

Abstract
INTRODUCTION
There is limited understanding of relationships between genotype, phenotype and other conditions contributing to health in neonates with medium-chain acyl-coenzyme A dehydrogenase deficiency (MCADD) identified through newborn screening.
METHODS
Retrospective analysis of comprehensive data from a cohort of 221 newborn-screened subjects identified as affected with MCADD in the Inborn Errors of Metabolism – Information System (IBEM-IS), a long term follow-up database of the Inborn Errors of Metabolism Collaborative, was performed.
RESULTS
The average age at notification of first newborn screen results to primary care or metabolic providers was 7.45 days. The average octanoylcarnitine (C8) value on first newborn screen was 11.2 umol/L (median 8.6, range 0.36–43.91). A higher C8 level correlated with an earlier first subspecialty visit. Subjects with low birth weight had significantly lower C8 values. Significantly higher C8 values were found in symptomatic newborns, in newborns with abnormal lab testing in addition to newborn screening and/or diagnostic tests, and in subjects homozygous for the c.985A>G ACADM gene mutation or compound heterozygous for the c.985A>G mutation and deletions or other known highly deleterious mutations. Subjects with neonatal symptoms, or neonatal abnormal labs, or neonatal triggers were more likely to have at least one copy of the severe c.985A>G ACADM gene mutation. C8 and genotype category were significant predictors of the likelihood of having neonatal symptoms. Neonates with select triggers were more likely to have symptoms and laboratory abnormalities.
CONCLUSIONS
This collaborative study is the first in the United States to describe health associations of a large cohort of newborn-screened neonates identified as affected with MCADD. The IBEM-IS has utility as a platform to better understand the characteristics of individuals with newborn-screened conditions and their follow-up interactions with the health system.
Keywords: medium-chain acyl-coenzyme A dehydrogenase deficiency, MCAD, octanoylcarnitine, ACADM, newborn screening, inborn error of metabolism
1. INTRODUCTION
Medium-chain acyl-coenzyme A dehydrogenase deficiency (MCADD) is an autosomal recessive mitochondrial fatty acid oxidation disorder. A recent report on the birth prevalence of disorders detectable through newborn bloodspot screening noted an overall MCADD birth prevalence of 5.3 (4.1–6.7, 99% CI) per 100,000 births across a variety of racial/ethnic groups (1). Impaired hepatic ketogenesis resulting in hypoketotic hypoglycemia, metabolic acidosis, liver disease, and lethargy can rapidly progress to coma and death when glycogen stores are depleted during catabolic physiological states (2). Undiagnosed, morbidity and mortality are considerable, but when the diagnosis is known, MCADD can be successfully managed and outcomes improved (3). Early detection of affected infants is important (4). Thus, MCADD is a core condition on the U.S. Department of Health and Human Services Advisory Committee on Heritable Disorders in Newborns and Children’s Recommended Uniform Screening Panel and newborn screening for this condition is done in all 50 U.S. states, the District of Colombia and Puerto Rico (5) (6). Despite an acknowledgement that this condition can have a significant health impact in early life, limited understanding exists of the complex interplay between genotype, biochemical phenotype, and other conditions associated with health outcomes of neonates with MCADD.
Accumulation of the medium-chain acylcarnitine species is characteristic of MCADD, with octanoylcarnitine (C8) as the prominent blood marker (7). Higher C8 values in blood spot newborn screening have been reported in association with homozygosity for the common c.985A>G pathogenic gene variant (8), or the presence of other severe pathogenic variants such as deletion, nonsense, or splice site mutations in the ACADM gene (9). Higher blood spot C8 values have also been reported in MCADD affected neonates, particularly those homozygous for the c.985A>G mutation, whose blood spots were collected sooner after birth (10).
This is the first multi-state, multi-center collaborative study in the United States to describe associations between newborn screen C8 values, ACADM genotype, clinical circumstances and symptoms, and clinical laboratory abnormalities in a large cohort of newborn-screened neonates identified as affected with MCADD.
Using data submitted by the Inborn Errors of Metabolism Collaborative, this study sought to learn if first newborn screen C8 values are related to gender, genotype, birth weight, or initial food source. We investigated whether C8 values are associated with the presence of neonatal triggers, the presence of neonatal symptoms, and the presence of neonatal abnormal lab results in addition to newborn screening or diagnostic tests. We also assessed if the presence of neonatal triggers is associated with the manifestation of neonatal symptoms and abnormal neonatal labs. We examined if there are correlations between first newborn screen C8 value and birth weight, and days of age at the first subspecialist visit, and if potential correlations between C8 value and the days of age at the first subspecialist visit differ between subjects with and without neonatal triggers, with and without neonatal symptoms, with and without neonatal abnormal labs. Finally, we sought associations between ACADM genotype and neonatal triggers, neonatal symptoms, and neonatal abnormal labs and examined if C8 values and genotype category predict whether a subject manifests neonatal symptoms.
2. METHODS
2.1. DATA
The research protocol was approved by the Institutional Review Boards of the Inborn Errors of Metabolism Collaborative (IBEMC) partners and informed consent was documented in the Inborn Errors of Metabolism – Information System (IBEM-IS) for all enrolled MCADD subjects (11) (12). The IBEMC data collection began in 2007 and is currently managed using REDCap electronic data capture tools hosted at the Michigan Public Health Institute (MPHI) (13).
Data for this study were extracted from the IBEM-IS on August 7, 2015. The inclusion criteria for this study were subjects having: 1) consented for data-sharing, 2) assignment of the condition MCADD, 3) abnormal newborn screening result, and 4) a first newborn screen C8 value. Newborn screen MCADD-related acylcarnitine values other than C8 were not included in this study due to insufficient data collection on those values in the IBEM-IS. Blood spot collection timing was unavailable for most subjects.
