
Figure 1.
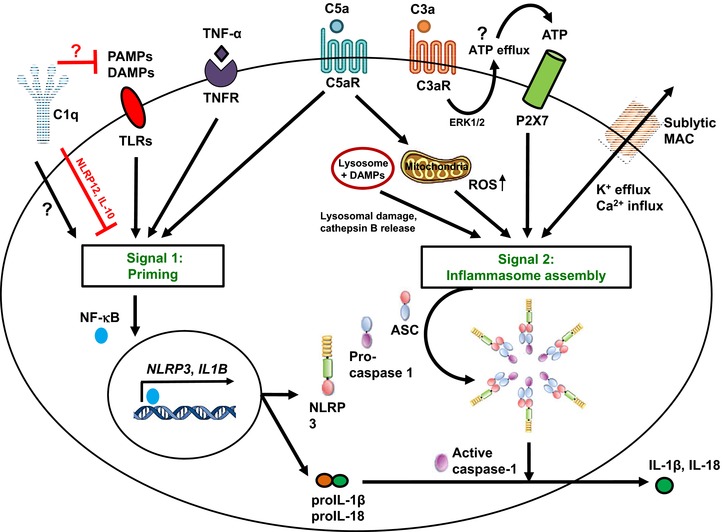
Mechanisms of complement‐mediated NLRP3‐inflammasome regulation. NLRP3 inflammasome activation requires two distinct signals. The “priming” Signal 1 is triggered by PAMP/DAMP recognition by PPRs (e.g. TLRs) and certain cytokines (TNF‐α) and drives NF‐κB nuclear translocation and NLRP3 and IL1B gene transcription. Signal 2 induces the assembly of NLRP3, ASC, and caspase‐1 supracomplexes to form an active NLRP3 inflammasome, where active caspase‐1 processes proIL‐1β/proIL‐18 into mature IL‐1β/IL‐18. The complement components C1q and C5aR1 (together with tumor necrosis factor receptor and/or TLR signaling) potentiate Signal 1. C5aR1 act as a priming signal to sustain inflammasome activation during the uptake of DAMPs, with a mechanism involving increased lysosomal damage and cathepsin B release. C5aR1 activation directly delivers Signal 2 for NLRP3 inflammasome activation, via induction of mitochondrial damage and intracellular accumulation of ROS. The C3aR regulates ATP efflux (via a not yet identified channel, denoted by a question mark) and subsequent autocrine P2X7 engagement, and sublytic MAC formation increases intracellular Ca2+ levels and mitochondrial membrane potential. Of note, C1q can increase canonical NLRP3 inflammasome activation in epithelial cells through a not yet defined mechanism (denoted by a question mark) but can also function as a negative regulator of NLRP3 inflammasome activation by sequestering DAMPs (such as cholesterol crystals) and inhibiting PPR signaling.