

Abstract
Meningococcal septic shock is associated with profound vasoplegia, early and severe myocardial dysfunction, and extended skin necrosis responsible for a specific clinical entity designated purpura fulminans (PF). PF represents 90% of fatal meningococcal infections. One characteristic of meningococcal PF is the myocardial dysfunction that occurs in the early phase of sepsis. Furthermore, hemodynamic studies have shown that the prognosis of meningococcal sepsis is directly related to the degree of impairment of cardiac contractility during the initial phase of the disease. To gain insight into a potential interaction of Neisseria meningitidis with the myocardial microvasculature, we modified a previously described humanized mouse model by grafting human myocardial tissue to SCID mice. We then infected the grafted mice with N. meningitides. Using the humanized SCID mouse model, we demonstrated that N. meningitidis targets the human myocardial tissue vasculature, leading to the formation of blood thrombi, infectious vasculitis, and vascular leakage. These results suggest a novel mechanism of myocardial injury in the course of severe N. meningitidis sepsis that is likely to participate in primary myocardial dysfunction.
INTRODUCTION
Neisseria meningitidis is a human pathogen responsible for life-threatening infections such as septic shock and meningitis, mainly in children and young adults (1). Meningococcal septic shock is associated with profound vasoplegia, early and severe myocardial dysfunction, and extended skin necrosis responsible for a specific clinical entity designated purpura fulminans (PF). PF represents 90% of fatal cases of severe meningococcal infection (2).
Approximately 60% of patients admitted to intensive care units for severe sepsis or septic shock present a clinical picture of cardiac dysfunction responsible for a mortality rate ranging from 70 to 90%. On the other hand, mortality is 20% in septic patients without cardiovascular involvement (3, 4). A specificity of meningococcal PF is the myocardial dysfunction that occurs in the early phase of the sepsis and is often present from the start (5, 6). In contrast, myocardial dysfunction appears during the late phase of sepsis with septic shock caused by other Gram-negative infections. Furthermore, hemodynamic studies have shown that the prognosis of meningococcal sepsis is directly related to the degree of impairment of cardiac contractility during the initial phase of the disease (7, 8).
N. meningitidis interacts with endothelial cells of the microvasculature throughout the body (9, 10). N. meningitidis forms microcolonies on the apical surface of endothelial cells and manipulates cell signaling pathways to disrupt cellular and endothelial tissue architectures; the bacteria eventually cross the vascular endothelium and colonize the surrounding tissue or the adjacent organ (10–12). A direct interaction of N. meningitidis with the myocardial tissue and vasculature has not been demonstrated but is strongly suspected. Indeed, postmortem examination showed the presence of meningococci in the myocardial tissue associated with histological features of myocarditis (13). To gain insight into a potential interaction of N. meningitidis with the myocardial microvasculature, we modified a previously described humanized mouse model by grafting human myocardial tissue to SCID mice (14). Using this humanized SCID mouse model, we demonstrated that N. meningitidis targets the human myocardial tissue vasculature, leading to the formation of blood thrombi, infectious vasculitis, and vascular leakage. These results suggest a novel mechanism of myocardial tissue injury in the course of severe N. meningitidis sepsis that is likely to participate in primary myocardial dysfunction.
MATERIALS AND METHODS
Bacterial strains.
We used the serogroup C meningococcal strain designated 2C4.3 and a nonpiliated isogenic derivative (N. meningitidis ΔpilE) that was constructed by inserting a kanamycin resistance cassette in the pilE gene locus (15). The N. meningitidis strain 2C4.3, also known as clone 12 of the clinical N. meningitidis strain LNP8013, is a naturally occurring pilin antigenic variant of the original clinical isolate LNP8013, which expresses a pilin variant responsible for a high adhesive phenotype. N. meningitidis strains were stored frozen at −80°C in Giolitti-Cantoni broth (GCB) supplemented with 20% glycerol.
Mice.
Six-week-old CB17/Icr-Prkdcscid/IcrIcoCrl SCID female mice were purchased from Charles River Laboratories (Saint Germain sur l'Arbresle, France). Experimental procedures were performed under sterile conditions and in accordance with the guidelines of the Institut National de la Santé et de la Recherche Médicale (INSERM). The experimental protocol was approved by the Animal Experimentation Ethics Committee of the Université Paris Descartes (study registered under number 00309.0.02).
Graft protocol.
