

Abstract
Background:
Monoclonal gammopathy of undeterminated significance is the most common form of plasma cell dyscrasia, usually considered as benign. In rare cases it may have a malignant course, sometimes limited to an organ such as peripheral nerves.
Methods:
We describe clinical, electrophysiological and pathological findings in a patient presenting a immunoglobulin G (IgG) paraproteinemic polyneuropathy clinically mimicking a chronic inflammatory demyelinating polyneuropathy.
Results:
Immuno-electron microscopy (immune-EM) demonstrated that the widenings of the myelin lamellae resulted from the infiltration of IgG between a significant number of myelin lamellae (with absence of inflammatory cells in the epineurium, endoneurium, and perineurium, and the lack signs of vasculitis). This patient was finally treated successfully with lenalidomide then mycophenolate mofetil.
Conclusions:
In polyneuropathies associated to a monoclonal gammopathy, a nerve biopsy may clinch the diagnosis. Immuno-EM may be required to determine the role of the pathological immunoglobulin in the destruction of the peripheral nerve parenchyma. Diagnosis of such a direct involvement of peripheral nerve can endorse more aggressive treatment of real efficiency.
Keywords: deposition, IgG, MGUS, nerve biopsy, neuropathy
1. Introduction
Monoclonal gammopathies (MG) are caused by a proliferation of monoclonal plasma cells or B lymphocytes: it is characterized by the proliferation and deposition of M proteins (or paraproteins) which are formed by a single heavy chain (M, G, or A) and a light chain (kappa or lambda).[1] Monoclonal gammopathy of undeterminated significance (MGUS) is the most common form of plasma cell dyscrasia (immunoglobulin G [IgG] MGUS accounting for up to 61% of the cases).[2] Its prevalence is 3.5% in the general adult population >50 years; its incidence increases with age (being >5% in patients aged >70 years).[3] MGUS is defined by the presence of a serum monoclonal component concentration ≤3 g/dL (0.6 g/dL > N > 2.5 g/dL), bone marrow plasma cell counts <10%, and the absence of signs/symptoms related to multiple myeloma (MM) or other lymphoproliferative disorders (whereas MGUS has a rate of malignant progression of approximately 1% per year); for IgG and IgA MGUS, Bence-Jones proteinuria has to be ≤1 g/24 h (normal value of proteinuria <0.15 g/24 h).[4–6]
We know that 5% to 10% of patients with otherwise unexplained polyneuropathy have an MG (mostly an IgM MG). Approximately 40% to 70% of these patients have IgM MG and antibodies against myelin-associated glycoprotein (MAG).[7] Neuropathy related to IgA or IgG MG are less common.[8] We report a case of paraproteinemic polyneuropathy characterized by unusual myelin lesions directly linked to IgG MGUS. On electron microscopy, the features were identical to those commonly described in IgM neuropathies with anti-MAG activity.
2. Case report
A 51-year-old patient (with a medical history of acute coronary syndrome and chronic tobacco smoking) complained of paresthesia of both hands for 18 months. Because entrapment of the ulnar nerve at elbow was initially suspected, a surgical treatment was proposed but gave no improvement. One year later, he experienced some falls as well as difficulties in writing. Six months later, on clinical examination we observed a mild distal motor deficit of the lower limbs (flexion and extension of feet and toes were weak) without amyotrophia. Deep tendon reflexes were absent at ankles. No pyramidal sign (as well as no sphincter disturbance) was found. There was a distal hypoesthesia of the lower limbs (limited to the feet) without gait disturbance (Overall Neuropathy Limitations Scale [ONLS] was 3/12).