2.2. VARIABLES
The IBEM-IS defines the neonatal period as the first 28 days of life. It does not require specification of whether recorded neonatal complications, symptoms and abnormal labs are clinically determined to be caused by or related to MCADD. For this analysis, we reclassified the documented neonatal complications, symptoms and abnormal labs into three types: 1) neonatal triggers, 2) neonatal symptoms, and 3) neonatal abnormal labs, and defined “neonatal” as those that were known to have occurred within the first 28 days of life. Neonatal triggers were defined as neonatal complications and interventions suggestive of underlying health complications determined by clinician authors as most likely to result in potential MCADD symptoms. Neonatal symptoms were defined by clinician authors as symptoms consistent with MCADD, many based on reports of symptoms manifested in individuals affected with MCADD (9) (14) (15) (16) (17) (18) (19) (20). Neonatal abnormal labs were defined by clinician authors as laboratory test abnormalities of potential concern in the context of MCADD (excluding newborn screening and MCADD diagnostic biochemical and molecular test results). The IBEM-IS data did not allow for determination of whether jaundice and hyperbilirubinemia during the neonatal period reflected physiologic versus pathogenic newborn conditions. To avoid possible over-estimation of the neonatal symptoms/abnormal labs classified as associated with MCADD, we excluded jaundice/hyperbilirubinemia from the analyses. Birth weight was treated both as a continuous variable and was also categorized as low birth weight (<=2.5 kg), or not. The source of neonatal nutrition was categorized as breastfed only, or not. Age at first visit, age at notification, and age at intervention are age of days since birth until the subject was first seen by a subspecialist, the subject’s primary care or metabolic provider was first notified of the newborn screening results, and the intervention for MCADD was initiated, respectively. C8 is the octanoylcarnitine value, measured in umol/L, on the first newborn screen.
To analyze genotype, alleles were categorized based on documented ACADM gene allele findings (Table 5). The categories were as follows: A) the c.985A>G mutation (21) (22), B) ACADM deletions, and mutations other than c.985A>G for which reports of decreased fatty acid oxidation in fibroblast studies or considerably decreased MCAD enzyme activity were found in published literature (23) (24) (25) (26) (27) (28) (29), C) all other allele findings not meeting criteria for A or B (4) (23) (25) (29) (30) (31) (32) (33) (34) (35), and D) empty or indecipherable entries in the allele data fields, as data are entered as free text. The dataset contained eight combinations of two alleles: AA (n=69), AB (n=18), BB (n=1), AC (n=49), AD (n=26), CC (n=12), CD (n=2), and DD (n=44). The BB group contained too few cases to compute stable statistics and was excluded from genotype analysis. The AD, CD, and DD groups were also excluded from analysis due to the lack of usable genotype information. The remaining four genotype categories were further collapsed into two categories for selected additional analyses as follows: 1) AA and AB, and 2) AC and CC.
Table 5.
aDifferent ACADM allele findings of the 221 study subjects
| Allele | Genotype Category Assigned | Reference |
|---|---|---|
| bc.985A>G (p.K329E or p.K304E) | A | (21) (22) |
| c.233T>C | B | (23) (24) |
| c.1102_1105delTTAG | B | (25) (26) |
| c.734C>T(p.S245L) | B | (23) (25) |
| c.1238G>A (p.R413H) | B | (25) |
| c.928G>A | B | (23) |
| c.362 C>T | B | (23) (27) |
| c.347G>A | B | (28) |
| cc.799G>A | B | (29) |
| N=2 additional different deletions, not found by published literature search, were recorded in the IBEM-IS | B | |
| c.127 G>A (p.E43K) | C | (25) |
| c.797A>G | C | (25) |
| c.1207A>G | C | (25) |
| c.600-18G>A | C | (25) (31) |
| c.554T>C | C | (25) |
| c.443G>A | C | (25) |
| c.757G>A | C | (25) |
| c.1115C>A (p.A372D) | C | (25) |
| c.526G>A | C | (25) |
| c.(−34)T>C | C | (25) |
| c.558T>A | C | (4) |
| dc.199T>C (Y67H; Y42H) | C | (23) |
| c.387+1delG | C | (30) |
| c.157C>T | C | (32) |
| c.583G>A | C | (33) |
| c.728G>A | C | (4) |
| c.617G>T (p.R206L) | C | (30) |
| c.447 G>A | C | (29) (34) |
| c.250C>T (p.L84F) | C | (25) |
| c.92G>A | C | (35) |
| N=28 additional different allele findings, not found by published literature search, were recorded in the IBEM-IS | C |
Excludes subjects with empty and indecipherable entries in the allele data fields (genotype category D)
Frequency of genotype category A allele is 231
Most frequently appearing genotype category B allele (n=6)
Most frequently appearing genotype category C allele (n=14).
2.3 STATISTICAL ANALYSES
The Mann-Whitney U test was used to assess differences in C8 values between each pair of groups, defined by: low birth weight or not, breastfed only or not, presence or absence of neonatal triggers, presence or absence of neonatal symptoms, presence or absence of neonatal abnormal labs, and the two collapsed genotype categories. The Kruskal-Wallis test was used to compare C8 values among the four genotype categories. Correlations between C8, birth weight and age at first visit were tested using the Spearman correlation test. Chi-square tests were used to test the associations between categorical variables.
To further examine whether the strength of the relationship between C8 and age at first visit differs between each pair of subgroups defined by, presence or absence of neonatal triggers, presence or absence of neonatal symptoms, and presence or absence of neonatal abnormal labs, we constructed three generalized linear models (negative binomial with log link function). In each model, age at first visit was the dependent variable, C8 and one of the above three binary variables, along with the interaction term, were the independent variables.
Logistic regression with neonatal symptoms as the binary outcome variable, and the collapsed genotype category, C8, and gender as predictors, was conducted to determine whether these variables predict the likelihood of developing neonatal symptoms.