We used the experimental model described by Yan et al. (14) that was initially designed to study the expression and role of endothelial cell adhesion molecules for white blood cell migration. Myocardial tissues were obtained from pediatric patients undergoing cardiac surgery with myomectomy for left ventricular outflow tract obstruction in the Centre Chirurgical Marie Lannelongue (Clamard, France). In accordance with French legislation, human myocardial samples were obtained from patients on whose behalf legal tutors gave informed consent for participation in the study. The myocardial tissue was maintained on ice-cold cardioplegia medium (Custodiol HTK; Franz Köhler Chemie GmbH). Myocardial tissues were grafted under skin on the backs of SCID mice (16). Briefly, mice were prepared for transplantation by shaving the hair of the back and abdominal areas after an intraperitoneal (i.p.) injection of ketamine 100 mg/kg and xylazine 10 mg/kg using a pair of sponge forceps. Human myocardial tissue was placed onto the wound bed. The transplants were held in place with up to 6 nonabsorbable monofilament sutures, and the skin was then sutured above the transplant. Grafted mice were used for N. meningitidis infection experiments 15 days after human myocardial tissue transplantation.
Infection protocol and bacterial counts.
In vivo, iron-binding proteins such as transferrin and lactoferrin restrict the amount of ferric iron available in body fluids to a level that does not support meningococcal growth (17). To counteract these iron-deficient conditions, N. meningitidis has several iron acquisition systems, such as transferrin binding proteins A and B that are expressed when bacteria are grown under iron-deficient conditions (18). These receptors are specific to human transferrin; therefore, bacterial strains were grown under iron-deficient conditions before each infectious challenge, and human holotransferrin was added to the bacterial inoculum to optimize bacterial growth in vivo (19, 20). N. meningitidis strains were grown overnight at 37°C on GCB agar plates prepared without iron and supplemented with deferoxamine (Desferal; Novartis) at a final concentration of 15 μM. Kanamycin (100 μg/ml) was added to the medium to grow the kanamycin-resistant ΔpilE strain. Bacterial colonies were harvested and cultured in RPMI medium with 1% bovine serum albumin (BSA) and 0.06 μM deferoxamine with gentle agitation to reach the exponential growth phase. Bacteria were then concentrated by centrifugation and resuspended in physiological saline containing 20 mg/ml of human holotransferrin to promote bacterial growth in vivo (2914HT; R&D Systems) (18). Preliminary experiments showed that the inoculum necessary to obtain a sustained bacteremia by intravenous injection of bacteria was 100-fold-higher than that needed by the intraperitoneal route (data not shown). Mice were infected intraperitoneally with 0.5 ml of this bacterial suspension. To assess bacteremia in infected animals, 10 μl of blood was sampled using a heparinized hematocrit glass tube either after puncture of the lateral tail vein or at the time of death by intracardiac puncture after intraperitoneal injection of a lethal dose of ketamine and xylazine.
Bacterial counts were performed by plating serial dilutions of blood or of myocardial tissue graft homogenates on GCB agar plates. Results are expressed as the number of CFU per milliliter of blood.
Histology and immunofluorescence.
Mice were killed 24 h after infection. Mouse myocardial tissue grafts and mouse heart were removed, fixed in 10% buffered formalin, and embedded in paraffin. Hematoxylin, eosin, and safranin staining and immunofluorescence labeling were performed on 5-μm tissue sections using a Leica Bond-Max automated system (Leica Microsystems, Wetzlar, Germany). Tissues were fixed overnight in phosphate-buffered saline (PBS)–4% paraformaldehyde (PFA). Cells were incubated with primary antibodies at the recommended concentration in PBS containing 0.3% BSA for 1 h. After cells were washed three times in the same buffer, Alexa-conjugated phalloidin and 4′,6′-diamidino-2-phenylindole ([DAPI] 0.5 μg/ml) were added to Alexa-conjugated secondary antibodies for 1 h. After additional washings, coverslips were mounted in Mowiol.
For fluorescence microscopy polyclonal anti-PilE antiserum was used. Anti-rabbit Alexa 56 was used as a secondary antibody. All samples were mounted in Vectashield mounting reagent (Vector Laboratories, Eurobio ABCys, France).
Image acquisition was performed on a structured illumination microscope (Apotome 2; Zeiss) with a 63× (numerical aperture [NA], 1.4) oil objective. Images were collected using Zen 2012 (Zeiss) software. Figures were created using Fiji (version 1.43) software and the FigureJ (version 1.) plug-in. Final figures were created in Photoshop (Adobe, USA).
RESULTS
Human myocardial tissue xenograft in SCID mice is revascularized by mouse vessels.
