

Abstract
Efforts to develop strategies for small molecule chemical probe discovery against the readers of the methyl-lysine (Kme) post-translational modification have been met with limited success. Targeted disruption of these protein-protein interactions via peptidomimetic inhibitor optimization is a promising alternative to small molecule hit discovery; however, recognition of identical peptide motifs by multiple Kme reader proteins presents a unique challenge in the development of selective Kme reader chemical probes. These selectivity challenges are exemplified by the Polycomb repressive complex 1 (PRC1) chemical probe, UNC3866, which demonstrates sub-micromolar off-target affinity toward the non-PRC1 chromodomains CDYL2 and CDYL. Moreover, since peptidomimetics are challenging subjects for structure-activity relationship (SAR) studies, traditional optimization of UNC3866 would prove costly and time-consuming. Herein, we report a broadly applicable strategy for the affinity-based, target-class screening of chromodomains via the repurposing of UNC3866 in an efficient, combinatorial peptide library. A first-generation library yielded UNC4991, a UNC3866 analog that exhibits a distinct selectivity profile while maintaining sub-micromolar affinity toward the CDYL chromodomains. Additionally, in vitro pull-down experiments from HeLa nuclear lysates further demonstrate the selectivity and utility of this compound for future elucidation of CDYL protein function.
Chemical probes are an integral piece in the puzzle of preclinical target validation.1,2,3 While molecular biology and genetic approaches elucidate important roles for biological targets, probes are uniquely capable of distinguishing between scaffolding effects and a functional activity of the target (i.e. catalytic or protein-protein interaction), and thus the potential for therapeutic intervention. Successfully assigning biological effects to target inhibition requires that chemical probes be extensively characterized for their on-target activity and selectivity. Developing these potent and selective chemical tools requires both identification of a synthetically tractable starting point and time-intensive hit-to-probe optimization.
In the case of many protein-protein interactions (PPIs), the simplest starting point for peptidic inhibitor development often involves determining the minimum peptide length required to retain binding to the target protein, but the optimization of peptidomimetic ligands is by no means straightforward.4 Peptidomimetic ligands are faced with the exceptional challenge of mimicking the unique geometries achieved by peptides, bridging the large protein surface grooves characteristic of many PPIs, while gaining improved cellular permeability and proteolytic stability relative to a fully peptidic compound.5,6 Additionally, peptide precursors are often low affinity ligands and tend to interact with multiple proteins. Lastly, peptide optimization is further hindered by the large size of these compounds which provides a multitude of regions to optimize, increasing the probability of missing synergistic modifications if all combinations are not evaluated.
Despite the potential challenges associated with peptidomimetic probe development, systematic study and optimization of peptides can eventually lead to the discovery of powerful chemical tools.7,8,9 Our lab recently reported the development of UNC3866 (Figure 1a), a cellularly active peptidomimetic chemical probe of the Polycomb repressive complex 1 (PRC1) chromodomains (CBX2, -4, -6, -7, and -8).10 As a subfamily of Kme reader proteins, chromodomains typify the surface-groove binding characteristic of many Kme readers.11,12,13 While the diversity of methylated marks interpreted by chromodomains is vast, many of the chromodomains bind the methylated histone consensus sequence ARKme3S.14,15,16,17 This common recognition motif interacts with the well-conserved three-stranded anti-parallel beta sheet and C-terminal alpha helix of the chromodomains to form a beta sandwich. For some chromodomains, induced fit binding of the histone peptide results in formation of the aromatic cage that is critical for Kme recognition. Unlike non-peptidomimetic small molecule ligands, UNC3866 is able to mimic the native substrate and provoke an induced-fit binding mode upon engaging the PRC1 chromodomains, resulting in a high affinity interaction (Kd ∼ 100 nM).18,19,20,21 Despite the success of UNC3866, the strategy applied toward the optimization of this compound was time-intensive and costly. Such an approach is not broadly applicable to efficient chemical probe discovery.
Figure 1.
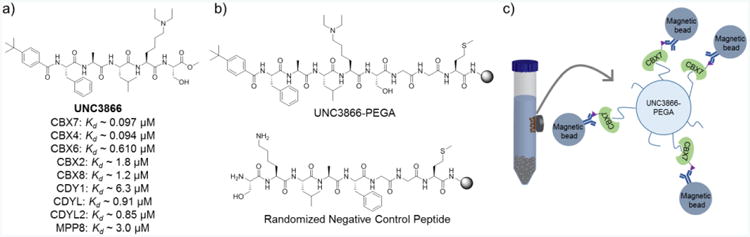
Chromodomain validation for combinatorial chemistry optimization. (A) Structure and selectivity profile of chemical probe UNC3866. The selectivity profile of UNC3866 enables combinatorial repurposing of its peptidic scaffold for inhibitors of non-PRC1 chromodomains. (B) Chemical structures of on-bead controls for magnetic enrichment assays. (C) Magnetic enrichment schematic wherein on-bead positive hits are coated by the His-tagged target chromodomains (ex. CBX7, in green). Subsequent incubation with magnetic beads coated with anti-His antibody selectively coats hit beads with magnetic beads and allows for their magnetic isolation.