All statistical significance tests were two-sided. Bonferroni correction was used to adjust for multiple comparisons to keep the overall significance level at α=0.05. All the statistical analyses were conducted in IBM SPSS Statistics for Windows, Version 23.0.
3. RESULTS
A total of 337 consented subjects were assigned the condition MCADD, 285 (85%) of whom had abnormal newborn screening. Of the 285 subjects, 223 had a first newborn screen C8 value documented in the IBEM-IS. Two subjects with first newborn screen C8 values presumed to be erroneously recorded (2406 umol/L and 1738 umol/L) were excluded. The remaining 221 subjects, all identified in the IBEM-IS as alive at the time of data extraction, constituted the dataset for this study.
Age of subjects at IBEMC enrollment ranged from infancy to 10 years (n=218). Average reported birth weight was 3.35 kg (n=213, Std. Dev.=0.53). Subjects’ first newborn screen C8 value averaged 11.2 umol/L (median=8.6, range 0.36–43.91, Std. Dev.=10.12). MCADD diagnostic testing was documented to be done for the majority of subjects in this cohort (Table 1).
Table 1.
Characteristics of the 221 study subjects
| Characteristic | N (Total=221) |
Percent |
|---|---|---|
| Age (in years) at IBEMC enrollment | ||
| < 1 | 100 | 45% |
| 1–2 | 55 | 25% |
| 3–5 | 39 | 18% |
| 6–10 | 24 | 11% |
| Missing | 3 | 1.4% |
| Race | ||
| American Indian or Alaska Native | 0 | 0% |
| Asian | 1 | 0.5% |
| Black or African American | 6 | 2.7% |
| Native Hawaiian or Other Pacific Islander | 0 | 0% |
| White | 189 | 86% |
| Two or More Races | 3 | 1.4% |
| Not Reported or Unknown | 22 | 10% |
| Ethnicity | ||
| Hispanic or Latino | 14 | 6% |
| Not Hispanic or Latino | 191 | 86% |
| Not Reported or Unknown | 16 | 7% |
| Gender | ||
| Male | 117 | 53% |
| Female | 104 | 47% |
| Biochemical diagnostic testing for MCADD | ||
| Any testing | ||
| Done | 202 | 91% |
| Not done | 2 | 1% |
| Plasma acylcarnitine profile | ||
| Done | 166 | 75% |
| Abnormal | 161 | 97% |
| Within normal limits | 4 | 2% |
| Urine organic acids | ||
| Done | 109 | 49% |
| Abnormal | 82 | 75% |
| Within normal limits | 20 | 18% |
| Urine acylglycine profile | ||
| Done | 16 | 7% |
| Abnormal | 15 | 94% |
| Within normal limits | 1 | 6% |
| Enzyme assay | ||
| Done | 0 | 0% |
| Fatty acid oxidation probe assay | ||
| Done | 5 | 2% |
| Abnormal | 5 | 100% |
| Genetic (DNA) testing for MCADD | ||
| Done | 186 | 84% |
| Not done | 21 | 10% |
Eligibility for enrollment in the IBEMC is dependent upon the individual having been given a diagnosis of an inborn error of metabolism, in this case, MCADD. Diagnostic methods and results were documented in the IBEM-IS for the majority of subjects with MCADD included in this study (Table 2).
Table 2.
MCADD diagnostic methods and results
| Genotype Category & Molecular Testing | Biochemical Diagnostic Testing for MCADD | ||||||
|---|---|---|---|---|---|---|---|
| aAbnormal | bNormal | cDone/No Info | dNot Done | eNo Info | Total N | ||
| AA, AB, AC, BB, OR CC | N | 130 | 3 | 11 | 2 | 3 | 149 |
| fC8 Median | 8.35 | 4.30 | 12.45 | 2.11 | 5.88 | ||
| fC8 Range | 0.36–43.91 | 0.40–13.40 | 1.74–30.59 | 1.03–3.19 | 1.04–28.79 | ||
| AD | N | 17 | 1 | 3 | 5 | 26 | |
| fC8 Median | 3.59 | 2.57 | 8.97 | 6.64 | |||
| fC8 Range | 0.72–38.00 | 6.20–13.88 | 0.72–26.12 | ||||
| CD | N | 1 | 1 | 2 | |||
| fC8 Median | 37.96 | 3.90 | |||||
| fC8 Range | |||||||
| DD Molecular Done |
N | 7 | 1 | 1 | 9 | ||
| fC8 Median | 12.45 | 12.55 | 7.06 | ||||
| fC8 Range | 1.67–26.90 | ||||||
| DD Molecular Not Done |
N | 11 | 10 | 21 | |||
| fC8 Median | 11.05 | 7.69 | |||||
| fC8 Range | 1.04–42.30 | 0.50–28.84 | |||||
| DD Molecular Unknown |
N | 4 | 2 | 8 | 14 | ||
| fC8 Median | 9.08 | 9.66 | 13.01 | ||||
| fC8 Range | 2.50–20.16 | 5.12–14.20 | 1.19–19.35 | ||||
|
| |||||||
| Total N | 170 | 5 | 27 | 2 | 17 | 221 | |
IBEM-IS data recorded at least one abnormal biochemical diagnostic test result
IBEM-IS data recorded at least one normal biochemical diagnostic test result, no abnormal biochemical diagnostic test result, with or without unknown/missing biochemical testing information
IBEM-IS data documented the completion of at least one biochemical diagnostic test, without documentation of actual result findings (normal or abnormal)
IBEM-IS data noted that biochemical diagnostic testing was not done
IBEM-IS data contained no information on whether biochemical diagnostic testing was done, nor any biochemical diagnostic testing results
C8 value (in umol/L) on first newborn screen
The majority of subjects had at least one clinical biochemical diagnostic laboratory test abnormality and/or two ACADM allele findings recorded in the IBEM-IS (N=189, 86%). Among the five subjects with normal biochemical diagnostic testing recorded, three had two ACADM allele findings, with genotype categories AA, CC, and CC. The two CC subjects had normal plasma acylcarnitine profiles, the AA subject had normal urine organic acids, and additional biochemical diagnostic testing results were not recorded for these three subjects. The remaining two subjects had one allele finding, both with normal urine organic acids and without other biochemical diagnostic testing results recorded.