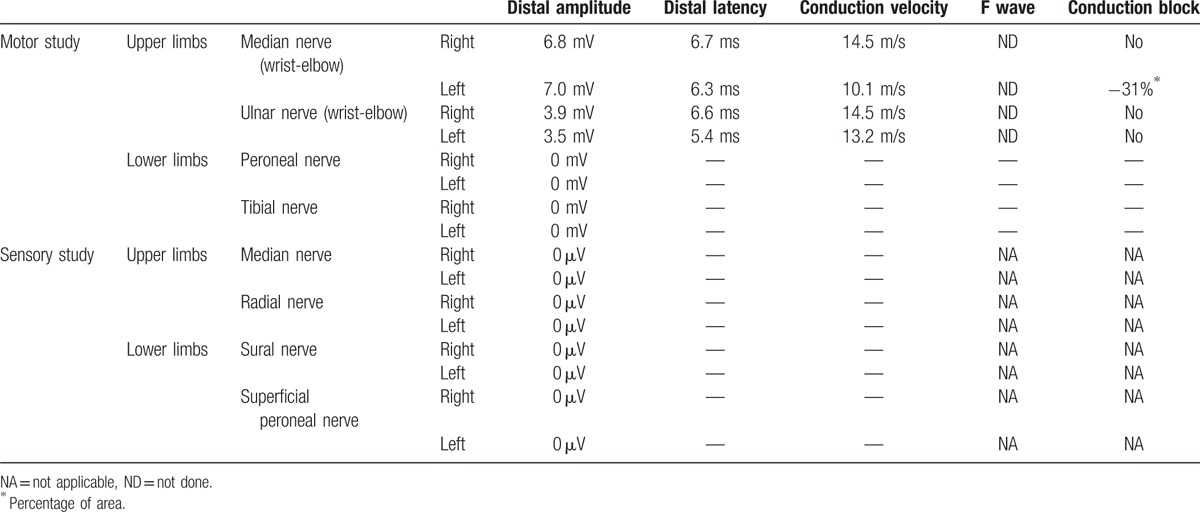
The electrophysiological study showed a severe primary demyelinating sensorimotor polyneuropathy, with no sensory nerve action potential in the 4 limbs and no compound muscle action potential in the lower limbs. In the upper limbs, we found severe slowing of the motor nerve conduction velocities with distal latencies and a conduction block on the left median nerve (wrist-elbow) (Table 1). Laboratory tests showed serum IgG-kappa monoclonal gammopathy with no plasma cell expansion on bone marrow aspiration. The kappa/lambda ratio was >5 (0.26 < N < 1.65). No cryoglobulinemia and no anti-MAG or anti-glycolipid antibodies were detected. Laboratory examination of endocrine function was normal. Cerebrospinal fluid protein was slightly elevated to 70 mg/dL (N < 45 mg/dL) with no leucocytes.
Table 1.
Electrophysiological study of motor and sensory nerves.
At that time, we diagnosed a mild form of chronic inflammatory demyelinating polyneuropathy (CIDP) associated with MGUS, and decided to treat him with intravenous immunoglobulins (IVIg; 0.4 g/kg/day for 5 days, every month). During the next months, despite several courses of IgIV we observed a worsening of the clinical picture. The patient finally presented an acute worsening leading to severe tetraparesis (with diaphragmatic palsy) needing intensive care (ONLS was 10/12). After having added oral corticosteroids (1 mg/kg/d) and one course of plasma exchanges, his motor strength progressively improved. We then continued corticosteroids and monthly IVIg. We introduced azathioprine (1 year), and then cyclophosphamide (6 months) as a steroid-sparing agent, without success. Four years after the first acute worsening, he again presented with rapidly progressive severe paraparesis (in a context of skin infection). As for the first worsening, the patient improved after we increased the steroid dosage (to 1 mg/kg/d). ONLS was 2/12, but at that time he also became dependant on IgIV: he required 150 g of IVIg every 11 days (Fig. 1).
Figure 1.