UNC3866 demonstrates off-target chromodomain activity that has been difficult to overcome, targeting the CDYL chromodomains and the chromodomain of MPP8 as determined more recently, albeit at a much reduced potency relative to CBX7 and CBX4 (∼8-fold and ∼30-fold selective, respectively). Since UNC3866 demonstrated the tractability of small peptidomimetics as cellularly active tool compounds for perturbing the reading function of chromodomain-containing proteins, we decided to capitalize on the off-target activities of UNC3866 and develop novel inhibitors of the CDYL proteins, as no other CDYL ligands have previously been reported. Rationally designing chromodomain selectivity was a daunting task due to the high structural similarity between the CDYL and CBX protein families. Efforts from the Hof group recently reported one route to selectively target an individual chromodomain within the CBX family, CBX6,21 by modifying a single amino acid in the parent ligand, but their approach did so at the cost of potency and does not appear to be a generalizable approach to the discovery of selective chromodomains inhibitors. In moving forward with chromodomain peptidomimetic inhibitors, we sought to develop a platform for the cost-effective and rapid discovery of these tools that would also allow for riskier substitutions that might dramatically alter the compound selectivity profile. Thus, the implementation of a combinatorial strategy for peptide optimization was explored.
Peptides and peptidomimetics are uniquely suited to split-and-pool synthesis due to their simple and efficient chemistries and the ease with which individual compounds can be sequenced via tandem mass spectrometry.22,23 Optimization of peptidomimetic ligands on-bead offers the opportunity to ask thousands of SAR questions simultaneously with minimal effort and cost. In addition, on-bead libraries are conducive to both batch-wise screening and iterative screening attempts to improve potency and selectivity. Exemplary work employing combinatorial ligand optimization has been conducted in the Kodadek lab through the successful screening of iterative libraries while reducing non-specific binding through stringent negative selections;24,25,26,27 yet, a truly target-class screening strategy designed to address issues of potency and selectivity simultaneously within a protein family has not been explored using one-bead one-compound (OBOC) libraries.
Target-class screening employs a cross-screening strategy wherein representative members of a protein family are screened against focused compound sets to develop compound SAR profiles and inform next-generation inhibitor design.28,29 When applied to OBOC libraries, cross-screening can more simply be applied as negative selection screens with proteins other than the primary target where beads with off-target activity are removed from the hit pool, leading to the enrichment of only the most selective ligands. While previous efforts have explored target-class profiling of enzymatic substrate-specificity using combinatorial libraries, this strategy does not seek to isolate ligands of predefined selectivity profiles.30,31 Effective application of homologous negative selection screens would be uniquely capable of identifying ligands of novel selectivity profiles across a protein class.
We designed a focused UNC3866-derivative library that was screened against CDYL2 followed by cross-screening of representative individual chromodomains (CBX7, CBX8, CBX5, and MPP8). We hypothesized that our screening strategy would yield new chromodomain ligands with novel selectivity profiles, dramatically reducing CBX7 potency while retaining or improving CDYL2 potency relative to UNC3866. Importantly, we used this library to validate the amenability of chromodomains to on-bead screening and develop a broadly applicable selectivity screening platform for OBOC libraries.
Results and Discussion
Our initial efforts began by validating on-bead assays for chromodomain screening. Since UNC3866 has known activity against most of the chromodomains we were interested in screening, we first synthesized a derivatized UNC3866 on PEGA resin (UNC3866-PEGA, Figure 1b). Simultaneously, a randomized peptide was synthesized as a negative control for assay validation. Current on-bead binding assays in our lab include two types of screens, one of which uses fluorescence to identify hits,32 and the other, magnetic enrichment.33 Magnetic enrichment enables rapid hit isolation via the introduction of a magnet to pull up hit beads, whereas the fluorescent assay requires individual bead picking under a fluorescent microscope to isolate hit beads. For swiftness and ease, we moved forward with the magnetic enrichment assay (Figure 1c). In brief, we incubated UNC3866-PEGA in the presence of His-tagged CBX7. Next, incubation with an anti-His antibody selectively coats those compound beads already bound by His-CBX7. This was followed by treatment with magnetic Protein G Dynabeads capable of binding the IgG fragment of the anti-His antibody, which also coat the positive hits. When subjected to these conditions, UNC3866-PEGA beads became magnetized while the randomized negative control peptide remained unmagnetized, and in the absence of CBX7, no magnetization was observed for either peptide, as expected. Selective magnetization of UNC3866-PEGA by CBX7 indicates that when the chromodomain binding site is occupied, the His-tag remains accessible to antibody binding and that the components of the assay are not non-specifically interfering with ligand enrichment. Analogous control magnetic enrichment assays were also run for the His-tagged chromodomains of MPP8, CDYL2, and CBX8 using UNC3866-PEGA. Despite the starkly different potencies of all of these proteins for UNC3866 in solution (Figure 1a), UNC3866-PEGA became magnetized in each assay while the random control did not.
With validated on-bead assays in-hand, we began designing a library to rapidly explore the chromodomain SAR around UNC3866 (Figure 2a). A constant five residue linker was first included between the PEGA resin and potential chromodomain ligands. We selected methionine to allow for cyanogen bromide (CNBr) C-terminal cleavage of hit compounds following screening.33 N-terminal to the methionine, a lysine was included to engender a more dispersed display of hydrophobic compounds and improve ionization for follow-up tandem mass spectrometry.34 Next, an invariant amino acid linker (βAla-Ala-Ala) was incorporated to provide a spacer between the methionine and the residues being varied in the library.
Figure 2.