Subjects’ average age at notification of the first abnormal newborn screen to a primary care or metabolic provider was 7.45 days (n=191, Std. Dev.=19.44). Average age since birth to initiation of intervention for MCADD was 8.11 days (n=197, Std. Dev.=19.41). Subjects averaged 16.77 days of age at the time of the first subspecialist visit (n=202, Std. Dev.=22.27).
The types and frequencies of select neonatal clinical characteristics and laboratory abnormalities in our dataset are summarized in Table 3.
Table 3.
Type and frequency of neonatal triggers, neonatal symptoms, and neonatal abnormal labs
| Neonatal Triggers, Symptoms and Abnormal Labs | Frequency of Appearance |
|---|---|
| aNeonatal Triggers (37 subjects) | |
| Poor Feeding | 20 |
| Prematurity (<37 weeks gestation) | 15 |
| Antibiotics | 12 |
| Respiratory distress | 7 |
| Infection/sepsis | 5 |
| Dehydration | 4 |
| Failure to thrive | 2 |
| Fever | 2 |
| Intralipids | 2 |
| Transient Tachypnea of the Newborn | 2 |
| Decreased oxygen saturation, fasted, loose stools, mild gastroesophageal reflux, poor growth, poor latch, vomiting, gavage feeding | 1 each |
| bNeonatal Symptoms (28 subjects) | |
| Lethargy | 13 |
| Distress | 9 |
| Tachypnea | 8 |
| Hypoglycemia | 5 |
| Hypothermia | 3 |
| Hypotonia | 3 |
| Irritability | 3 |
| Cardiomyopathy | 2 |
| Sleepy | 2 |
| Apnea, hepatomegaly, limp, metabolic acidosis, Echo: mild left hypertrophy, pallor, seizure, sweaty, tachycardia, temperature instability | 1 each |
| cNeonatal Abnormal Labs (21 subjects) | |
| Hypoglycemia | 20 |
| Elevated liver function tests | 7 |
| Metabolic acidosis | 6 |
| Elevated uric acid | 3 |
| Low Co2 | 2 |
| Hyperuricemia | 2 |
| Elevated C reactive protein, abnormal carnitine level, abnormal CMP, elevated BUN, elevated CK, elevated creatinine, hyperammonemia, ketonuria, slight elevation ALT | 1 each |
neonatal complications and interventions suggestive of underlying health complications in the data determined by clinician authors as most likely to result in potential MCADD symptoms.
neonatal symptoms in the data determined by clinician authors as consistent with MCADD, many based on reports of symptoms manifested in individuals affected with MCADD. IBEM-IS data entry does not require clinician specification of whether a subject’s symptoms were ultimately attributed to or related to the particular IBEM diagnosis.
neonatal laboratory test abnormalities in the data determined by clinician authors to be of potential concern in the context of MCADD (excluding newborn screening and MCADD diagnostic biochemical and molecular test results).
C8 values and age at first visit were negatively correlated (p = 0.001, Spearman’s rho = −0.227) indicating subjects seen by subspecialists sooner had higher C8 levels. Generalized linear regression results confirmed the negative correlation; for every one unit increase in C8, age at first visit decreased by 2% (p < 0.001). While the correlation was stronger (more negative) in subjects who had neonatal symptoms or neonatal abnormal labs than for subjects who did not, the difference in the magnitude of the correlation was not statistically significant.
There was no difference in C8 values between males and females (mean rank=112 vs. 110, p=0.76), nor between breastfed-only subjects and those whose neonatal diet contained other types of nutrition such as formula, total parenteral nutrition, and/or intralipids, plus or minus breast milk (mean rank=90 vs. 84, p=0.43). Subjects with birth weight less than or equal to 2.5 kg had significantly lower C8 values than the rest (mean rank=73 vs. 109, p=0.04). Significantly higher C8 values were found in subjects with neonatal symptoms (mean rank=114 vs. 86, p=0.008), and in subjects with neonatal abnormal labs (mean rank=59 vs. 41, p=0.003). There was no significant difference in C8 values between subjects with and without neonatal triggers (mean rank=107 vs. 98, p=0.388) (Table 4), although subjects with neonatal triggers were more likely to have neonatal symptoms and neonatal abnormal labs (p<0.001 in both tests, results not shown).
Table 4.
