

ABSTRACT
As the research and use of immunotherapies is expanding, isolating ideal combinational strategies has become the next goal for many investigators. Vaccine therapies are also becoming one of the many combinational strategies being utilized in conjunction with immunostimulatory antibodies such as checkpoint blockade or adjuvants to stimulate immune responses. Here we review aspects of the immune responses that remain to be considered for designing future targeted therapies given the recent findings of the role of out of order T cell activation signaling. Specifically, we review some considerations in generating primary T cell responses under conditions of strong immunostimulatory signals based on recent studies completed by our group and others.
KEYWORDS: memory T cell expansion, primary antigen-specific response, T cell activation
Factors impacting primary immune responses
The goal of most therapeutic vaccine approaches is to induce robust primary antigen-specific responses which then lead to strong antigen-specific secondary responses upon rechallenge. However, there are many parameters that may affect the induction of a successful primary response. These include: the nature of the antigen, the method of administration, the duration of antigen exposure, the age of the recipient which markedly affects the number and activity of naive T cells and the inflammatory milieu that accompanies the response which can drive both antigen-presenting cell (APC) and T cell responses. It is now well documented that the generation of a successful primary antigen-specific T cell response requires the successive completion of certain steps that proceed in a critical order. These have been called “Signals” by which the naïve antigen-specific T cell interacts with the APC (Fig. 1A). “Signal 1” occurs when the T cell receptor (TCR) on the naïve CD4+ or CD8+ T cell interacts with the APC where the antigen is presented in the context of MHC Class I or Class II molecules. “Signal 2” results when costimulatory receptors such as CD28 bind to their ligands in response to signal 1 (i.e. B7 proteins present on the licensed APC). “Signal 3” then occurs when a component of the IL2 receptor, CD25, is upregulated following Signal 1, forming the high affinity IL2 receptor complex which allows for greater proliferative and activation responses after exposure to cytokines such as IL2 even at low levels.1 This results in a selective advantage for the antigen-primed T cell to respond to cytokines and clonally expand. The successful completion of these 3 steps allow for T effector cell generation as well as the subsequent programming of the T cell to become a long-lived antigen-specific memory T cell critical for the generation of recall responses to the antigen. It has been demonstrated that the sequence of these signals, particularly Signal 1 and Signal 2, are absolutely critical for an antigen-specific T cell activation. This represents a critical control checkpoint in immune activation. Signal 1 without Signal 2 has been demonstrated to result in unresponsiveness or antigen-specific anergy of the T cell.1-3 Signal 1 occupancy is also critical for increased proliferation and upregulation of CD25 to allow for increased responsiveness to IL2.4,5 The vast majority of studies thus far have focused on the impact of Signals 1 and 2. However, the role of Signal 3 alone on T cell function, particularly with regard to naïve T cells, has remained elusive until the recent studies by Sckisel et al. and Urban et al.6,7
Figure 1.
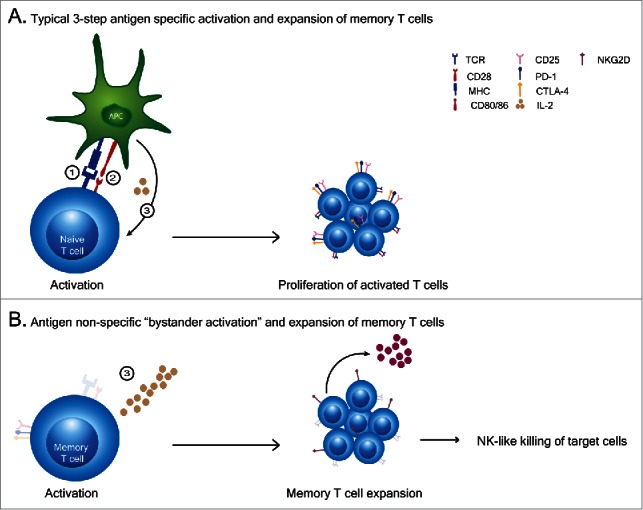
(T)cell activation patterns. (A) A typical primary T cell activation requires 3 signals to occur in sequence and depends on: 1). antigen recognition by TCR engagement and the strength of the signal received, 2). ligation of co-stimulatory molecules to their ligands and 3). cytokine mediated expansion and differentiation of cells to effector cells. (B) Cytokine exposure alone causes an antigen non-specific “bystander activation” expansion of memory T cells which exhibit broad lytic capabilities in comparison to antigen specfic activated T cells.