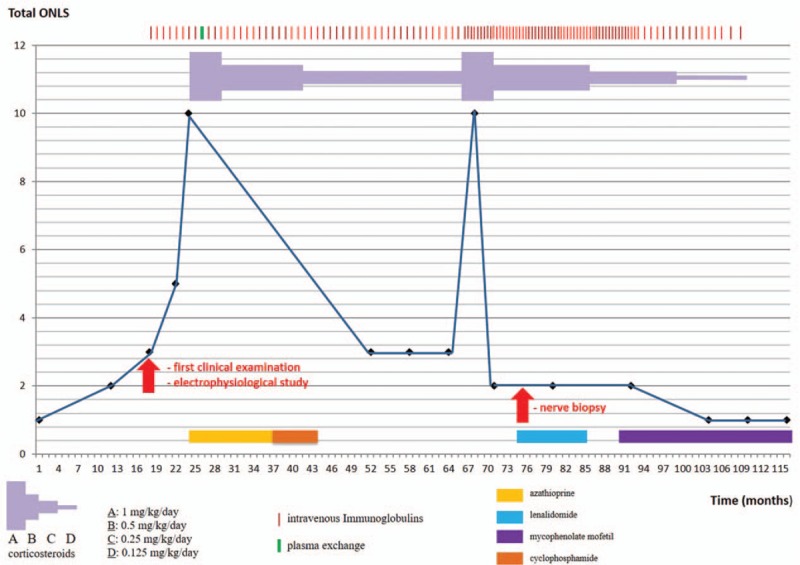
Course of the polyneuropathy of the patient: evaluation with total ONLS. ONLS = Overall Neuropathy Limitations Scale.
Because of the atypical clinical course of the CIDP, a sural nerve biopsy was performed. Paraffin sections were examined using standard methods. Another fragment was fixed in buffered glutaraldehyde, embedded in epon, and prepared for light and electron microscopic (EM) examination. A few other fragments were embedded in London Resin White for an immuno-EM study. Tissues were immunostained using rabbit antisera monospecific to human IgA, IgG, IgM, kappa, and lambda light chains. These antibodies were revealed by adding IgG-coated colloidal particles (12 nm in diameter at 1:25 dilution).[9] No abnormal cells were seen in the endoneurium, epineurium, or perineurium. There was no vasculitis. A significant reduction in the number of myelinated fibers (approximately 50%), mainly large ones, was observed. Approximately all the remaining myelinated fibers were abnormal: most had myelin sheaths that were too thin relative to axonal diameters, indicative of demyelination (Fig. 2). Most of these myelin sheaths examined by EM exhibited severe abnormalities of myelin compaction: there were numerous abnormal regular widenings of the myelin lamellae (WML) which were localized across all sheaths. The WML were regularly spaced (33.9 ± 1.4 nm vs 15.2 ± 1.9 nm for spacing in normal myelin) (Fig. 3). The dense intraperiodic lines appeared to be dissociated. At high magnification these WML appeared to be filled with a grayish granular material which was anti-IgG, anti-kappa light chain positive (Fig. 4). Because of the abnormal compaction of myelin lamellae, mutations of the myelin protein zero (MPZ) gene were systematically excluded by Sanger sequencing analysis.
Figure 2.
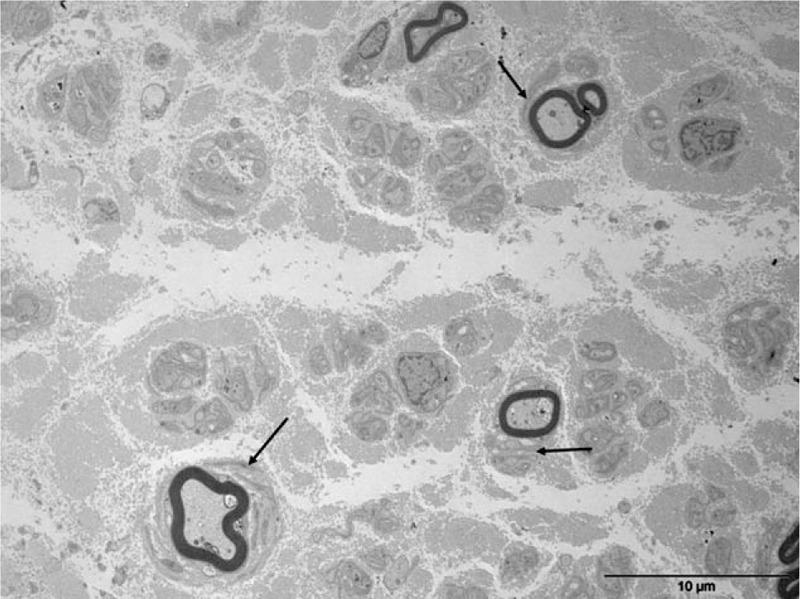
Electron micrograph, transverse section. Severe loss of myelinated fibers. Several thin myelin sheaths are surrounded by some proliferations of Schwann cell like “onion bulbs” (arrows) which are in favor of a demyelinating-remyelinating process.