Comparison of the first newborn screen C8 value by select characteristics
| Factor | Mean C8 | Median C8 | Std. Dev. From Mean | Mean Rank |
|---|---|---|---|---|
| Gender | ||||
| Female (n=104) | 10.15 | 8.32 | 8.25 | 109.61 |
| Male (n=117) | 12.13 | 8.69 | 11.48 | 112.24 |
| Breastfed Only | ||||
| Yes (n=97) | 12.16 | 10.28 | 10.24 | 89.66 |
| No (n=76) | 10.96 | 8.24 | 10.36 | 83.61 |
| Low Birthweight | ||||
| Yes (n=13) | 7.51 | 2.85 | 10.54 | 73 |
| No (n=200) | 11.58 | 8.93 | 10.07 | 109.21 |
| Neonatal Triggers | ||||
| Yes (n=37) | 13.24 | 11.7 | 11.80 | 106.84 |
| No (n=161) | 11.00 | 8.6 | 9.77 | 97.81 |
| Neonatal Symptoms | ||||
| Yes (n=28) | 16.32 | 13.35 | 11.99 | 114.36 |
| No (n=152) | 10.48 | 8.37 | 9.62 | 86.11 |
| Neonatal Abnormal Labs | ||||
| Yes (n=21) | 18.46 | 14.93 | 12.42 | 59.45 |
| No (n=68) | 10.12 | 8.32 | 9.45 | 40.54 |
| Genotype (Four Categories) | ||||
| AA (n=69) | 15.76 | 13.36 | 10.76 | 96.00 |
| AB (n=18) | 11.89 | 11.00 | 10.24 | 80.72 |
| AC (n=49) | 5.73 | 3.65 | 6.51 | 49.24 |
| CC (n=12) | 4.63 | 3.37 | 4.31 | 44.67 |
| Genotype (Two Categories) | ||||
| AA&AB (n=87) | 14.96 | 13.30 | 10.71 | 92.84 |
| AC&CC (n=61) | 6.83 | 3.72 | 6.13 | 48.34 |
C8 values significantly differed among the four genotype categories (p<0.001). Post hoc pairwise comparisons showed C8 values significantly higher in the AA genotype group than in the AC (adjusted p<0.001) and CC (adjusted p=0.001) groups; and C8 values significantly higher in the AB genotype group than in the AC group (adjusted p=0.046). Although the data suggested a difference in C8 values between the AB genotype and the CC genotype groups, the test failed to reach statistical significance (adjusted p=0.144), which may be due to low power. C8 values were significantly higher in the AA&AB group than in the AC&CC group (p<0.001). Figure 1 shows the mean, median, and rank of C8 value for the four-category and two-category genotype variables.
Figure 1.
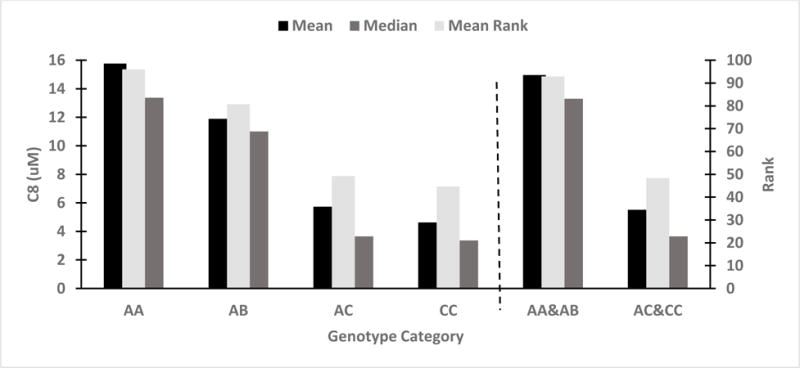
C8 comparisons by genotype category
For subjects with recorded and decipherable ACADM allele findings in the IBEM-IS, the c.985A>G mutation was most frequently appearing. The c.199T>C allele was the next most frequently appearing, with twelve subjects compound heterozygous for this allele and the c.985A>G mutation, and two subjects heterozygous for this allele and two other C type alleles. Subjects with the c.985A>G/c.199T>C genotype had first newborn screen C8 values ranging from 1.28–5.43 umol/L) and either no or unknown triggers, symptoms or abnormal labs in the first 28 days of life. The other two subjects with the c.199T>C/other C type genotype had first newborn screen C8 values of ≤0.6 umol/L, and had no triggers, symptoms or abnormal labs in the first 28 days of life.
Additionally, the proportions of subjects with genotype category AA or AB were significantly higher in subjects who had neonatal symptoms, neonatal abnormal labs, or neonatal triggers than the proportions in subjects recorded as asymptomatic (91% vs 52%, p=0.001, Phi=0.298)., without abnormal labs (88% vs 58%, p=0.032, Phi=0.275),, or without triggers during neonatal period (81% vs 55%, p=0.009, Phi=0.227). Further analysis demonstrated that both the two-category genotype variable and C8 were significant predictors of the likelihood of having neonatal symptoms. For every one unit increase in C8 value, the odds of having neonatal symptoms increased by 6% (p=0.016, EXP(B)=1.06). The odds of having neonatal symptoms in subjects with AA or AB genotype was 4.93 times of that in subjects with AC, or CC (p=0.050, EXP(B)=4.93).
4. CONCLUSIONS
This collaborative study is the first in the United States to describe health associations of a large cohort of newborn-screened neonates with MCADD. The IBEM-IS platform was designed to allow examination of complex associations between newborn screening results, clinical conditions and laboratory findings of individuals with inborn errors of metabolism. The IBEM-IS data increases our understanding of interactions with public health and clinical systems related to the notification and follow-up of abnormal newborn screening results for children with rare inborn errors of metabolism. The opportunity to examine a large cohort of newborn screened individuals with MCADD expands understanding of factors associated with their health in the first 28 days of life.
In 2014, the Society of Inherited Metabolic Disorders identified MCADD as one of several critical conditions requiring immediate notification of the health care provider upon ascertainment of an abnormal newborn screening result (36). Recently, the Advisory Committee on Heritable Disorders in Newborns and Children recommended that presumptive positive screening results for time-critical conditions be immediately reported to the child’s health care provider and by no later than 5 days of life (37). Our subjects had a longer documented mean age (7.45 days) at the time of newborn screen result notification indicating that improvement in timely notification must remain a priority to minimize risks of symptom initiation in these vulnerable newborns. Initiation of MCADD intervention occurred at a mean age of 8 days in our cohort. The close proximity in time of notification and intervention reflects the priority of clinicians to intervene in the care of newborns with possible MCADD as soon as possible.
In an attempt to determine if poor initiation of breast-feeding might be a risk factor for neonates with this condition, we specifically queried whether the diet of the neonates was associated with C8 values. Despite the failure to observe an association between high C8 values and exclusive breast-feeding in this data analysis, neonates who are exclusively breast-fed and in whom initiation of feeding is problematic may well be at additional risk for decompensation. Our observations cannot rule out poor breastfeeding initiation as a risk factor without additional information about this specific issue.