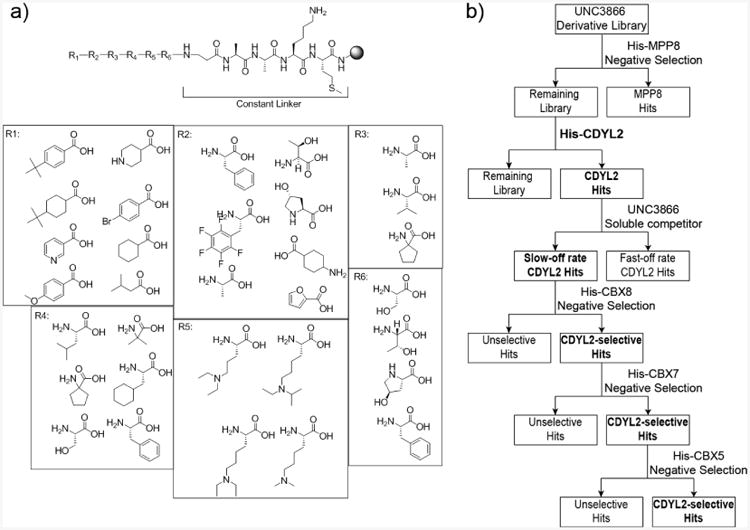
One-bead-one-compound library design and screening. (A) UNC3866-derivative library was designed to test structural hypotheses at all six positions of the parent compound. A constant linker containing methionine was included to allow for hit cleavage via cyanogen bromide treatment. (B) Library screening cascade used to selectively target CDYL2 which includes negative selections of homologous chromodomains (MPP8, CBX8, CBX7, and CBX5) on CDYL2 hit beads. Soluble UNC3866 was used to select for only higher potency ligands.
Designing a targeted library required a balance between including conservative substitutions and pursuing riskier modifications because the combination of these features seemed most likely to yield significant differences in selectivity. Importantly, we needed to avoid an overly conservative library as high hit rates tend to prevent effective selection of the best hits for follow-up studies. Since the three central residues of UNC3866, A-L-KEt2 (R3-R4-R5 respectively), participate in backbone hydrogen bonds critical for beta strand formation, we did not investigate modifications of the peptide backbone. Instead, exploration of these residues focused on optimizing the hydrophobic interactions of the side chains and exploring the steric constraints within the hydrophobic channel of the chromodomains. Additionally, at the R3 and R4 positions, residues capable of inducing new, potentially beneficial geometries were included. Only modest alterations to the methyl-lysine mimetic at position R5 were included due to previous characterization of modifications at this position. Our prior efforts demonstrated that the hydrophobic contacts of the N-terminal tert-butyl benzoyl group are essential for the initial binding of UNC3866 to CBX710 so we chose to include both cyclic and acyclic aliphatic caps along with aromatic caps at position R1 to assess individual chromodomain preferences at this critical position. Lastly, substitutions were made at the serine and phenylalanine positions (R6 and R2 respectively) to diversify the explored area of chemical space for new and unanticipated routes to selectivity. Risky, more diverse substitutions were incorporated at these two positions with the expectation that they may also significantly limit the hit rate.
Once we concluded the library design, the 14,112 compound library was synthesized via split-and-pool synthesis using standard Fmoc solid phase synthesis and amine functionalized PEGA resin. Recent efforts in the Kodadek lab have extensively characterized methods for on-bead screening that minimize the number of false positive hit beads,35,36 a pervasive issue in screening OBOC libraries. By screening libraries with a theoretical redundancy of at least three-fold, they demonstrated that redundant hits were almost always true positive ligands. Consequently, we synthesized the library at approximately 15-fold redundancy. The Kme mimetics at position R5 were synthesized from Fmoc lysine via reductive amination while all other amino acids and carboxylic acids were purchased. The beads were split for amide coupling reactions and pooled for Fmoc deprotections. The final library was pooled and Boc/tBu deprotected under acidic conditions and washed extensively before picking 10 random beads to confirm the integrity of the library. These beads were CNBr cleaved from the resin and subjected to tandem mass spectrometry for sequencing. Of the 10 beads chosen, 9 were able to be sequenced and identified (Supporting Information Figure S1), suggesting that we could reasonably infer that the library was of sufficient integrity to proceed with screening.
Subsequently, we conducted iterative on-bead screening of the UNC3866-derivative library with the initial goal of identifying ligands with improved potency and selectivity for CDYL2 (Figure 2b). Of the chromodomains previously validated for on-bead screening, the chromodomain of MPP8 shares the highest sequence identity with that of CDYL2 so we hypothesized that screening MPP8 first would remove many of the unselective beads. Hundreds of MPP8 hit compounds and beads non-specifically interacting with components of the assay were isolated by magnet and removed from the library. The remaining unmagnetized library beads were presumed to have minimal affinity for MPP8 and so they were stripped of any residual protein and washed extensively prior to screening CDYL2.
CDYL2 was screened next in the cascade and several hundred beads were magnetized, posing a potential concern for follow-up characterization. During the original validation of the magnetic enrichment assay we had observed that UNC3866-PEGA magnetization could be competed off by soluble UNC3866 over a period of 15-90 minutes. Thus, we sought to decrease the hit rate and selectively enrich for compounds more potent than our starting ligand by treating the CDYL2 bound hits with soluble UNC3866 (10 μM). The beads that remained magnetized after 120 minutes (slow-off rate CDYL2 hits) were isolated and stripped of protein and magnetic beads for subsequent screening. This time-dependent, soluble competitor strategy removed roughly 50-75% of the original CDYL2 hit beads.
Proceeding forward with the selectivity screening cascade (Figure 2b), the slow-off rate and presumably most potent CDYL2 hits were screened sequentially against His-tagged CBX8, CBX7, and CBX5 via the same protocol as MPP8. For each screen, the beads that bound to these off-target chromodomains were removed from the hit pool and the unmagnetized, more CDYL2 selective beads were isolated and used in each successive screen. Taken as a whole, this target-class screening strategy was designed to isolate ligands within the library that maintained the highest activity for CDYL2 and demonstrated significantly reduced affinity for MPP8, CBX7, CBX8, and CBX5.