Signal 3 alone results in antigen-nonspecific memory T cell expansion
There are critical differences between cell surface molecules of naïve and memory T cells with regard to the IL2 receptor complex subunit expression. The high affinity receptor complex to IL2 consists of 3 subunits: CD25, CD122 and CD132. T cells, both naive and memory, constitutively express CD122 and CD132 (the common gamma chain) forming a low affinity receptor complex but allowing responsiveness to high concentration of cytokines that signal through the common gamma chain (i.e., IL2, IL7, IL15).8,9 However, with the expression of CD132 on the naive T cell pool, CD122 has shown to be expressed at higher levels on T cells of memory phenotype compared to naïve T cells.9,10 This provides memory T cells (CD44hi/CD62Llo in the mouse and CD45RO in human T cells) with an advantage for increased cytokine responsiveness. Memory cells having undergone 2 rounds of selection already by the immune system (thymic education and antigen exposure in the periphery allowing them to become memory phenotype) are thus “safer” to expand.
We and others have previously observed that exposure of memory T cells to a strong inflammatory signal such as IL2 or other cytokines that interact via the CD122/CD132 complex results in an antigen-nonspecific activation of these cells (Fig. 1B). These cytokine induced and expanded memory T cells have a unique phenotype and function. They do not upregulate CD25 and therefore do not express the high affinity IL2 receptor complex. This necessitates continuous exposure to high amounts of cytokine in order to sustain their survival and function. These cells, once activated, have been shown to express perforin and NKG2D and have the ability to lyse target cells in an MHC-unrestricted manner through recognition of the NKG2D ligands which serve as “stress-induced” ligands on cells.11,12 In essence, tissue resident memory CD8+ T cells exposed to a strong inflammatory stimulus become NK-cell like with regard to cytokine dependence and broad killing capability. These cells, which can exist in almost every tissue as long-lived tissue-resident memory T cells, perform an auxiliary function which serves to augment target cell killing but only in conditions of high cytokine exposure. These are the classically described “bystander” T cells seen in viral infections. Once the cytokine is withdrawn, these cells contract rapidly. We have previously demonstrated that these cytokine activated T killer cells are responsible for many of the anti-tumor effects associated with anti-CD40 and IL2 in mice.11,13 This represents a relatively safe mechanism by which T cells, which have already undergone positive and negative selection in the thymus and then peripheral selection by antigen, become memory cells that can expand and function with decreased likelihood of autoreactivity upon activation in a highly inflammatory environment such as during viral infections. These bystander activated T cells amplify immune attack and form an important bridge between the innate and adaptive immune responses. A critical question which remains elusive is whether a primary immune response is capable of being generated during these inflammatory periods.