Figure 3.
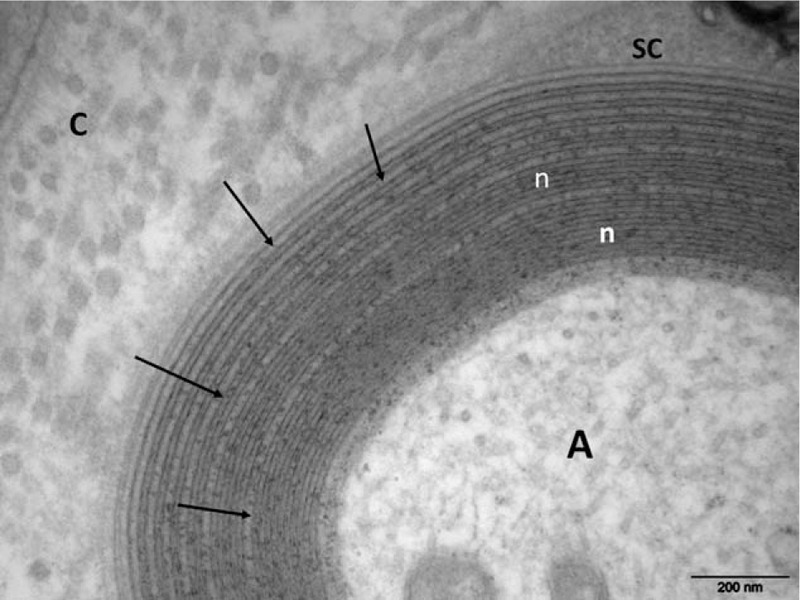
Electron micrograph, transverse section. At high magnification numerous regular widenings of the myelin lamellae are well seen (arrows). Otherwise, the myelin compaction is normal (n). A = axon, C = collagen, SC = Schwann cell cytoplasm.
Figure 4.
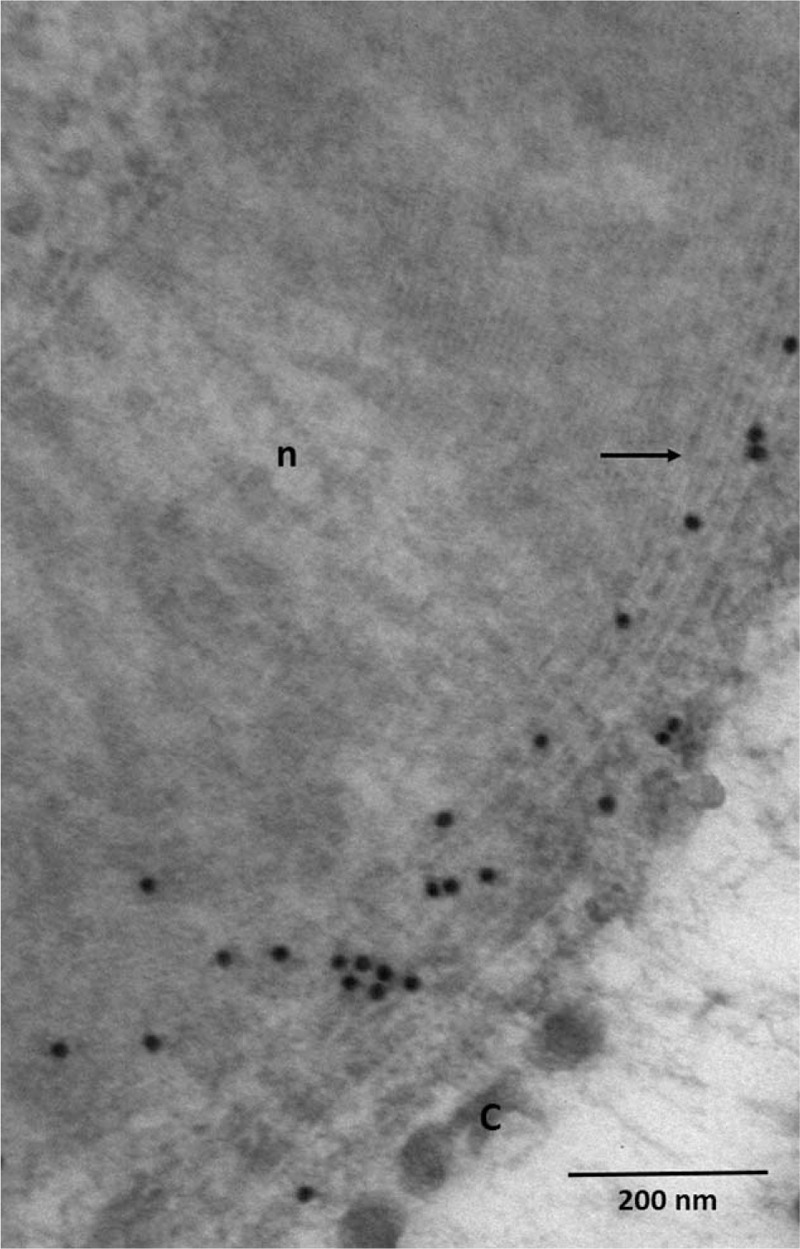
Immunoelectron micrograph, transverse section. Presence of IgG (black granules) at the level of widenings of the myelin lamellae (arrows). C = collagen, n = normally compacted myelin.
Because of the presence of IgG-kappa, we decided to introduce lenalidomide for 1 year (without consequence on IgIV frequency and steroid dependence). Despite stabilization (ONLS was 2/12), we finally switched this treatment to mycophenolate mofetil (MMF). During the next months, we observed a sustained clinical improvement (ONLS was 1/12). We were able to decrease the frequency of the courses of IgIV (1 every 4 weeks, and then 1 every 6 weeks) and the steroid dosage, and then to stop both steroids and IgIV. The only current treatment is MMF (Fig. 1).
A relapsing form of inflammatory polyradiculoneuropathy induced by intramyelinic deposition of IgG-kappa in a context of IgG MGUS was diagnosed.
3. Discussion
The prevalence of symptomatic neuropathy in patients with MGUS ranges from 1% to 36%, and is higher in MGUS associated with IgM than with IgG or IgA paraproteins.[10,11] As in our patient, paraproteinemic neuropathy affects men who classically present with a chronic sensorimotor symmetrical neuropathy similar to CIDP.[11,12] Some authors have described more sensory involvement in paraproteinemic neuropathy than in idiopathic CIDP.