Following these six screens, ∼400 hit beads were isolated, washed, cleaved from the solid-support by cyanogen bromide treatment, and subjected to tandem mass spectrometry. To triage such a large number of hits, we excluded in our analyses any compounds without a redundant hit. Moreover, the highest redundancy hits were given priority for follow-up studies. Setting a threshold for high redundancy at 4 redundant beads, the screening strategy isolated 8 high redundancy compounds (Figure 3; also see Supporting Information Figure S2). All of the redundant hits in Figure 3 contained a conserved alanine at the -2 position from lysine (position R3), despite alanine comprising only a third of the total library. The alanine of UNC3866 fits tightly within the induced channel formed between CBX7 and the ligand in the co-crystal structure. Conservation of this alanine was unsurprising and indicated that these compounds were unlikely to be false positives. The C-terminal residue (position R6) of all of the redundant hits retained an alcohol substituent, favoring serine or threonine. At position R2, CDYL2 highly favored phenylalanine which is conserved in 6 of the 8 redundant hit structures. The trends observed at positions R2, R3, and R6 show little deviation from the structure of UNC3866 and indicated that CBX7 and CDYL2 share similar binding preferences at these positions.
Figure 3.
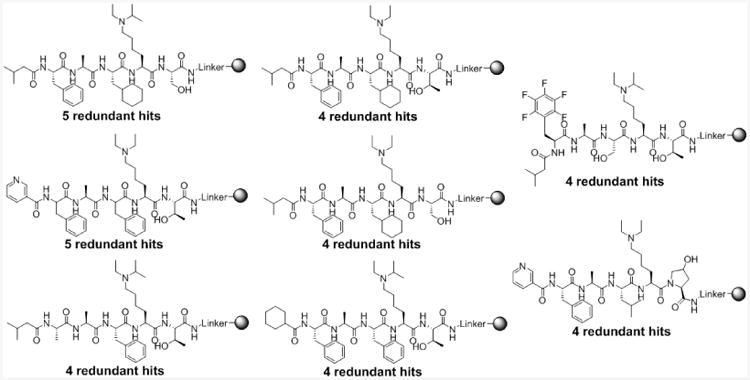
Highest redundancy CDYL2-selective hits. Greater than 400 hit beads were magnetically enriched for CDYL2, but not for MPP8, CBX8, CBX7, or CBX5. Of these, eight hits had a redundancy of more than 3-fold.
At the position of the lysine mimetic (R5), both the diethyl and ethyl-isopropyl Kme mimetics were equally enriched in the highest redundancy hits. Since UNC3866 contains diethyl-lysine and both mimetics were equally favored by CDYL2, this residue seemed unlikely to be dictating selectivity, though possible synergistic effects cannot be ruled out. The hits began to differ more at the R4 position which was tolerant to a variety of substituents, but demonstrated an overall preference for hydrophobic side chains, namely phenylalanine and cyclohexylalanine. Lastly, the N-terminal capping position (R1) contained the most variability among the highest redundancy hits. The isobutyl, cyclohexyl, and pyridyl caps represent the most distinct changes from UNC3866, particularly with our knowledge of how essential the tert-butyl phenyl moiety of UNC3866 is for CBX7 binding, suggesting that our previous hypothesis was correct: chromodomain selectivity could be modulated at the N-terminal capping position.
To quantitatively evaluate the effectiveness of our screening strategy and ensure that the top hits were at least equipotent to UNC3866 for CDYL2, we resynthesized a subset of the CDYL2 redundant hits for confirmation in a secondary fluorescence polarization (FP) assay. Four compounds were selected that displayed each type of capping residue and various internal peptide modifications relative to UNC3866. All four ligands were resynthesized with a C-terminal alkyne and a beta-alanine linker. Using click chemistry, fluorescein was ligated to the C-terminus of the peptides yielding the final FP ligands (Figure 4a). Dissociation constants (Kd's) of these fluorescent ligands for CDYL2, MPP8, CBX7, CBX8, and CBX5 were determined in the FP assay to better assess selectivity (Figure 4b; Table 1; also see Supporting Information Figure S3). Gratifyingly, all four compounds were most potent against CDYL2. UNC4979 had reduced affinity for CDYL2 relative to UNC3866-FAM (Supporting Information Figure S3), but the remaining three compounds were all equipotent or more potent than UNC3866-FAM. All four resynthesized hits, at a minimum, reduced CBX7 activity by 15-fold as compared to UNC3866-FAM, and CBX8 activity by 8-fold. Interestingly, the pyridyl N-terminal cap of UNC4982 led to a 2-fold enhancement in MPP8 activity relative to UNC3866-FAM, while the cyclohexyl and isobutyl caps reduced MPP8 activity. Overall, UNC4980 and UNC4981 displayed the most optimal CDYL2 profiles. The N-terminal caps of the two compounds have clear similarities, both being aliphatic and hydrophobic, yet are distinct from UNC3866 which also has high affinity for CDYL2, and this suggests higher tolerance at the capping position as compared to the other chromodomains. Ultimately, the FP results confirmed the success of our on-bead screening strategy for the isolation and identification of potent and selective CDYL2 ligands.
Figure 4.
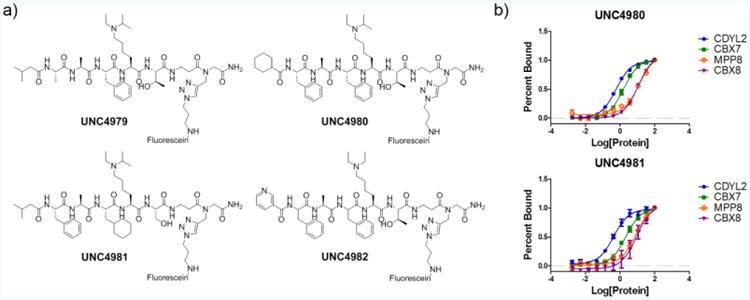
(A) Structures of resynthesized peptides with C-terminal fluorescein labels installed via click chemistry. These ligands were then used in FP assays to quantify binding for the chromodomains of CDYL2, MPP8, CBX7, CBX8, CBX5, CDYL, and CDY1. (B) Select FP binding affinity curves for the two most promising CDYL2 ligands.