C8 values >0.3 umol/L along with additional results of MCADD-related ratios have been considered by some as indicative of MCADD by neonatal screening (20) (38). A worldwide collaborative project looking at the clinical validation of the cutoff target range of C8 in tandem mass spectrometry newborn screening describes disorder ranges for acylcarnitines and related ratios in MCADD, creating a tool for assessing screening results (39). Although all subjects in our cohort had a first newborn screen C8 value ≥0.36 umol/L, data available did not uniformly include additional newborn screening acylcarnitine values or ratios, preventing inclusion in our data analysis. The significant associations we found between the first newborn screen C8 value and low birth weight, symptoms, and clinical lab abnormalities for neonates with MCADD highlight the importance of providing quantitative screening result data to clinicians caring for children with abnormal newborn screen results for MCADD. Clinicians receiving such results should view very high C8 values as a signal for increased concern for symptomatic presentation of the condition.
Overrepresentation of infants with flagged newborn screening acylcarnitine values among infants in neonatal intensive care or with very low birth weight has been reported (10). Distribution of blood spot C8 concentrations did not vary greatly by birth weight in another study (40). In our study, neonates with MCADD in the low birth weight (<=2.5 kg) group had significantly lower C8 values. Although some of our low birth weight subjects had neonatal triggers, most had neither neonatal symptoms nor neonatal abnormal labs, and 7 out of 13 were homozygous for the c.985A>G ACADM mutation. These findings suggest that low birth weight itself may be a factor associated with lower newborn screen C8 values than might otherwise be expected given risk factors such as a deleterious genotype. The relative lower C8 values in this group may be due to these babies with low birth weight already receiving medical care with adequate prevention of fasting to prevent MCADD-associated complications causing elevations of C8. Data collection regarding the timing of first newborn screen sampling was a relatively recent addition to the IBEM-IS. Therefore, data on age at first newborn screen bloodspot collection was unavailable for most of our subjects. This is an important limitation to the conclusions of our study, given the findings of others regarding the relationship of C8 values and age at sample timing for infants with MCADD, as previously noted (10).
Our findings support the work of others demonstrating significant associations between higher C8 newborn screen values and homozygosity for the c.985A>G mutation as well as higher C8 newborn screen values in the presence of other severe ACADM mutations. Conservative categorization of B genotype alleles may have influenced our results. Some C genotype alleles may be more deleterious than currently categorized, solely supported by our literature search, a strategy chosen to minimize the risk that our conclusions overemphasize the association between deleterious mutations and increasing C8 values.
Although this study includes information about a very large number of children with MCADD, there are important limitations in considering our conclusions. This cohort does not represent the full denominator of newborn-screened children diagnosed with MCADD in the catchment area of the participating IBEMC centers. Study limitations include the potential for selection bias in subject enrollment. We also accepted the premise that all subjects assigned the condition MCADD in the IBEM-IS are truly affected, and most but not all subjects in this study had IBEM-IS documentation of at least one biochemical diagnostic testing abnormality and/or two ACADM allele findings. While the IBEM-IS does not mandate documentation of the rationale for individual diagnostic testing decisions and practices, we note that of the 10% of subjects with ACADM molecular testing recorded as “not done” at the time of data extraction, all are minors and all had MCADD biochemical diagnostic testing performed. Over half of them had at least one biochemical diagnostic testing abnormality recorded, and the remainder of those subjects had wide ranging (0.50–28.84 umol/L) first newborn screen C8 values but no MCADD biochemical diagnostic testing results documented in the IBEM-IS. Finally, though the data collection tools were designed to primarily elicit fixed responses, rare fields require free-text responses (for example, genotype) and few fields obligate data entry, yielding the potential for partial or otherwise inaccurate entry of information. There is also the potential for data entry errors in the IBEM-IS.
Additional work by the IBEMC is needed to further clarify and understand the significance of the 30 additional different ACADM alleles documented in the IBEM-IS for which published literature referencing the finding was not found. Such work ultimately may or may not support the genotype allele categorization strategy used in this study, and could potentially improve understanding of genotype-phenotype correlations. Importantly, this cohort of newborn-screened subjects allows for the observation of longer-term health outcomes for individuals with MCADD identified early in life.
Highlights.
Retrospective analysis of 221 newborn-screened subjects with MCAD deficiency (76)
NBS C8 and genotype were significant predictors of having neonatal symptoms (75)
Symptomatic neonates were more likely to have at least one copy of 985A>G mutation (82)
Neonates with select triggers were more likely to have symptoms (64)
The IBEM-IS is a platform to better understand newborn-screened conditions (74)
Acknowledgments
Research reported in this publication was supported by the Eunice Kennedy Shriver National Institute of Child Health and Development (NICHD), National Institutes of Health under award number 5R01HD069039. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This research was facilitated by the Newborn Screening Translational Research Network (“NBSTRN”), which is funded by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (HHSN275201300011C).
This research was facilitated by the Health Resource and Services Administration (HRSA) Maternal and Child Health Bureau (MCHB) Regional Genetic and Newborn Screening Services Collaboratives, Heritable Disorders Program through grants to: Region 2 - New York Mid-Atlantic Consortium for Genetic and Newborn Screening Services (NYMAC) (H46MC24094), Region 4 Midwest Genetics and Newborn Screening Collaborative (H46MC24092), Region 5 Heartland Genetic Services Collaborative (H46MC24089), and Region 6 - Mountain States Genetics Regional Collaborative (H46MC24095).
Study data were collected and managed using REDCap (Research Electronic Data Capture) tools hosted at the Michigan Public Health Institute (MPHI) (13).
