
Figure 1.
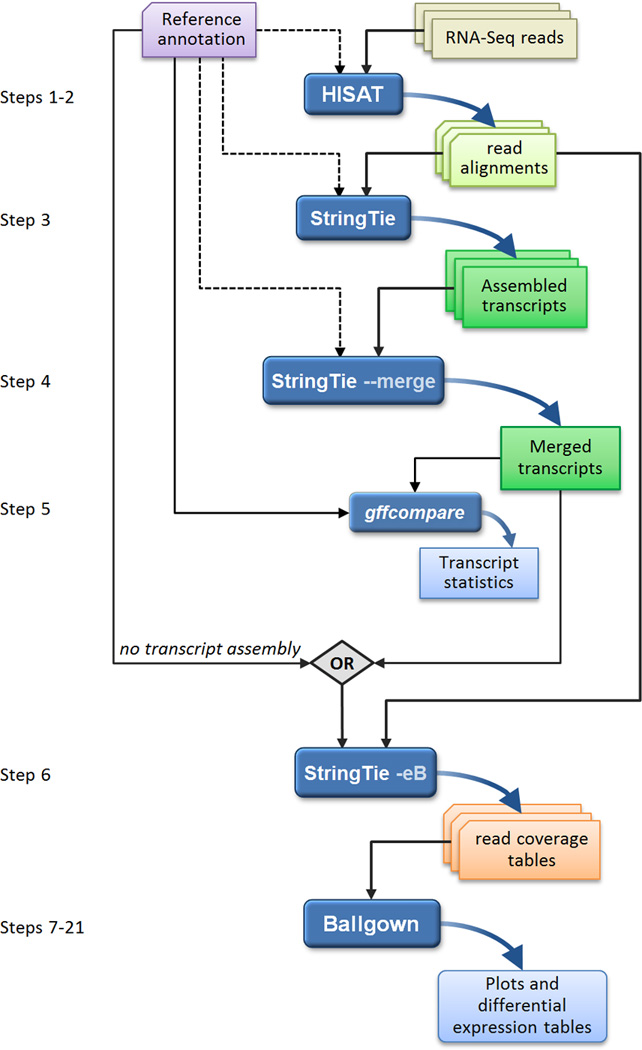
An overview of the "new Tuxedo" protocol. In an experiment involving multiple RNA-seq data sets, reads are first mapped to the genome using HISAT (Steps 1–2). Annotation of reference genes and transcripts can be provided as input, but this is optional, as indicated by the dotted line. The alignments are then passed to StringTie (Step 3), which assembles and quantifies the transcripts in each sample. (In the alternative protocol, the alignments from Step 2 are passed directly to Step 6, skipping all assembly steps. Step 6 will then estimate abundance only for known, annotated transcripts.) After initial assembly, the assembled transcripts are merged together (Step 4) by a special StringTie module, which creates a uniform set of transcripts for all samples. StringTie can use annotation in both of these steps, as shown by the dotted lines. The gffcompare program then compares the genes and transcripts to the annotation and reports statistics on this comparison (Step 5). In Step 6, StringTie processes the read alignments and either the merged transcripts or the reference annotation (through the diamond labeled "OR"). Using this input, StringTie re-estimates abundances where necessary and creates new transcript tables for input to Ballgown. Ballgown then compares all transcripts across conditions and produces tables and plots of differentially expressed genes and transcripts (Steps 7–21). Black and curved blue lines in the figure represent input to and output from the programs, respectively. Optional inputs are represented by dotted lines.