Table 1.
Kd values (μM) determined via FP for resynthesized hits.a
| CDYL2 | CBX7 | CBX8 | MPP8 | CBX5 | CDYL | CDY1 | |
|---|---|---|---|---|---|---|---|
| UNC3866-FAM | 0.710 ±0.425 | 0.093 ±0.027 | 0.666 ±0.009 | 4.787 ±0.538 | Not tested | 1.011 ±0.203 | 6.414 ±0.920 |
| UNC4979 | 5.426 ±1.266 | >20 | >20 | >20 | >20 | 15.23 ±1.328 | >20 |
| UNC4980 | 0.430 ±0.070 | 2.280 ±0.748 | 7.632 ±1.828 | 8.019 ±0.594 | >20 | 1.262 ±0.487 | 3.589 ±1.247 |
| UNC4981 | 0.577 ±0.058 | 1.623 ±0.350 | 5.561 ±0.110 | 12.27 ±2.122 | >20 | 1.000 ±0.463 | 7.897 ±1.320 |
| UNC4982 | 0.362 ±0.040 | 2.165 ±0.299 | >20 | 2.255 ±0.446 | 19.347 ±3.447 | 1.312 ±0.269 | 3.057 ±0.187 |
The data for the average Kd values was calculated from 3 replicate runs. Error is reported as the standard deviation of the 3 values.
All four compounds were also evaluated for their affinity against CDYL and CDY1, two other chromodomain containing proteins that are members of the CDY-family. CDYL shares greater than 70% sequence homology with CDYL2. Only two residues in the histone-CDYL interface differ from CDYL2 which accounts for their overall similar activity profiles (Table 1). In contrast, CDY1 differs from CDYL2 at the residues interacting with the capping position (R1), and these differences clearly mediate a significant loss in potency of all four compounds for CDY1.
Analogs of UNC4980 and UNC4981 were synthesized as C-terminal primary amides without the linker and fluorophore (Figure 5) to differentiate any effects either may have on chromodomain binding. These ligands were evaluated against CDLY2, CDYL, and CBX7 by isothermal titration calorimetry (ITC) which requires no label for Kd determination (Supporting Information Figure S4). UNC4990 (Kd,[CDYL2] = 0.57 ± 0.13 μM) and UNC4991 (Kd,[CDYL2] = 0.64 ± 0.03 μM) demonstrated highly similar potencies and selectivity profiles as their fluorescent counterparts, confirming that the C-terminal linker and fluorescein of the FP ligands do not contribute to ligand binding in a significant way. These smaller ligands are promising for continued optimization as cell permeable ligands.
Figure 5.
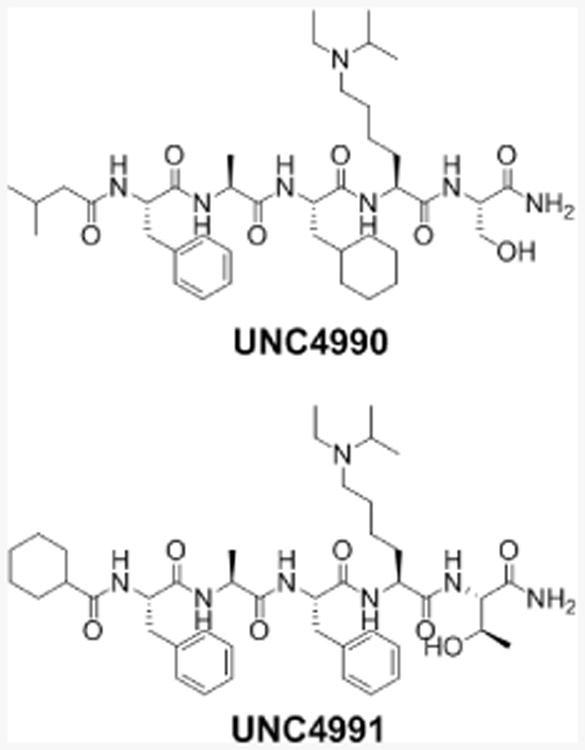
Unlabelled ligands used for ITC experiments and as soluble competitors in pulldown experiments.
To elucidate the structural mechanism of the 5-fold selectivity of UNC4991 for CDYL2 with respect to CBX7, we built a model of the CDYL2 chromodomain in complex with UNC4991. The models of both the protein and the compound are based on the x-ray structure of the UNC3866:CBX7 complex (PDB 5EPJ). The CDYL2/CBX7 sequence homology (47%) is high enough to yield a plausible CDYL2 model. Moreover, due to the high structural similarity between UNC4991 and UNC3866, it was safe to infer that UNC4991 would bind chromodomains similarly to UNC3866. To enable an unbiased comparative analysis of the protein-ligand interactions between CDYL2/CBX7 and UNC4991/UNC3866, we performed all-atom molecular dynamics (MD) simulations in explicit solvent for all four protein-ligand complexes. The MD simulations were long enough (300 ns each) to sufficiently sample side chain rearrangements on the ligand-protein interface. The analysis suggests that the major driver of the observed UNC4991 selectivity for CDYL2 is non-polar interactions between the cyclohexyl cap and a hydrophobic cage formed by phenylalanine 47, histidine 43, glutamate 46 and valine 5 (Figure 6a). The cap participates in up to 20 van der Waals (vdW) contacts with the surrounding cage and maintains most of these interactions throughout the simulation. The respective hydrophobic cage in CBX7, formed by leucine 53, arginine 52 and aspartate 50, is significantly smaller due to strain induced by proline 51 (Figure 6b). UNC4991's cyclohexyl cap does not align well into this smaller cage and makes significantly fewer vdW contacts with CBX7. To a lesser extent, the selectivity of UNC4991 for CDYL2 may be influenced by the isopropyl group on the alkylated lysine residue. The simulations of UNC4991 in complex with CDYL2 and CBX7 show that on average it makes more vdW contacts within the slightly larger lysine aromatic cage of CDYL2 than in CBX7.
