
Figure 5.6.15.
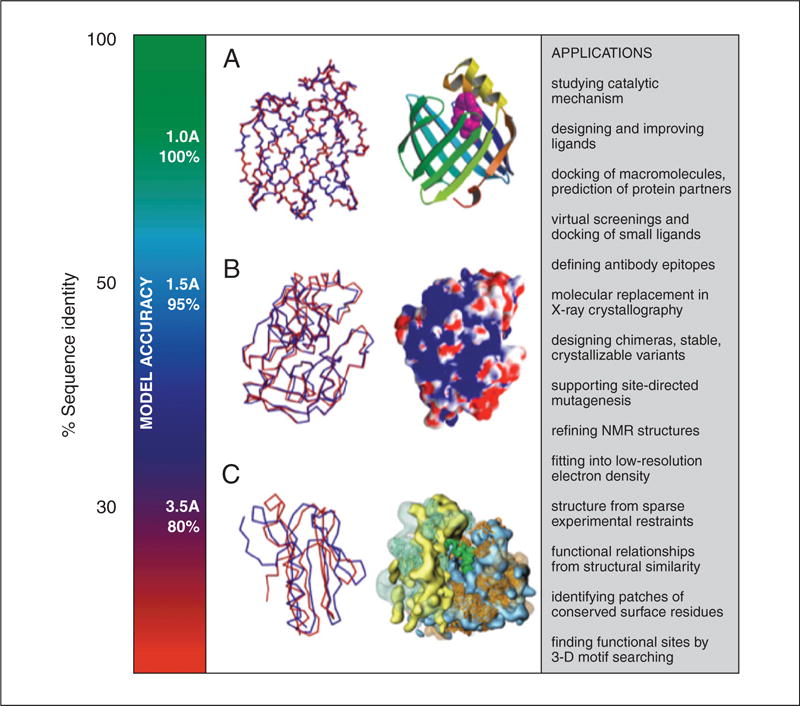
Accuracy and application of protein structure models. The vertical axis indicates the different ranges of applicability of comparative protein structure modeling, the corresponding accuracy of protein structure models, and their sample applications. (A) The docosahexaenoic fatty acid ligand (violet) was docked into a high accuracy comparative model of brain lipid-binding protein (right), modeled based on its 62% sequence identity to the crystallographic structure of adipocyte lipid-binding protein (PDB code 1adl). A number of fatty acids were ranked for their affinity to brain lipid-binding protein consistently with site-directed mutagenesis and affinity chromatography experiments (Xu et al., 1996), even though the ligand specificity profile of this protein is different from that of the template structure. Typical overall accuracy of a comparative model in this range of sequence similarity is indicated by a comparison of a model for adipocyte fatty acid binding protein with its actual structure (left). (B) A putative proteoglycan binding patch was identified on a medium-accuracy comparative model of mouse mast cell protease 7 (right), modeled based on its 39% sequence identity to the crystallographic structure of bovine pancreatic trypsin (2ptn) that does not bind proteoglycans. The prediction was confirmed by site-directed mutagenesis and heparin-affinity chromatography experiments (Matsumoto et al., 1995). Typical accuracy of a comparative model in this range of sequence similarity is indicated by a comparison of a trypsin model with the actual structure. (C) A molecular model of the whole yeast ribosome (right) was calculated by fitting atomic rRNA and protein models into the electron density of the 80S ribosomal particle, obtained by electron microscopy at 15 Å resolution (Spahn et al., 2001). Most of the models for 40 out of the 75 ribosomal proteins were based on template structures that were approximately 30% sequentially identical. Typical accuracy of a comparative model in this range of sequence similarity is indicated by a comparison of a model for a domain in L2 protein from B. stearothermophilus with the actual structure (1rl2).