N. meningitidis does not interact with nonhuman cells. In order to study the consequences of a potential interaction between N. meningitidis and myocardial microvasculature, we modified a previously described humanized mouse model by grafting human myocardial tissue to SCID mice. Human myocardial tissues were obtained from pediatric patients undergoing cardiac surgery with myomectomy for left ventricular outflow tract obstruction, and tissues were grafted less than 2 h after surgical removal. Our first goal was to assess whether the vascularization of human myocardial tissues connects with that of the mouse as in the case for human skin grafts. As shown in Fig. 1A, the macroscopic aspect of the graft at day 14 was satisfactory, showing well-vascularized tissue. Histological microscopic analysis of the graft showed normal vascularization with healthy cardiomyocytes surrounding the vessels. The architecture of cardiac cells and cardiac tissue was preserved, and there was no evidence of inflammatory cell infiltrates in the graft tissue (Fig. 1B). The vessels present in the graft are all microvessels with a diameter of less than 300 μm, and all of them presented the structure of a vein or a capillary with thin vascular walls without a layer of smooth muscle cells. We conclude that human myocardial tissues xenotransplanted into SCID mice are efficiently revascularized and that the tissue displays characteristics of human myocardial tissue.
FIG 1.
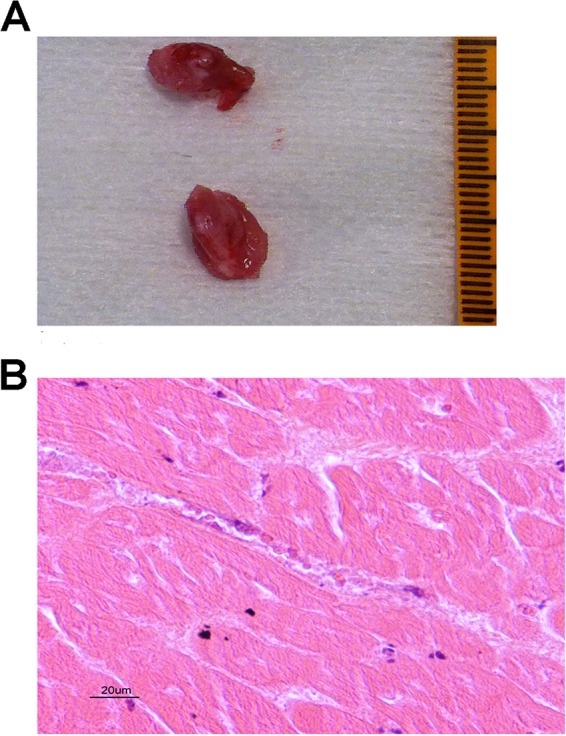
(A) Viability of the myocardial tissue xenograft, macroscopic aspect. (B) Viability of the myocardial tissue xenograft, microscopic aspect. Eosin-hematoxylin staining of the control graft shows normal myocardial muscular cells and normal myocardial muscle architecture. Graft vessels of human origin are perfused by red cells of mouse origin.
Neisseria meningitidis colonizes the myocardium venous and capillary microvessels.
Grafted mice were subsequently infected using 106 CFU i.p. as this inoculum produced reproducibly high and sustained bacteremia in infected animals. Twenty-four hours after infection, animals were sacrificed. At this time, the explanted grafts were colored and well perfused, with the aspect of healthy tissue.
Human myocardial tissue grafts were sampled and analyzed. All the vessels were colonized by N. meningitidis, while extracellular bacteria adhering to the intraluminal surface of endothelial cells involved in microcirculation in myocardial tissue were seen in all human myocardial tissue grafts sampled from infected animals (3/3 myocardial tissue grafts obtained from three different experiments) (Fig. 2; see also Fig. 4 and 5). All the colonized vessels were capillaries or venules with thin walls. N. meningitidis was dispersed as a layer of several strata of bacteria uniformly distributed on the inner face of the vessel. Bacteria underlined the entire endoluminal surface of vessels or in some circumstances completely occluded the vascular lumen (Fig. 2A and B).
FIG 2.