Figure 6.
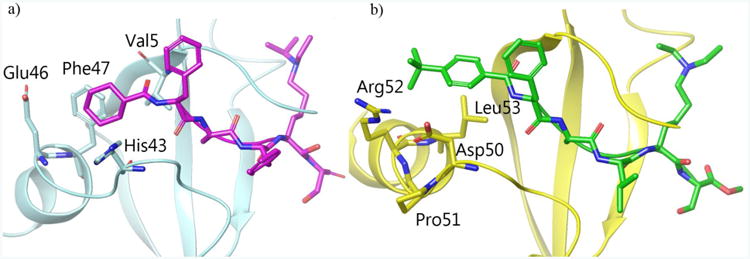
Structural comparisons of CDYL2 and CBX7 binding modes. (A) Docked structure of UNC4991 bound to CDYL2. The cyclohexyl cap binds within a hydrophobic cage that facilitates up to 20 van der Waals contacts whereas (B) the co-crystal structure of UNC3866 bound to CBX7 (PDB 5EPJ) depicts a smaller hydrophobic cage comprised of Asp50, Arg52, and Leu53.
We next synthesized a biotinylated derivative of UNC4991 to evaluate the ability of this reagent to chemiprecipitate CDYL2, CDYL, and CBX7 from cellular lysates and better gauge the selectivity of the compound in a more complex system (Figure 7a). Streptavidin coated magnetic Dynabeads were incubated with biotinylated UNC4991 and added to HeLa nuclear extracts in the presence or absence of 100 μM UNC4991 (Figure 7b). Western blot analysis indicated successful pulldown of both CDYL2 and CDYL. We attributed the observed chemiprecipitation to interaction specifically with the chromodomains since pull-down was almost completely outcompeted by the addition of soluble UNC4991. Importantly, CBX7 was not pulled-down under the same conditions which concurs with the on-bead screening results and confirms that UNC4991 interacts more potently with endogenous CDYL2 and CDYL.
Figure 7.
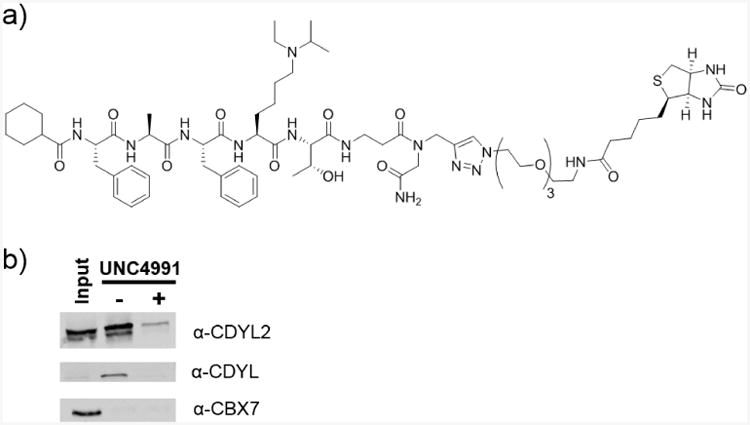
Chemiprecipitation of CDYL proteins. (A) Structure of C-terminally biotinylated derivative of UNC4991 for chemiprecipitation from HELA nuclear lysates and (B) Western blot analysis demonstrating that UNC4991-Biotin selectively chemiprecipitates CDYL2 and CDYL from HeLa nuclear lysates (middle lane, −UNC4991). Chemiprecipitation is effectively blocked in the presence of 100 μM soluble UNC4991 (right lane, + UNC4991).
Conclusions
Combinatorial repurposing of UNC3866 toward the discovery of new chromodomain ligands led to the rapid discovery of peptidomimetic inhibitors UNC4979, UNC4980, UNC4981, and UNC4982, which demonstrate improved selectivity for CDYL2. Our on-bead screening strategy effectively utilized homolog negative selections and a soluble competitor ligand (UNC3866) to enrich for ligands that have the desired selectivity profile and improved affinity for CDYL2. The selectivity profiles predicted for the resulting hits based on the on-bead screening assays were recapitulated by in vitro characterization in orthogonal binding assays, and ligand binding for other members of the CDY family of chromodomains (CDYL, CDY1) was also evaluated. Removal of the C-terminal derivatization to yield small molecule peptidomimetics on par with the size of UNC3866, a cell permeable chemical probe, maintained CDYL2 and CDYL affinity. Lastly, C-terminal biotinylation of UNC4991 generated a CDYL2/CDYL chemiprecipitation tool that could also be utilized for future characterization of these proteins and their interaction partners. UNC4991 is, therefore, a selective and potent CDYL2/CDYL in vitro chemical tool and future combinatorial chemistry or medicinal chemistry efforts to further optimize this series of ligands may yield cellular chemical probes capable of elucidating new biological roles for the CDYL proteins. Limiting false positive hit rates and selectively enriching only the most potent and selective inhibitors has remained a challenge with targeted OBOC libraries, but our platform suggests a broadly applicable path forward to reduce high hit rates and isolate improved compounds via homolog cross-screening.