References
- 1.Feuchtbaum L, Carter J, Dowray S, Currier RJ, Lorey F. Birth prevalence of disorders detectable through newborn screening by race/ethnicity. Genet Med. 2012 Nov;14(11):937–45. doi: 10.1038/gim.2012.76. [DOI] [PubMed] [Google Scholar]
- 2.Matern D, Rinaldo P. Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 2000. Apr 20, [updated 2015 Mar 5] 1993–2016. Available from http://www.ncbi.nlm.nih.gov/books/NBK1424/ [PubMed] [Google Scholar]
- 3.Wilson CJ, Champion MP, Collins JE, Clayton PT, Leonard JV. Outcome of medium chain acyl-CoA dehydrogenase deficiency after diagnosis. Arch Dis Child. 1999 May;80(5):459–62. doi: 10.1136/adc.80.5.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andresen BS, Lund AM, Hougaard DM, et al. MCAD deficiency in Denmark. Mol Genet Metab. 2012 Jun;106(2):175–88. doi: 10.1016/j.ymgme.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 5.U.S. Department of Health and Human Services Advisory Committee on Heritable Disorders in Newborns and Children. Recommended Uniform Screening Panel. www.hhs.gov. [Online] [Cited: Mar. 25, 2016] http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/recommendedpanel/index.html.
- 6.NewSTEPs. Screened Conditions Report. www.newsteps.org. [Online] [Cited: Mar. 25, 2016] https://data.newsteps.org/newsteps-web/reports/screenedConditions/list.action.
- 7.Millington DS, Kodo N, Norwood DL, Roe CR. Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis. 1990;13(3):321–4. doi: 10.1007/BF01799385. [DOI] [PubMed] [Google Scholar]
- 8.Rhead WJ. Newborn screening for medium-chain acyl-CoA dehydrogenase deficiency: a global perspective. 2006 Apr-Jun;29(2–3):370–7. doi: 10.1007/s10545-006-0292-1. [DOI] [PubMed] [Google Scholar]
- 9.Arnold GL, Saavedra-Matiz CA, Galvin-Parton PA, et al. Lack of genotype-phenotype correlations and outcome in MCAD deficiency diagnosed by newborn screening in New York State. Mol Genet Metab. 2010 Mar;99(3):263–8. doi: 10.1016/j.ymgme.2009.10.188. [DOI] [PubMed] [Google Scholar]
- 10.Zytkovicz TH, Fitzgerald EF, Marsden D, et al. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin Chem. 2001 Nov;47(11):1945–55. Nov. [PubMed] [Google Scholar]
- 11.Berry SA, Jurek AM, Anderson C, Bentler K, Region 4 Genetics Collaborative Priority 2 Workgroup The inborn errors of metabolism information system: a project of the Region 4 Genetics Collaborative Priority 2 workgroup. Genet Med. 2010 Dec;12(12 Suppl):S215–9. doi: 10.1097/GIM.0b013e3181fe5d23. Dec. [DOI] [PubMed] [Google Scholar]
- 12.Berry SA, Leslie ND, Edick MJ, Hiner S, Justice K, Cameron C. Inborn Errors of Metabolism Collaborative: large-scale collection of data on long-term follow-up for newborn-screened conditions. Genet Med. 2016 May 19; doi: 10.1038/gim.2016.57. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009 Apr;42(2):377–81. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iafolla AK, Thompson RJ, Jr, Roe CR. Medium-chain acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr. 1994 Mar;124(3):409–15. doi: 10.1016/s0022-3476(94)70363-9. Mar. [DOI] [PubMed] [Google Scholar]
- 15.Christodoulou J, Hoare J, Hammond J, Ip WC, Wilcken B. Neonatal onset of medium-chain acyl-coenzyme A dehydrogenase deficiency with confusing biochemical features. J Pediatr. 1995 Jan;126(1):65–8. doi: 10.1016/s0022-3476(95)70504-x. [DOI] [PubMed] [Google Scholar]
- 16.Gramer G, Haege G, Fang-Hoffmann J, et al. Medium-Chain Acyl-CoA Dehydrogenase Deficiency: Evaluation of Genotype-Phenotype Correlation in Patients Detected by Newborn Screening. JIMD Rep. 2015;23:101–12. doi: 10.1007/8904_2015_439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Derks TG, Reijngoud DJ, Waterham HR, et al. The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: clinical presentation and outcome. J Pediatr. 2006 May;148(5):665–670. doi: 10.1016/j.jpeds.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 18.Wilcken B, Hammond J, Silink M. Morbidity and mortality in medium chain acyl coenzyme A dehydrogenase deficiency. Arch Dis Child. 1994 May;70(5):410–2. doi: 10.1136/adc.70.5.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcì M, Ajovalasit P. Medium-Chain Acyl-CoA Dehydrogenase Deficiency in an Infant with Dilated Cardiomyopathy. Cardiol Res Pract. 2009;2009:281389. doi: 10.4061/2009/281389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pourfarzam M, Morris A, Appleton M, Craft A, Bartlett K. Neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency. Lancet. 2001 Sep 29;358(9287):1063–4. doi: 10.1016/S0140-6736(01)06199-2. [DOI] [PubMed] [Google Scholar]
- 21.Matsubara Y, Narisawa K, Miyabayashi S, Tada K, Coates PM. Molecular lesion in patients with medium-chain acyl-CoA dehydrogenase deficiency. Lancet. 1990 Jun 30;335(8705):1589. doi: 10.1016/0140-6736(90)91413-5. [DOI] [PubMed] [Google Scholar]