In any case, it may be difficult to confirm the direct implication of MGUS if the patient presents the typical electrophysiological and clinical hallmark of a CIDP (as in our case). The poor response to the conventional treatment of CIDP (such as IVIg, corticosteroids, or therapeutic plasma exchange) may be a “red flag”; in our patient, the therapeutic response to IVIg and corticosteroids was initially positive (but with IVIg and corticosteroids dependence) with a remittent-recurrent course of the disease. On revisiting the evidence in view of the lack of response to treatment, we decided to perform a nerve biopsy which showed unusual myelin lesions directly linked to IgG MGUS.
Immuno-EM demonstrated that the WML resulted from the infiltration of IgG between a significant numbers of myelin lamellae (with the absence of inflammatory cells in the epineurium, endoneurium, and perineurium, and the lack signs of vasculitis). In our experience, endoneurial deposits of immunoglobulins may be observed in any kind of paraproteinemic neuropathy (IgM, IgG, or IgA).[13,14] As these WML are very small, it is impossible to identify them by light microscopy on sections taken from nerve fragments either embedded in paraffin or in epon. EM examination is often indispensable in the context of a polyneuropathy associated with a monoclonal gammopathy. First, it can reveal various types of lesions directly linked to the monoclonal gammopathy. Second, immuno-EM may characterize specific endoneurial deposits. Immunoglobulin deposits in the interstitial tissue out of Schwann cells have been also detected.[15] The pathological lesions of our patient can account for the electrophysiological findings of progressive slowing of conduction velocities due to the abnormalities of myelin sheaths.