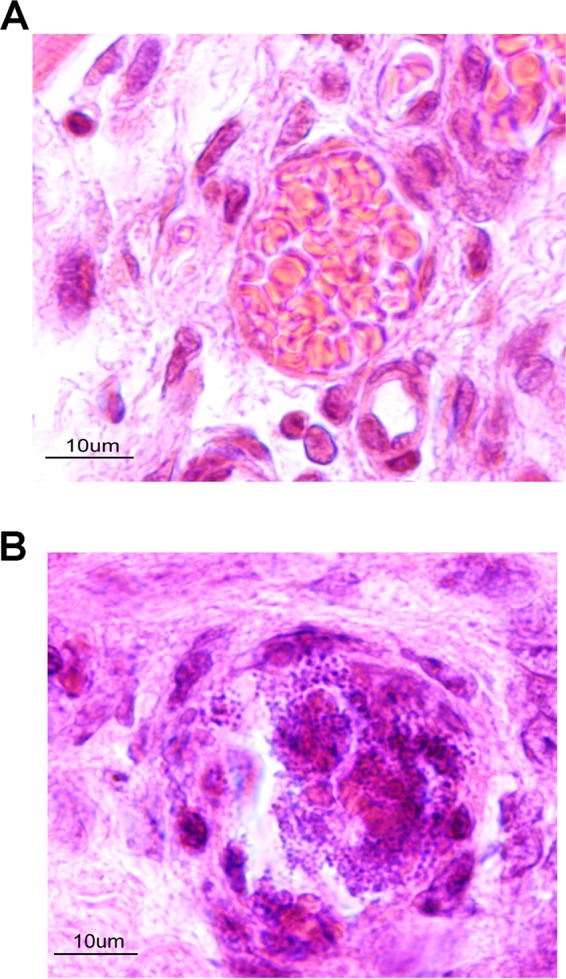
(A) Control graft of the noninfected animal showing a normal vessel of human origin in the graft tissue with circulating red blood cells of mouse origin in its lumen. (B) Graft vessel from an infected animal with a central infectious clot occupying the entire lumen of the vessel and a large amount of bacteria localized inside the clot and on the vessel's internal surface.
FIG 4.

(A) A vessel occluded by a septic thrombus composed of meningococcus and a clot of fibrin and platelets. Arrow 1 indicates the central clot mainly composed of red cells; arrow 2 indicates the ring of meningococci surrounding the clot; arrow 3 indicates meningococci outside the vascular lumen. (B) A vessel occluded by a septic thrombus composed of meningococcus and a clot of fibrin and platelets with vascular lumen filed by the meningococci. Arrow 1 indicates the central clot mainly composed of red cells; arrow 2 indicates the ring of meningococci surrounding the clot; arrow 3 indicates a rupture in the vascular endothelium, with red cells and bacteria passing in the surrounding tissue. (C) Multiple-field (4- by 2-field) reconstruction from Apotome 2 images. Scale bar, 20 μm. Images are as follows: upper left, tissue imaging using transmitted light; upper right, circling of the blood vessels; lower left, DAPI (blue) and 2C43 antibody (red) staining, with white arrows indicating the presence of N. meningitidis in the surrounding tissue; lower right, magnification of the boxed section in the lower left image with a central clot circled by a ring of N. meningitidis bacteria.
FIG 5.

(A) Massive infiltration of two vessels by polynuclear cells that results in the loss of their architecture and the surrounding tissue. (B) Multiple-field (4- by 2-field) reconstruction from Apotome 2 images. Scale bar, 20 μm. Images are as follows: upper left, tile scan tissue imaging using transmitted light; upper right, circling of the blood vessels; lower left, DAPI (blue) and 2C43 antibody (red) staining; lower right, magnification of the boxed area in the lower left image with a massive infiltration of the vessel by PMN. White arrows indicate N. meningitidis bacteria present in the surrounding tissue.
It should be pointed out that hematoxylin, eosin, and safranin staining of sections containing both human myocardial tissue grafts and subcutaneous mouse paniculus carnosus demonstrated that N. meningitidis interacted specifically with human graft vessels (Fig. 3). Furthermore, hearts of infected animals were sampled at the same time as the grafts and prepared according to the same protocol and did not show any vascular colonization by N. meningitidis, as expected (data not shown). Altogether these data demonstrated a specific interaction of N. meningitidis with the human myocardial vessels.
FIG 3.
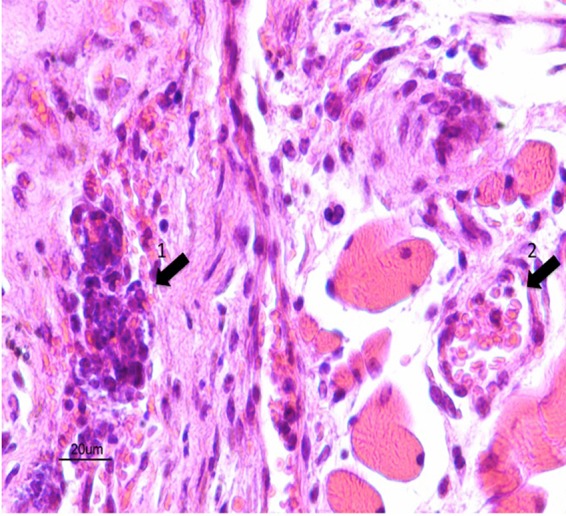
Arrow 1 is in the human graft showing a human vessel infiltrated by polynuclear cells and a total loss of the vessel architecture; arrow 2 is in surrounding mouse tissue showing a mouse vessel of normal aspect, emphasizing the human specificity of N. meningitidis infection and vasculitis.