Methods
All methods are described in the Supporting Information.
Supplementary Material
Acknowledgments
We thank N. Dicheva and the UNC Michael Hooker Proteomics Center for assistance with and use of their AbSciex 5800 MALDI-TOF/TOF. We thank the Structural Genomics Consortium for providing CBX7, CBX8, MPP8, and CBX5 protein constructs and M. Bedford for providing CDYL2, CDYL, and CDY1 protein constructs. We also thank M. Waters and A. Koenig for assistance with the CBX5 FP assay. We thank J. Lee for providing HeLa nuclear extracts. The research described here was supported by the National Institute of General Medical Sciences, U.S. National Institutes of Health (NIH, grant R01GM100919) and the University Cancer Research Fund, University of North Carolina at Chapel Hill.
Abbreviations
- Kme
methyl-lysine
- PRC1
Polycomb repressive complex 1
- CDYL2
Chromodomain Y-like protein 2
- CDYL
Chromodomain Y-like protein
- SAR
structure-activity relationship
- PPI
protein-protein interaction
- CBX7
chromobox protein homolog 7
- CBX4
chromobox protein homolog 4
- CBX6
chromobox protein homolog 6
- CBX2
chromobox protein homolog 2
- CBX8
chromobox protein homolog 8
- CDY1
Testis-specific chromodomains protein Y 1
- MPP8
M-phase phosphoprotein 8
- His
6× histidine
- Kme3
trimethyl-lysine
- Kd
dissociation constant
- OBOC
one-bead-one-compound
- PEGA
poly[acryloyl-bis(aminpropyl)polyethylene glycol]
- CNBr
cyanogen bromide
- βAla
beta-alanine
- KEt2
diethyl-lysine
- Fmoc
fluorenylmethoxycarbonyl
- Boc
tert-butoxycarbonyl
- tBu
tert-butyl
- FP
fluorescence polarization
- ITC
isothermal titration calorimetry
- MD
molecular dynamics
- vdW
van der Waals
- FAM
fluorescein
Footnotes
Supporting Information. Supporting figures, a detailed description of the chemical synthesis and characterization, and the materials and experimental methods are described in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes: The authors declare no competing financial interest.
References
- 1.Frye SV. The art of the chemical probe. Nature Chemical Biology. 2010;6(3):159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- 2.Bunnage ME, Chekler ELP, Jones LH. Target validation using chemical probes. Nature Chemical Biology. 2013;9(4):195–199. doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- 3.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME. The promise and peril of chemical probes. Nature Chemical Biology. 2015;11(8):536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hruby VJ. Prospects for peptidomimetic drug design. Drug Discovery Today. 1997;2(5):165–167. [Google Scholar]
- 5.Vagner J, Qu H, Hruby VJ. Peptidomimetics, a synthetic tool of drug discovery. Current Opinion in Chemical Biology. 2008;12(3):292–296. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craik DJ, Fairlie DP, Liras S, Price D. The Future of Peptide-based Drugs. Chemical Biology & Drug Design. 2013;81(1):136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 7.Hock FJ, Wirth K, Albus U, Linz W, Gerhards HJ, Wiemer G, Henke S, Breipohl G, Konig W, Knolle J, et al. Hoe 140 a new potent and long acting bradykinin-antagonist: in vitro studies. British Journal of Pharmacology. 1991;102(3):769–73. doi: 10.1111/j.1476-5381.1991.tb12248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broqua P, Riviere PJM, Conn PM, Rivier JE, Aubert ML, Junien JL. Pharmacological Profile of a New, Potent, and Long-Acting Gonadotropin-Releasing Hormone Antagonist: Degarelix. Journal of Pharmacology and Experimental Therapeutics. 2002;301(1):95–102. doi: 10.1124/jpet.301.1.95. [DOI] [PubMed] [Google Scholar]
- 9.Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nature Reviews Drug Discovery. 2004;3(3):215–225. doi: 10.1038/nrd1331. [DOI] [PubMed] [Google Scholar]
- 10.Stuckey JI, Dickson BM, Cheng N, Liu Y, Norris JL, Cholensky SH, Tempel W, Qin S, Huber KG, Sagum C, Black K, Li F, Huang XP, Roth BL, Baughman BM, Senisterra G, Pattenden SG, Vedadi M, Brown PJ, Bedford MT, Min J, Arrowsmith CH, James LI, Frye SV. A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nature Chemical Biology. 2016;12(3):180–7. doi: 10.1038/nchembio.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cavalli G, Paro R. Chromo-domain proteins: linking chromatin structure to epigenetic regulation. Current Opinion in Cell Biology. 1998;10(3):354–60. doi: 10.1016/s0955-0674(98)80011-2. [DOI] [PubMed] [Google Scholar]
- 12.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. Embo Reports. 2006;7(4):397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eissenberg JC. Structural biology of the chromodomain: form and function. Gene. 2012;496(2):69–78. doi: 10.1016/j.gene.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410(6824):120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 15.Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes & Development. 2003;17(15):1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Escamilla-Del-Arenal M, da Rocha ST, Spruijt CG, Masui O, Renaud O, Smits AH, Margueron R, Vermeulen M, Heard E. Cdyl, a New Partner of the Inactive X Chromosome and Potential Reader of H3K27me3 and H3K9me2. Molecular and Cellular Biology. 2013;33(24):5005–5020. doi: 10.1128/MCB.00866-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang Y, Horton JR, Bedford MT, Zhang X, Cheng X. Structural Insights for MPP8 Chromodomain Interaction with Histone H3 Lysine 9: Potential Effect of Phosphorylation on Methyl-Lysine Binding. Journal of Molecular Biology. 2011;408(5):807–814. doi: 10.1016/j.jmb.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simhadri C, Daze KD, Douglas SF, Quon TT, Dev A, Gignac MC, Peng F, Heller M, Boulanger MJ, Wulff JE, Hof F. Chromodomain antagonists that target the polycomb-group methyllysine reader protein chromobox homolog 7 (CBX7) Journal of Medicinal Chemistry. 2014;57(7):2874–83. doi: 10.1021/jm401487x. [DOI] [PubMed] [Google Scholar]