Although there is a lack of consensus on the treatment of IgG and IgA paraproteinemic neuropathies (only 1 randomized clinical trial showing a modest benefit of therapeutic plasma exchange),[16] some patients may respond to immunosuppressant or immunomodulatory treatments such as plasma exchanges and cyclophosphamide (combined with prednisolone, IVIg, and corticosteroids).[17] MGUS is considered as a premalignant disorder: IgG and IgA MGUS may be precursor conditions of MM, whereas light-chain MGUS can be a precursor condition of light-chain MM. Assessment of the risk of progression of patients with asymptomatic MG is based on various factors (including clonal burden, cytogenetic abnormalities, and light-chain production), but plasma cell clones may occasionally be responsible for severe organ damage (such as in the peripheral nerve) through the production of M-protein which deposits in tissues or has autoantibody activity (although the hemopathy may not appear to be biologically malignant).[18] Such patients have to be considered to have MM requiring therapy similar to patients with symptomatic disease.[19]
Lenalidomide (and its analog thalidomide) is an immunomodulatory drug widely used in the treatment of MG in cases of refractory MM.[20] For this reason, we decided to use lenalidomide; however, after many months, we only observed a clinical stabilization, and so tried MMF which induced a dramatic and long-lasting improvement. MMF acts by inhibiting inosine monophosphate dehydrogenase, thereby limiting the proliferation of B and T lymphocytes.[21] It is approved for clinical use in the prevention of acute allograft rejection following organ transplantation and hematopoietic stem cell transplantation, but it has shown efficacy in autoimmune diseases as well. MMF was initially developed as a replacement of azathioprine and (as azathioprine) it may also be an effective therapy for patients with naive or refractory CIDP,[22] in conjunction with its steroid-sparing action.[21] Although MMF has antimyeloma activity in vitro,[23] we are not aware of other cases of improvement of paraproteinemic neuropathy in the medical literature after such a treatment. In our patient, this improvement was thought to be a consequence of the cumulative effect of lenalidomide and MMF.
In polyneuropathies associated with MG (of whatever type), a nerve biopsy may clinch the diagnosis. Immuno-EM may be required to determine the role of the pathological immunoglobulin in the destruction of the peripheral nerve parenchyma. Diagnosis of such a direct involvement of peripheral nerve can endorse more aggressive treatment of real efficiency.
Footnotes
Abbreviations: CIDP = chronic inflammatory demyelinating polyneuropathy, EM = electron microscopy, Ig = immunoglobulin, IVIg = intravenous immunoglobulins, MAG = myelin-associated glycoprotein, MG = monoclonal gammopathy, MGUS = monoclonal gammopathy of undeterminated significance, MM = multiple myeloma, MMF = mycophenolate mofetil, ONLS = Overall Neuropathy Limitations Scale, WML = widenings of the myelin lamellae.
Authorship: SM and J-MV drafted the manuscript; JF managed the clinical follow-up of the patient; J-MV and LR performed the pathological study; and JF critically reviewed the manuscript for intellectual content.
Ethics committee approval: Informed consent was obtained from the patient, but this case report did not require an ethics committee approval according to the current laws in our Hospital.
Disclosure: The authors have no disclosures to report; the authors have no conflicts of interest to disclose.