Signal 3 alone paralyzes naïve CD4+ T cell responses
We previously observed that after successful initial anti-tumor effects by CD8+ T cells, following strong systemic immunostimulatory regimens in mice (i.e. anti-CD40/IL2, anti-CD40/IL15 or CpG/IL12 regimens), surviving mice actually displayed impaired responses upon tumor rechallenge compared to mice that received irradiated tumor vaccine alone.14 When mice received the tumor vaccine with the immunostimulatory regimens this impaired response remained unchanged. It was also observed that while the CD8+ T cells, primarily of memory/effector phenotype, were markedly expanding, the CD4+ T cells were decreasing in numbers and undergoing apoptosis. Furthermore, the total numbers of naïve CD4+ or CD8+ cells did not alter, just their percentages. This was attributed to interferon-gamma which was a critical mediator of the anti-tumor effects but also responsible for the contraction of the memory CD4+ T cell compartment.14 Thus, the short-term benefit of successful anti-tumor effects mediated by the bystander CD8+ T cells was also concurrent with impairment of antigen-specific secondary responses. Next, we sought to examine the effects of these and other immunostimulatory regimens on the naïve T cell compartment. We observed that mice undergoing strong immunostimulatory regimens were completely unable to mediate primary allogeneic responses in a mixed lymphocyte reaction (MLR). Importantly, isolated T cells from cancer patients undergoing high dose IL2 treatments also were completely unable to mount a response following an MLR, linking the mouse and human effects.6 Once treatments were discontinued, however, the primary allogeneic responses recovered from their “paralysis.” Sorting purified memory and naïve CD4+ and CD8+ T cells revealed that the CD4+ naïve T cell compartment was completely paralyzed even to anti-CD3 and anti-CD28 or mitogen stimulation.6 It was also noted that this paralysis occurred during systemic LPS administration. Microarray analysis indicated that suppressor of cytokine signaling-3 (SOCS3) was induced during the immunotherapy regimens in the CD4+ T cells. While SOCS3 has previously been demonstrated to be induced by IL6 and other inflammatory cytokines, purified CD4+ T cells but not CD8+ T cells exposed to high dose IL2 resulted in the induction of SOCS3 along with paralysis indicating direct effects of the regimens on the CD4+ T cell populations.15 Use of antisense to SOCS3 on the CD4+ T cells or examination of SOCS3 knockout mice resulted in partial reversal of the paralysis and restoration of the CD4+ T cell responses. Overall, if mice were immunized with allogeneic cells and received immunostimulatory regimens, there was a significant impairment of CD8+ T cell antigen-specific responses upon rechallenge indicating the critical role of naïve CD4+ T cells in this process. This appears to represent a critical control or “emergency brake” that the immune system employs to control CD4+ T cell responses in instances of strong inflammation or signal 3 alone (Fig. 2). It is important to note that despite the extensive species and genetic differences between mouse and humans, that there are remarkable parallels in this pathway for controlling T cell responses. Interestingly, SOCS1 has been demonstrated to impair primary CD8+ T cell responses and was shown to signal via type 1 interferon.7 Altogether, this implicates that the SOCS family members are critical for controlling naïve T cell responses and provides yet another advantage for the memory T cell pool over the naïve during cytokine responses.
Figure 2.
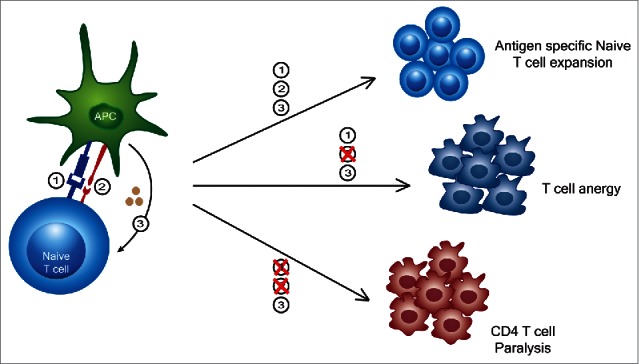
Out of sequence (T)cell activation signaling leads to variable responses. While a typical 3 signal activation results in an antigen specific T cell expansion, a lack of signal 2 can lead to T cell anergy. More recently, it has been shown that signal 3 alone not only causes an expansion of memory T cells into broadly lytic CD8+ T cells, but also causes a transient paralysis of CD4+ T cells thereby suppressing their activation and expansion to memory.
In conclusion, strong inflammatory responses, either during infections or immunostimulatory regimens, even with adjuvant use, may impair primary antigen responsiveness. CD4+ T cells appear to be particularly susceptible to this paralysis. Vaccination regimens may need to take these parameters into consideration in order to optimize responsiveness and efficacy. This temporary paralysis also raises many questions as to whether permanent tolerance can result if the TCR is engaged during SOCS3 induction resulting in a lack of co-stimulation.
Abbreviations
- TCR
T cell receptor
- APC
Antigen presenting cell
- SOCS
Suppressor of cytokine signaling
Disclosure of potential conflicts of interest
The authors reported no conflicts of interest.