N. meningitidis myocardium vascular colonization induced intravascular thrombosis and vascular endothelial destruction as well as vasculitis.
Two different types of vascular lesions were observed in the myocardial tissue xenograft. The most frequent type of vascular lesions (type I) were vessel occlusions by infectious clots (75% on a count of 100 vessels), which was mainly observed in vessels having an average diameter of 20 μm. These clots are composed of the aggregation of fibrin, blood cells, and bacteria (Fig. 2). Only a few vessels of the graft did not present any histological lesion (8% on a count of 100 vessels). These septic clots had a very peculiar organization. The red blood cells and fibrin clots were central to the vessels' lumens and were surrounded by a layer of bacteria adhering to the vascular endothelium (Fig. 4). The clots' structural organization suggests that the clot formation followed a centripetal process from the endothelium to the center of the vessel. This organization suggests that the bacterial interaction with the endothelium triggered the clotting process leading to the blood clot formation.
A second type of vascular lesion (type II) was observed less frequently (17% on a count of 100 vessels) and was characterized by smaller vessels with an average size of 15 μm (count of 100 vessels). These lesions corresponded to vasculitis with massive infiltration by polymorphonuclear cells (PMN), with destruction of the vascular wall and disappearance of the vascular lumen (Fig. 5A and B). Unlike what was seen in the type I lesions, few bacteria or bacterial clots are visible in these lesions. This loss of vascular wall integrity results in vascular leakage, leading to red cells infiltrating the perivascular space (Fig. 4B). It should be pointed out that in only 8% of the vessels were no lesions observed.
In many instances, but especially in type II lesions, N. meningitidis was found outside the vascular lumen and in the myocardial tissue surrounding the vessels, accompanied by red blood cells, which indicated an endothelial rupture (Fig. 4A and B). These images shown in Fig. 4 resemble those described in purpuric skin lesions that are characteristic of the meningococcal purpura fulminans (9). Moreover, some bacteria were localized between the vessels and most frequently in close proximity to polymorphonuclear cells (Fig. 4C and 5B). The colonization of the surrounding tissues by N. meningitidis is clearly visible adjacent to the vessels (Fig. 4C and 5B).
N. meningitidis adhesion to endothelial cells of the coronary microvascularization and infectious vasculitis require type IV pilus.
Type IV pili have a central role in meningococcal pathogenesis (15). In vitro and in vivo meningococcal adhesion to human endothelial cells has been shown to require type IV pili. We subsequently tested the ability of a capsulated nonpiliated derivative of N. meningitidis 2C4.3 (ΔpilE strain) to target the human myocardial tissue graft vasculature. As shown in Fig. 6, the nonpiliated strain failed to target the human myocardial tissue graft, as demonstrated by the absence of bacteria sticking to the vessel endothelium. In addition, no thrombosis or vascular lesions were observed in the graft microvasculature when grafts were infected by the nonpiliated strain, thus demonstrating that the histologic lesions observed in the vasculature of grafts infected with wild-type N. meningitidis are type IV pilus dependent.
FIG 6.

In the micrographs on the left, the upper panel is an image of a control graft with no infection, the central panel is an image of a graft infected with an isogenic mutant (N. meningitidis ΔpilE) and showing normal graft vessels and no inflammatory cells, and the lower panel is an image of a graft infected with wild-type strain 2C4.3 and showing N. meningitidis underlining the vascular endothelium (black arrow). The data in graph A indicate similar levels of bacteremia in mice at 1 (H1) and 6 (H6) hours after infection with the wild-type 2C4.3 N. meningitidis strain or with the ΔpilE derivative of 2C4.3 (NS, not significant; two mice per group). The data in graph B show that the bacterial load of the myocardial graft was lower in mice infected with the nonpiliated strain 24 h after infection (one sample of myocardial tissue analyzed for both conditions).