- 22.Yokota I, Indo Y, Coates PM, Tanaka K. Molecular basis of medium chain acyl-coenzyme A dehydrogenase deficiency. An A to G transition at position 985 that causes a lysine-304 to glutamate substitution in the mature protein is the single prevalent mutation. J Clin Invest. 1990 Sep;86(3):1000–3. doi: 10.1172/JCI114761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andresen BS, Dobrowolski SF, O’Reilly L, et al. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet. 2001 Jun;68(6):1408–18. doi: 10.1086/320602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Touw CM, Smit GP, de Vries M, et al. Risk stratification by residual enzyme activity after newborn screening for medium-chain acyl-CoA dehyrogenase deficiency: data from a cohort study. Orphanet J Rare Dis. 2012 May 25;7:30. doi: 10.1186/1750-1172-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith EH, Thomas C, McHugh D, et al. Allelic diversity in MCAD deficiency: the biochemical classification of 54 variants identified during 5 years of ACADM sequencing. Mol Genet Metab. 2010 Jul;100(3):241–50. doi: 10.1016/j.ymgme.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Hjelm LN, Chin EL, Hegde MR, Coffee BW, Bean LJ. A simple method to confirm and size deletion, duplication, and insertion mutations detected by sequence analysis. J Mol Diagn. 2010 Sep;12(5):607–10. doi: 10.2353/jmoldx.2010.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nielsen KB, Sørensen S, Cartegni L, et al. Seemingly neutral polymorphic variants may confer immunity to splicing-inactivating mutations: a synonymous SNP in exon 5 of MCAD protects from deleterious mutations in a flanking exonic splicing enhancer. Am J Hum Genet. 2007 Mar;80(3):416–32. doi: 10.1086/511992. Erratum in: Am J Hum Genet. 2007 Apr;80(4):816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.ter Veld F, Mueller M, Kramer S, et al. A novel tandem mass spectrometry method for rapid confirmation of medium- and very long-chain acyl-CoA dehydrogenase deficiency in newborns. PLoS One. 2009 Jul 30;4(7):e6449. doi: 10.1371/journal.pone.0006449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andresen BS, Bross P, Udvari S, et al. The molecular basis of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in compound heterozygous patients: is there correlation between genotype and phenotype? Hum Mol Genet. 1997 May;6(5):695–707. doi: 10.1093/hmg/6.5.695. [DOI] [PubMed] [Google Scholar]
- 30.Maier EM, Liebl B, Röschinger W, et al. Population spectrum of ACADM genotypes correlated to biochemical phenotypes in newborn screening for medium-chain acyl-CoA dehydrogenase deficiency. Hum Mutat. 2005 May;25(5):443–52. doi: 10.1002/humu.20163. [DOI] [PubMed] [Google Scholar]
- 31.Grünert SC, Wehrle A, Villavicencio-Lorini P, et al. Medium-chain acyl-CoA dehydrogenase deficiency associated with a novel splice mutation in the ACADM gene missed by newborn screening. BMC Med Genet. 2015 Jul 30;16:56. doi: 10.1186/s12881-015-0199-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andresen BS, Bross P, Jensen TG, et al. A rare disease-associated mutation in the medium-chain acyl-CoA dehydrogenase (MCAD) gene changes a conserved arginine, previously shown to be functionally essential in short-chain acyl-CoA dehydrogenase (SCAD) Am J Hum Genet. 1993 Sep;53(3):730–9. [PMC free article] [PubMed] [Google Scholar]
- 33.Waddell L, Wiley V, Carpenter K, et al. Medium-chain acyl-CoA dehydrogenase deficiency: genotype-biochemical phenotype correlations. Mol Genet Metab. 2006 Jan;87(1):32–9. doi: 10.1016/j.ymgme.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 34.Yokota I, Coates PM, Hale DE, Rinaldo P, Tanaka K. Molecular survey of a prevalent mutation, 985A-to-G transition, and identification of five infrequent mutations in the medium-chain Acyl-CoA dehydrogenase (MCAD) gene in 55 patients with MCAD deficiency. Am J Hum Genet. 1991 Dec;49(6):1280–91. [PMC free article] [PubMed] [Google Scholar]
- 35.Koster KL, Sturm M, Herebian D, Smits SH, Spiekerkoetter U. Functional studies of 18 heterologously expressed medium-chain acyl-CoA dehydrogenase (MCAD) variants. J Inherit Metab Dis. 2014 Nov;37(6):917–28. doi: 10.1007/s10545-014-9732-5. [DOI] [PubMed] [Google Scholar]
- 36.Society for Inherited Metabolic Disorders. SIMD Position Statement: Identifying abnormal newborn screens requiring immediate notification of the health care provider. www.simd.org. [Online] Aug. 21, 2014[Cited: Mar. 25, 2016] http://www.simd.org/Issues/SIMD%20NBS%20Critical%20Conditions%20policy%20statement.pdf.
- 37.NewSTEPs. Update: Suggested Recommendations for Timeliness in Newborn Screening. www.newsteps.org. [Online] Mar. 2015[Cited: Mar. 25, 2016] https://www.newsteps.org/news-and-education/news/update-suggested-recommendations-timeliness-newborn-screening.
- 38.Chace DH, Hillman SL, Van Hove JL, Naylor EW. Rapid diagnosis of MCAD deficiency: quantitative analysis of octanoylcarnitine and other acylcarnitines in newborn blood spots by tandem mass spectrometry. Clin Chem. 1997 Nov;43(11):2106–13. [PubMed] [Google Scholar]
- 39.McHugh D, Cameron CA, Abdenur JE, et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet Med. 2011 Mar;13(3):230–54. doi: 10.1097/GIM.0b013e31820d5e67. [DOI] [PubMed] [Google Scholar]
- 40.Carpenter K, Wiley V, Sim KG, Heath D, Wilcken B. Evaluation of newborn screening for medium chain acyl-CoA dehydrogenase deficiency in 275 000 babies. Arch Dis Child Fetal Neonatal Ed. 2001 Sep;85(2):F105–9. doi: 10.1136/fn.85.2.F105. [DOI] [PMC free article] [PubMed] [Google Scholar]