References
- [1].Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK and Mescher MF. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. JI 1999; 162:3256-62 [PubMed] [Google Scholar]
- [2].Yamamoto T, Hattori M, and Yoshida T. Induction of T-cell activation or anergy determined by the combination of intensity and duration of T-cell receptor stimulation, and sequential induction in an individual cell. Immunology 2007; 121:383-91; PMID:17376194; http://dx.doi.org/ 10.1111/j.1365-2567.2007.02586.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schwartz RH. T cell anergy: is there a common molecular mechanism? JEM 1996; 184(1):1-8; http://dx.doi.org/ 10.1084/jem.184.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bitegye C, Hannier S, Guérif S, Valitutti S, Demotz S. Tuning of T Cell clone size and activation threshold by control of CD25 expression through mitogen-activated protein kinase pathways. Int Arch Allergy and Immunol 2002; 127:322-32; http://dx.doi.org/ 10.1159/000057750 [DOI] [PubMed] [Google Scholar]
- [5].Acuto O and Frederique M. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol 2003; 3:939-51; PMID:14647476; http://dx.doi.org/ 10.1038/nri1248 [DOI] [PubMed] [Google Scholar]
- [6].Sckisel GD, Bouchlaka MN, Monjazeb AM, Crittenden M, Curti BD, Wilkins DEC, Alderson KA, Sungur CM, Ames E, Mirsoian A, et al.. Out-of-Sequence signal 3 paralyzes primary CD4+T-cell-dependent immunity. Immunity 2015; 43:240-50; PMID:26231116; http://dx.doi.org/ 10.1016/j.immuni.2015.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Urban SL, Welsh RM. Out-of-Sequence signal 3 as a mechanism for virus-induced immune suppression of CD8 T cell responses. PLoS Pathogens 2014; 10:e1004357; PMID:25255454; http://dx.doi.org/ 10.1371/journal.ppat.1004357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schwartz RH. T cell anergy. Annu Rev Immunol 2003; 21:305-34; PMID:12471050; http://dx.doi.org/ 10.1146/annurev.immunol.21.120601.141110 [DOI] [PubMed] [Google Scholar]
- [9].Geginat J, Sallusto F, Lanzavecchia A. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4(+) T cells. JEM 2001; 194:1711-9; http://dx.doi.org/ 10.1084/jem.194.12.1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Berard M and Tough DF. Qualitative differences between naïve and memory T cells. Immunology 2002; 106:127-38; PMID:12047742; http://dx.doi.org/ 10.1046/j.1365-2567.2002.01447.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tietze JK, Wilkins DEC, Sckisel GD, Bouchlaka MN, Alderson KL, Weiss JM, Ames E, Bruhn KW, Craft N, Wiltrout RH, et al.. Delineation of antigen-specific and antigen-nonspecific CD8+ memory T-cell responses after cytokine-based cancer immunotherapy. Blood 2012; 119:3073-83; PMID:22251483; http://dx.doi.org/ 10.1182/blood-2011-07-369736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ehl S, Hombach J, Aichele P, Hengartner H and Zinkernagel RM. Bystander activation of cytotoxic T cells: Studies on the mechanism and evaluation of in vivo significance in a transgenic mouse model. JEM 1997; 185:1241-51; http://dx.doi.org/ 10.1084/jem.185.7.1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Monjazeb AM, Tietze JK, Grossenbacher SK, Hsiao HH, Zamora AE, Mirsoian A, Koehn B, Blazar BR, Weiss JM, Wiltrout RH, et al.. Bystander activation and anti-tumor effects of CD8+ T cells following interleukin-2 based immunotherapy is independent of CD4+ T cell help. PLoS ONE 2014; 9:e102709; PMID:25119341; http://dx.doi.org/ 10.1371/journal.pone.0102709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Berner V, Liu H, Zhou Q, Alderson KL, Sun K, Weiss JM, Back TC, Longo DL, Blazar BR, Wiltrout RH, et al.. IFN-gamma mediates CD4+ T-cell loss and impairs secondary antitumor responses after successful initial immunotherapy. Nat Med 2007; 13:354-60; PMID:17334371; http://dx.doi.org/ 10.1038/nm1554 [DOI] [PubMed] [Google Scholar]
- [15].Palmer DC and Restifo NP. Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol 2009; 30:592-602; PMID:19879803; http://dx.doi.org/ 10.1016/j.it.2009.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]