

Abstract
Astrocytes react to brain injury in a heterogeneous manner with only a subset resuming proliferation and acquiring stem cell properties in vitro. In order to identify novel regulators of this subset, we performed genomewide expression analysis of reactive astrocytes isolated 5 days after stab wound injury from the gray matter of adult mouse cerebral cortex. The expression pattern was compared with astrocytes from intact cortex and adult neural stem cells (NSCs) isolated from the subependymal zone (SEZ). These comparisons revealed a set of genes expressed at higher levels in both endogenous NSCs and reactive astrocytes, including two lectins—Galectins 1 and 3. These results and the pattern of Galectin expression in the lesioned brain led us to examine the functional significance of these lectins in brains of mice lacking Galectins 1 and 3. Following stab wound injury, astrocyte reactivity including glial fibrillary acidic protein expression, proliferation and neurosphere‐forming capacity were found significantly reduced in mutant animals. This phenotype could be recapitulated in vitro and was fully rescued by addition of Galectin 3, but not of Galectin 1. Thus, Galectins 1 and 3 play key roles in regulating the proliferative and NSC potential of a subset of reactive astrocytes. GLIA 2015;63:2340–2361
Keywords: glia proliferation, neurosphere, genomewide analysis
Introduction
Reactive gliosis is a widespread reaction of glial cells to pathological processes in the brain. It involves astrocytes, NG2 glia, and microglia that mediate beneficial and adverse effects, such as wound closure and scar formation, respectively (Kettenmann et al., 2011). Recent work indicates a striking heterogeneity in the reaction of each type of glial cells (Anderson et al., 2014; Burda and Sofroniew, 2014; Dimou and Götz, 2014). This could be best demonstrated by live in vivo imaging, which revealed a surprisingly heterogeneous reaction of astrocytes reacting to stab wound injury in the adult murine cerebral cortex gray matter (GM), with some astrocytes hardly reacting at all, others polarizing toward the injury site and yet others proliferating and generating two daughter astrocytes (Bardehle et al., 2013). Furthermore, clonal analysis demonstrated that the differential reaction of astrocyte subtypes is seemingly related to their distinct developmental origin (Martín‐López et al., 2013). In view of this heterogeneity, it is now important to address the mechanisms regulating the reaction of these distinct astrocyte subsets after brain injury.
Astrocytes resuming cell division after lesion are of particular importance, as proliferation is the only means to increase astrocyte numbers at the injury site in the cerebral cortex GM (Bardehle et al., 2013). Indeed, proliferating astrocytes are critical for restricting the injury size and the number of infiltrating cells and inflammation, since their elimination has been shown to aggravate brain damage after lesion (Burda and Sofroniew, 2014). Interestingly, astrocyte proliferation in the GM is highly injury‐dependent and does not occur upon amyloid plaque deposition or even pronounced neuronal cell death, in spite of profound microglia activation and proliferation (Behrendt et al., 2013; Sirko et al., 2013). Instead, it is selectively elicited upon injury involving alterations of the blood brain barrier, such as traumatic, ischemic, and demyelinating injuries (Behrendt et al., 2013; Dimou and Götz, 2014; Gadea et al., 2008; Götz and Sirko, 2013; Kamphuis et al., 2012). These injury‐specific differences led to the identification of signals regulating reactive astrocyte proliferation, including endothelin‐1, sonic hedgehog and fibroblast growth factor (FGF) signaling (Gadea et al., 2008; Kang et al., 2014; Sirko et al., 2013; Zamanian et al., 2012). To obtain a more comprehensive view on the key regulators of reactive astrocyte proliferation, we set out to examine the pattern of gene expression in reactive astrocytes at the peak of their proliferation following stab wound injury in comparison to nonproliferative astrocytes in the intact adult cerebral cortex GM.
As a subset of proliferating reactive astrocytes acquire neural stem cell (NSC) potential after injury, monitored by the ability to form multipotent, self‐renewing neurospheres (Buffo et al., 2008; Grande et al, 2013; Sirko et al., 2013), this prompts the question how much of the gene expression changes of reactive astrocytes may be shared with NSCs. Only genomewide expression analysis comparing reactive astrocytes, NSCs and nonreactive astrocytes allow determining the degree of similarity between NSCs and reactive astrocytes and the extent of injury‐specific gene expression.
A small number of candidates shared by reactive astrocytes and endogenous NSCs have already been identified and tested, including glial fibrillary acidic protein (GFAP), Nestin, Musashi, DSD1‐proteoglycan, and Tenascin‐C (for review, see Götz et al., 2015; Robel et al., 2011; Sirko et al., 2009). However, these proteins also appear in injury conditions without reactive proliferation of astrocytes and/or neurosphere formation (Kamphuis et al., 2012; Robel at al., 2011), thus emphasizing the need for additional molecular insights. Toward this aim, we compared genomewide expression of astrocytes reacting to stab wound with astrocytes from the intact adult GM, as well as an existing expression profile of endogenous NSCs located in the adult SEZ (Beckervordersandforth et al., 2010).