DISCUSSION
The model allowed, for the first time, an analysis of the intimate relations between N. meningitidis, coronary microcirculation, and myocardial tissue in vivo. Our results showed that N. meningitidis adhered to myocardial microvessel walls and induced septic vasculitis of the coronary microcirculation. Moreover, N. meningitidis induced septic thrombosis of the microvessels, followed by destruction of the vascular endothelium. The damage observed resembled that observed in the skin of patients with meningococcemia. Vascular lesions were similar to those described in patients' purpuric skin lesions specific to purpura fulminans, including thrombosis of vessels, perivascular infiltrates, and vascular leakage (Fig. 4) (10). Inflammatory cells, including mononuclear cells and neutrophils, surrounded capillaries located in the superficial and middermis and infiltrated their walls (Fig. 2A and B). A mutant of N. meningitidis, defective for the pilin PilE, did not cause any vasculitis, demonstrating that type IV pilin is required for vasculitis.
Our results give experimental evidence that a direct interaction between N. meningitidis and the heart takes place. To our knowledge, this represents the first report of myocardial tissue colonization by bacteria. Moreover, our findings corroborate the observations made by Monsalve et al. obtained from postmortem examination of three patients suffering from meningococcemia that showed the presence of myocarditis associated with the presence of Gram-negative diplococci (13). From this study it was hypothesized that N. meningitidis colonizes the myocardial tissue and induces tissue inflammation. Considering our results, it is likely that the mechanism of this colonization is an infectious vasculitis of the coronary microvascularization. Destruction of the vascular endothelium allowed N. meningitidis to enter the myocardial tissue, generating neutrophil inflow and subsequent local inflammation.
Diverse mechanisms underlying septic myocardial depression have been described previously (21, 22). Sepsis-induced cardiomyopathy appears to be an excessive physiological adaptation of the heart function to the sepsis and not a myocardial tissue injury. Indeed, clinical studies have shown a specific hemodynamic profile of the sepsis-induced cardiac depression, i.e., a transient and global defect of the heart function, sometimes profound, followed by full recovery (23). It was shown that myocardial ischemia and necrosis are not the mechanisms of myocardial septic dysfunction, the main hypothesis being a massive signaling to the heart muscle. This signaling would be responsible for hibernation-like conditions where the myocardial muscle downregulates its function in order to reduce energy expenditure, thus preventing activation of cardiomyocyte cell death pathways (22, 24). Endotoxins, Toll-like receptors (TLR), and cytokines mediate this signaling. Cytokines are produced and liberated from activated immune cells after contact with bacterial compounds. The signaling is systemic, via cytokines, as well as local, via TLR4 activation, leading to myocardial septic dysfunction (22, 24). TLR4 must be present on macrophage and neutrophils to cause myocyte dysfunction during endotoxemia (25, 26). Interleukin-6 has been shown to have an important role in myocardial depressant activity in children with meningococcal septic shock (6). Our results, showing a massive presence of N. meningitidis in the coronary microvascularization as well as in myocardial tissue, suggest that N. meningitidis septicemia may be associated with strong local signaling. Massive stimulation of the TLRs by N. meningitidis, emphasized by the local septic thrombi and increased concentration of N. meningitidis lipopolysaccharide (LPS) analog due to endothelium adherence of the bacteria, could generate such antigenic stimulation, resulting in strong innate immune signaling and myocardial dysfunction. Polynuclear infiltration of the myocardial muscle has been demonstrated to be of critical importance in the TLR4-mediated contractile dysfunction induced by sepsis (26, 27). Polynuclear infiltration is present in almost 20% of the vessels analyzed in our model and may also participate in myocardial dysfunction. This model will help provide a better understanding of the septic myocardial depression induced by N. meningitidis, but mouse and human differences in complement, binding receptors, and signaling receptors would probably limit its exploitation at the molecular level.
In the case of severe meningococcal infection, our results showed vascular damage of the coronary microcirculation, presence of massive amount of LPS, and infiltration of polymorphonuclear cells and bacteria in the myocardial tissue. Indeed, the histological lesions of the coronary microcirculation observed in our model mimic the lesions observed at the skin level in purpura fulminans. The results of our ex vivo experiments suggest that this damage participates in the specificity of the N. meningitidis-associated sepsis-induced cardiomyopathy that takes place early in the course of the disease and is usually severe. The exact nature of the signaling and its consequences would need further investigation, but the trigger of the vascular damage appears to be linked to the presence of type IV pili. Indeed, the role of type IV pili in the disorganization of the cell-cell junctions and in the opening of a paracellular route in brain capillaries has already been shown to allow N. meningitidis to cross the blood-brain barrier and invade the meninges and in the skin capillaries to induce the skin lesions typical of the disease (Fig. 6) (10, 12). All together, these results may explain the early and profound septic cardiac dysfunction associated with N. meningitidis septic shock.