References
- 1.Ramchandren S, Lewis RA. An update on monoclonal gammopathy and neuropathy. Curr Neurol Neurosci Rep 2012; 12:102–110. [DOI] [PubMed] [Google Scholar]
- 2.Kyle RA. Diagnostic criteria of multiple myeloma. Hematol Oncol Clin North Am 1992; 6:347–358. [PubMed] [Google Scholar]
- 3.Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med 2006; 354:1362–1369. [DOI] [PubMed] [Google Scholar]
- 4.International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121:749–757. [PubMed] [Google Scholar]
- 5.Rossi F, Petrucci MT, Guffanti A, et al. Proposal and validation of prognostic scoring systems for IgG and IgA monoclonal gammopathies of undetermined significance. Clin Cancer Res 2009; 15:4439–4445. [DOI] [PubMed] [Google Scholar]
- 6.Joint Task Force of the EFNS the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst 2010; 15:185–195. [DOI] [PubMed] [Google Scholar]
- 7.Kuijf ML, Eurelings M, Tio-Gillen AP, et al. Detection of anti-MAG antibodies in polyneuropathy associated with IgM monoclonal gammopathy. Neurology 2009; 73:688–695. [DOI] [PubMed] [Google Scholar]
- 8.Vallat JM, Jauberteau MO, Bordessoule D, et al. Link between peripheral neuropathy and monoclonal dysglobulinemia: a study of 66 cases. J Neurol Sci 1996; 137:124–130. [DOI] [PubMed] [Google Scholar]
- 9.Keita M, Magy L, Richard L, et al. LR white post-embedding colloidal gold method to immunostain MBP, P0, NF and S100 in glutaraldehyde fixed peripheral nerve tissue. J Peripher Nerv Syst 2002; 7:128–133. [DOI] [PubMed] [Google Scholar]
- 10.Nobile-Orazio E, Casellato C, Di Troia A. Neuropathies associated with IgG and IgA monoclonal gammopathy. Rev Neurol (Paris) 2002; 158:979–987. [PubMed] [Google Scholar]
- 11.Simmons Z, Albers JW, Bromberg MB, et al. Long-term follow-up of patients with chronic inflammatory demyelinating polyradiculoneuropathy, without and with monoclonal gammopathy. Brain 1995; 118:359–368. [DOI] [PubMed] [Google Scholar]
- 12.Magy L, Chassande B, Maisonobe T, et al. Polyneuropathy associated with IgG/IgA monoclonal gammopathy: a clinical and electrophysiological study of 15 cases. Eur J Neurol 2003; 10:677–685. [DOI] [PubMed] [Google Scholar]
- 13.Vallat JM, Tabaraud F, Sindou P, et al. Myelin widenings and MGUS-IgA: an immunoelectron microscopic study. Ann Neurol 2000; 47:808–811. [PubMed] [Google Scholar]
- 14.Vallat JM, Magy L, Richard L, et al. Contribution of electron microscopy to the study of neuropathies associated with an IgG monoclonal paraproteinemia. Micron 2008; 39:61–70. [DOI] [PubMed] [Google Scholar]
- 15.Vallat JM, Magy L, Sindou P, et al. IgG neuropathy: an immunoelectron microscopic study. J Neuropathol Exp Neurol 2005; 64:386–390. [DOI] [PubMed] [Google Scholar]
- 16.Dyck PJ, Low PA, Windebank AJ, et al. Plasma exchange in polyneuropathy associated with monoclonal gammopathy of undetermined significance. N Engl J Med 1991; 325:1482–1486. [DOI] [PubMed] [Google Scholar]
- 17.Stork AC, Lunn MP, Nobile-Orazio E, et al. Treatment for IgG and IgA paraproteinaemic neuropathy. Cochrane Database Syst Rev 2015; 3:CD005376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duchesne M, Mathis S, Corcia P, et al. Value of nerve biopsy in patients with latent malignant hemopathy and peripheral neuropathy: a case series. Medicine (Baltimore) 2015; 94:e394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van De Donk N, De Weerdt O, Eurelings M, et al. Malignant transformation of monoclonal gammopathy of undetermined significance: cumulative incidence and prognostic factors. Leuk Lymphoma 2001; 42:609–618. [DOI] [PubMed] [Google Scholar]
- 20.Bird JM, Owen RG, D'Sa S, et al. Guidelines for the diagnosis and management of multiple myeloma 2011. Br J Haematol 2011; 154:32–75. [DOI] [PubMed] [Google Scholar]
- 21.Mathis S, Vallat JM, Magy L. Novel immunotherapeutic strategies in chronic inflammatory demyelinating polyneuropathy. Immunotherapy 2016; 8:165–178. [DOI] [PubMed] [Google Scholar]
- 22.Bedi G, Brown A, Tong T, et al. Chronic inflammatory demyelinating polyneuropathy responsive to mycophenolate mofetil therapy. J Neurol Neurosurg Psychiatry 2010; 81:634–636. [DOI] [PubMed] [Google Scholar]
- 23.Takebe N, Cheng X, Fandy TE, et al. IMP dehydrogenase inhibitor mycophenolate mofetil induces caspase-dependent apoptosis and cell cycle inhibition in multiple myeloma cells. Mol Cancer Ther 2006; 5:457–466. [DOI] [PubMed] [Google Scholar]