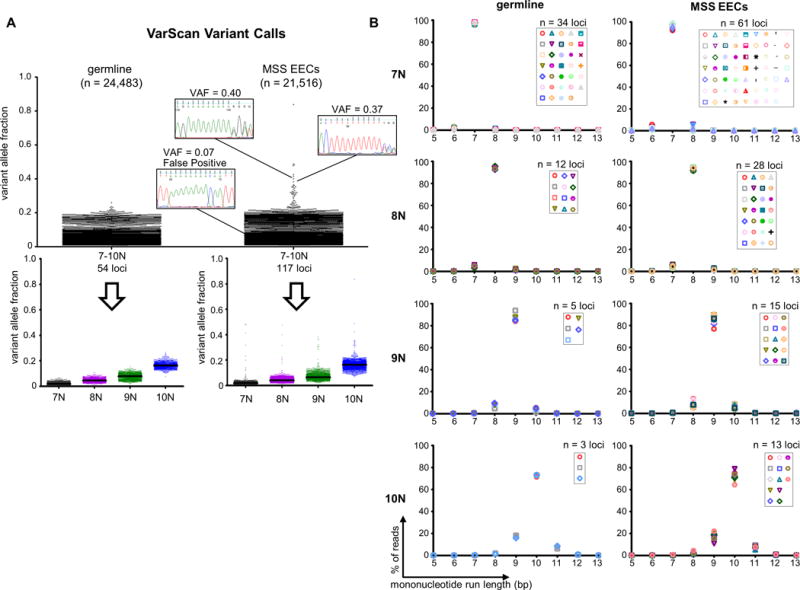
(A) Dot plots show the VarScan variant allele fractions (VAFs) for mononucleotide run indel calls in germline DNA samples and microsatellite stable endometrioid endometrial cancers (MSS EECs). Total number of calls (n) is given in parenthesis. Sanger sequencing using high fidelity Phusion polymerase for representative true and false positive calls is shown. Because the mononucleotide repeats analyzed are monomorphic, the vast majority of variants are presumed to be false positives. Bottom panels show the calls stratified by reference sequence mononucleotide run length, and illustrate increasing VAF (sequencing noise) with increasing run length. Black lines indicate medians. (B) Distributions for the repeat length for the combined reads from all germline or MSS EECs samples. Each marker represents a different repeat. Graphs illustrate that allele distributions are similar for mononucleotide runs of the same length. See also Supp. Table S1.