Conclusion.
We describe for the first time a model of human cardiac muscle tissue xenograft in SCID mice providing a new tool to study intimate interaction of human pathogens with the human heart. Our work demonstrates the tropism of N. meningitidis for human coronary microcirculation in vivo and its pilus-mediated specificity. This interaction with human coronary microcirculation may play a role in the profound and early cardiac dysfunction that classically goes with severe meningococcal sepsis. Our model will allow a better understanding of the pathogenesis of septic heart dysfunction and will open the path to specific treatments of this major complication of severe meningococcal sepsis.
ACKNOWLEDGMENTS
We thank Virginie Lambert, Catherine Rucker, Gérard Pivert, Omar Aimer, John Rohde, and Isabelle Le Maner for technical assistance and advice.
We declare that we received no funding, that we have no conflicts of interest, and that no previous part of this work was presented elsewhere.
REFERENCES
- 1.Stephens DS, Greenwood B, Brandtzaeg P. 2007. Epidemic meningitis, meningococcaemia and Neisseria meningitidis. Lancet 369:2196–2210. doi: 10.1016/S0140-6736(07)61016-2. [DOI] [PubMed] [Google Scholar]
- 2.de Greeff SC, de Melker HE, Schouls LM, Spanjaard L, van Deuren M. 2008. Pre-admission clinical course of meningococcal disease and opportunities for the earlier start of appropriate intervention: a prospective epidemiological study on 752 patients in the Netherlands, 2003-2005. Eur J Clin Microbiol Infect Dis 27:985–992. doi: 10.1007/s10096-008-0535-1. [DOI] [PubMed] [Google Scholar]
- 3.Vieillard-Baron A, Caille V, Charron C, Belliard G, Page B, Jardin F. 2008. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med 36:1701–1706. doi: 10.1097/CCM.0b013e318174db05. [DOI] [PubMed] [Google Scholar]
- 4.Makwana N, Baines PB. 2005. Myocardial dysfunction in meningococcal septic shock. Curr Opin Crit Care 11:418–423. doi: 10.1097/01.ccx.0000176699.51456.13. [DOI] [PubMed] [Google Scholar]
- 5.Thiru Y, Pathan N, Bignall S, Habibi P, Levin M. 2000. A myocardial cytotoxic process is involved in the cardiac dysfunction of meningococcal septic shock. Crit Care Med 28:2979–2983. doi: 10.1097/00003246-200008000-00049. [DOI] [PubMed] [Google Scholar]
- 6.Pathan N, Hemingway CA, Alizadeh AA, Stephens AC, Boldrick JC, Oragui EE, McCabe C, Welch SB, Whitney A, O'Gara P, Nadel S, Relman DA, Harding SE, Levin M. 2004. Role of interleukin 6 in myocardial dysfunction of meningococcal septic shock. Lancet 363:203–209. doi: 10.1016/S0140-6736(03)15326-3. [DOI] [PubMed] [Google Scholar]
- 7.Boucek MM, Boerth RC, Artman M, Graham TP Jr, Boucek RJ Jr. 1984. Myocardial dysfunction in children with acute meningococcemia. J Pediatr 105:538–542. doi: 10.1016/S0022-3476(84)80416-3. [DOI] [PubMed] [Google Scholar]
- 8.Pathan N, Faust SN, Levin M. 2003. Pathophysiology of meningococcal meningitis and septicaemia. Arch Dis Child 88:601–607. doi: 10.1136/adc.88.7.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernard SC, Simpson N, Join-Lambert O, Federici C, Laran-Chich MP, Maïssa N, Bouzinba-Ségard H, Morand PC, Chretien F, Taouji S, Chevet E, Janel S, Lafont F, Coureuil M, Segura A, Niedergang F, Marullo S, Couraud PO, Nassif X, Bourdoulous S. 2014. Pathogenic Neisseria meningitidis utilizes CD147 for vascular colonization. Nat Med 20:725–731. doi: 10.1038/nm.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Join-Lambert O, Lecuyer H, Miller F, Lelievre L, Jamet A, Furio L, Schmitt A, Pelissier P, Fraitag S, Coureuil M, Nassif X. 2013. Meningococcal interaction to microvasculature triggers the tissular lesions of purpura fulminans. J Infect Dis 208:1590–1597. doi: 10.1093/infdis/jit301. [DOI] [PubMed] [Google Scholar]