- 19.Ren C, Morohashi K, Plotnikov AN, Jakoncic J, Smith SG, Li J, Zeng L, Rodriguez Y, Stojanoff V, Walsh M, Zhou MM. Small-molecule modulators of methyl-lysine binding for the CBX7 chromodomain. Chemistry & Biology. 2015;22(2):161–8. doi: 10.1016/j.chembiol.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren C, Smith SG, Yap K, Li S, Li J, Mezei M, Rodriguez Y, Vincek A, Aguilo F, Walsh MJ, Zhou MM. Structure-Guided Discovery of Selective Antagonists for the Chromodomain of Polycomb Repressive Protein CBX7. ACS Medicinal Chemistry Letters. 2016 doi: 10.1021/acsmedchemlett.6b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milosevich N, Gignac MC, McFarlane J, Simhadri C, Horvath S, Daze KD, Croft CS, Dheri A, Quon TT, Douglas SF, Wulff JE, Paci I, Hof F. Selective Inhibition of CBX6: A Methyllysine Reader Protein in the Polycomb Family. ACS Medicinal Chemistry Letters. 2016;7(2):139–44. doi: 10.1021/acsmedchemlett.5b00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lam KS, Lebl M, Krchňák V. The “One-Bead-One-Compound” Combinatorial Library Method. Chemical Reviews. 1997;97(2):411–448. doi: 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- 23.Kodadek T. The rise, fall and reinvention of combinatorial chemistry. Chemical Communications. 2011;47(35):9757–9763. doi: 10.1039/c1cc12102b. [DOI] [PubMed] [Google Scholar]
- 24.Gao Y, Amar S, Pahwa S, Fields G, Kodadek T. Rapid Lead Discovery Through Iterative Screening of One Bead One Compound Libraries. ACS Combinatorial Science. 2015;17(1):49–59. doi: 10.1021/co500154e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trader DJ, Simanski S, Kodadek T. A Reversible and Highly Selective Inhibitor of the Proteasomal Ubiquitin Receptor Rpn13 Is Toxic to Multiple Myeloma Cells. Journal of the American Chemical Society. 2015;137(19):6312–6319. doi: 10.1021/jacs.5b02069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarkar M, Liu Y, Morimoto J, Peng H, Aquino C, Rader C, Chiorazzi N, Kodadek T. Recognition of Antigen-Specific B-Cell Receptors from Chronic Lymphocytic Leukemia Patients by Synthetic Antigen Surrogates. Chemistry & Biology. 2014;21(12):1670–1679. doi: 10.1016/j.chembiol.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gocke AR, Udugamasooriya DG, Archer CT, Lee J, Kodadek T. Isolation of Antagonists of Antigen-Specific Autoimmune T Cell Proliferation. Chemistry & Biology. 2009;16(11):1133–1139. doi: 10.1016/j.chembiol.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nature Reviews Drug Discovery. 2009;8(9):724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 29.Heilker R, Wolff M, Tautermann CS, Bieler M. G-protein-coupled receptor-focused drug discovery using a target class platform approach. Drug Discovery Today. 2009;14(5–6):231–240. doi: 10.1016/j.drudis.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Trinh TB, Xiao Q, Pei D. Profiling the Substrate Specificity of Protein Kinases by On-Bead Screening of Peptide Libraries. Biochemistry. 2013;52(33):5645–5655. doi: 10.1021/bi4008947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao Q, Luechapanichkul R, Zhai Y, Pei D. Specificity Profiling of Protein Phosphatases toward Phosphoseryl and Phosphothreonyl Peptides. Journal of the American Chemical Society. 2013;135(26):9760–9767. doi: 10.1021/ja401692t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olivos HJ, Bachhawat-Sikder K, Kodadek T. Quantum dots as a visual aid for screening bead-bound combinatorial libraries. Chembiochem. 2003;4(11):1242–5. doi: 10.1002/cbic.200300712. [DOI] [PubMed] [Google Scholar]
- 33.Astle JM, Simpson LS, Huang Y, Reddy MM, Wilson R, Connell S, Wilson J, Kodadek T. Seamless bead to microarray screening: rapid identification of the highest affinity protein ligands from large combinatorial libraries. Chemistry & Biology. 2010;17(1):38–45. doi: 10.1016/j.chembiol.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao Y, Kodadek T. Synthesis and Screening of Stereochemically Diverse Combinatorial Libraries of Peptide Tertiary Amides. Chemistry & Biology. 2013;20(3):360–369. doi: 10.1016/j.chembiol.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doran TM, Gao Y, Mendes K, Dean S, Simanski S, Kodadek T. Utility of redundant combinatorial libraries in distinguishing high and low quality screening hits. ACS Combinatorial Science. 2014;16(6):259–70. doi: 10.1021/co500030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mendes K, Ndungu JM, Clark LF, Kodadek T. Optimization of the Magnetic Recovery of Hits from One-Bead–One-Compound Library Screens. ACS Combinatorial Science. 2015;17(9):506–517. doi: 10.1021/acscombsci.5b00090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.