- 11.Coureuil M, Lécuyer H, Scott MG, Boularan C, Enslen H, Soyer M, Mikaty G, Bourdoulous S, Nassif X, Marullo S. 2010. Meningococcus hijacks a β2-adrenoceptor/β-arrestin pathway to cross brain microvasculature endothelium. Cell 143:1149–1160. doi: 10.1016/j.cell.2010.11.035. [DOI] [PubMed] [Google Scholar]
- 12.Coureuil M, Mikaty G, Miller F, Lécuyer H, Bernard C, Bourdoulous S, Duménil G, Mège RM, Weksler BB, Romero IA, Couraud PO, Nassif X. 2009. Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science 325:83–87. doi: 10.1126/science.1173196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monsalve F, Rucabado L, Salvador A, Bonastre J, Cuñat J, Ruano M. 1984. Myocardial depression in septic shock caused by meningococcal infection. Crit Care Med 12:1021–1023. doi: 10.1097/00003246-198412000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Yan HC, Juhasz I, Pilewski J, Murphy GF, Herlyn M, Albelda SM. 1993. Human/severe combined immunodeficient mouse chimeras. An experimental in vivo model system to study the regulation of human endothelial cell-leukocyte adhesion molecules. J Clin Invest 91:986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nassif X, Lowy J, Stenberg P, O'Gaora P, Ganji A, So M. 1993. Antigenic variation of pilin regulates adhesion of Neisseria meningitidis to human epithelial cells. Mol Microbiol 8:719–725. doi: 10.1111/j.1365-2958.1993.tb01615.x. [DOI] [PubMed] [Google Scholar]
- 16.Barrandon Y, Li V, Green H. 1988. New techniques for the grafting of cultured human epidermal cells onto athymic animals. J Investig Dermatol 91:315–318. doi: 10.1111/1523-1747.ep12475646. [DOI] [PubMed] [Google Scholar]
- 17.Calver GA, Kenny CP, Kushner DJ. 1979. Inhibition of the growth of Neisseria meningitidis by reduced ferritin and other iron-binding agents. Infect Immun 25:880–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simonson C, Brener D, DeVoe IW. 1982. Expression of a high-affinity mechanism for acquisition of transferrin iron by Neisseria meningitidis. Infect Immun 36:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holbein BE. 1980. Iron-controlled infection with Neisseria meningitidis in mice. Infect Immun 29:886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holbein BE. 1981. Enhancement of Neisseria meningitidis infection in mice by addition of iron bound to transferrin. Infect Immun 34:120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaky A, Deem S, Bendjelid K, Treggiari MM. 2014. Characterization of cardiac dysfunction in sepsis: an ongoing challenge. Shock 41:12–24. doi: 10.1097/SHK.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 22.Rudiger A, Singer M. 2007. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med 35:1599–1608. doi: 10.1097/01.CCM.0000266683.64081.02. [DOI] [PubMed] [Google Scholar]
- 23.Parker MM, Shelhamer JH, Bacharach SL, Green MV, Natanson C, Frederick TM, Damske BA, Parrillo JE. 1984. Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med 100:483–490. doi: 10.7326/0003-4819-100-4-483. [DOI] [PubMed] [Google Scholar]
- 24.Rudiger A, Singer M. 2013. The heart in sepsis: from basic mechanisms to clinical management. Curr Vasc Pharmacol 11:187–195. doi: 10.2174/1570161111311020008. [DOI] [PubMed] [Google Scholar]
- 25.Arslan F, de Kleijn DP, Pasterkamp G. 2011. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol 8:292–300. doi: 10.1038/nrcardio.2011.38. [DOI] [PubMed] [Google Scholar]
- 26.Tavener SA, Long EM, Robbins SM, McRae KM, Van Remmen H, Kubes P. 2004. Immune cell Toll-like receptor 4 is required for cardiac myocyte impairment during endotoxemia. Circ Res 95:700–707. doi: 10.1161/01.RES.0000144175.70140.8c. [DOI] [PubMed] [Google Scholar]
- 27.Baumgarten G, Knuefermann P, Schuhmacher G, Vervölgyi V, von Rappard J, Dreiner U, Fink K, Djoufack C, Hoeft A, Grohé C, Knowlton AA, Meyer R. 2006. Toll-like receptor 4, nitric oxide, and myocardial depression in endotoxemia. Shock 25:43–49. doi: 10.1097/01.shk.0000196498.57306.a6. [DOI] [PubMed] [Google Scholar]