Materials and Methods
Animals
The experiments were performed with 2–3 months old C57BL/6J mice (Charles River Laboratories; Sulzfeld, Germany) and the transgenic lines in which enhanced green fluorescent protein (eGFP) is driven by the aldehyde dehydrogenase 1 family member L1 Tg(Aldh1l1‐eGFP)OFC789Gsat (Heintz, 2004) or human GFAP TgN(hGFAP‐EGFP)GFEA (Nolte et al., 2001) and Lgals1− / −Lgals3− / − mice on C57BL/6J background (Colnot et al., 1998). Animals were allocated to experimental groups regarding their genotype and kept under standard conditions with access to water and food ad libitum. Animal handling and experimental procedures were performed in accordance with German and European Union guidelines and were approved by the State of upper Bavaria. All efforts were made to minimize suffering and number of animals used.
Surgical Procedures
Stab wound in the GM of the somatosensory cerebral cortex was performed as previously described (Buffo et al., 2008). Briefly, animals were anaesthetized and, using a stereotactic apparatus, subjected to 0.7 mm deep and 1–1.2‐mm long stab wound. Proliferating cells were labeled with 5′‐bromo‐desoxyuridine (BrdU; Sigma Aldrich) added to the drinking water (1 mg/mL water containing 1% sucrose) for 5 days or by single intraperitoneal BrdU injection (10 mg/100 g body weight) 2 h prior analysis. Operated animals were divided into two groups: one group sacrificed at 5 day past injury (dpi) and one group composed of animals sacrificed at increasing times after injury (1, 3, and 7 dpi).
Tissue Dissociation and Purification of Cells Using Fluorescence‐Activated Cell Sorting
Cortical cells from a tissue volume (∅ 0.35 cm) punched from the lesioned somatosensory GM of GFAP‐eGFP mice at 5 dpi or the corresponding region of noninjured Aldh1L1‐eGFP mice were dissociated as described previously (Buffo et al., 2008), sorted by a fluorescence‐activated cell sorting [FACS] Aria (BD) and processed for ribonucleic acid (RNA) isolation, as described by Beckervordersandforth et al. (2010) and in Supp. Info. M1.
Analysis of the Microarray Data
Analysis of differential gene expression was performed on normalized log2‐transformed intensities using Bioconductor packages implemented in CARMAweb (Rainer et al., 2006), including limma t‐test and Benjamin–Hochberg multiple testing corrections with false discovery rate (FDR) <10%. To generate the injury‐induced signature of reactive astrocytes, only genes with significant (FDR < 10%) regulation were selected. For direct comparison of average expression in adult NSCs, the datasets from Beckervordersandforth et al. (2010) were reprocessed along with samples in this study. Genes upregulated in both the aNSCs and reactive astrocytes were defined by limma t‐test P‐value < 0.01 and fold change >2× in both comparisons versus nonreactive astrocytes. The downregulated gene set (215 probe sets) in aNSCs and reactive astrocytes was defined similarly (FDR < 10%, fold change >2× down). Regulated gene sets were analyzed for statistically enriched biological processes (Gene Ontology; GO terms) and signal transduction pathways using the Genomatix Pathway System (GePS). Microarray data was submitted to GEO (GSE18765 and GSE66370).
Immunohistochemistry and In Situ Hybridization
After transcardial perfusion with phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS, brains were removed, postfixed in 4% PFA for 4 h at 4°C and processed for immunostainings or in situ hybridization (ISH), as described previously (Brill et al., 2009; Sirko et al. 2013). Nuclei were visualized with 4′,6′‐diamidino‐2‐phenylindole (DAPI; 0.1 µg/mL; Sigma Aldrich). For antibodies and probes see Supp. Info. M2,3.
Neurosphere and Reactive Gliosis Cultures
Neurospheres were prepared from cells of the injured GM of C57BL/6J (n = 6) and Lgals1− / −Lgals3− / − (n = 6) mice at 5 dpi as described in Sirko et al. (2013) (see also Supp. Info. M4). For some experiments, we used 50 µg/mL of human recombinant Galectin 1 (Gal‐1) and/or Galectin 3 (Gal‐3), which were purified from Escherichia coli cultures and added at the beginning of the experiment and after 3 day in vitro (div). Galectin activity was measured prior by the hemagglutination assay (Supp. Info. M3). For the adherent cultures, cells from the lesioned area were dissociated and cultured in 1mL medium (DMEM/F‐12, 1% B27 Supplement, 1% penicillin‐streptomycin, 1 mL of 45%‐Glucose, and 10% FBS [both Life Technologies]) on poly‐d‐lysine coated coverslips. At 2 div, the culture medium was replaced with 1 mL of freshly prepared medium containing 5′‐ethynyl‐2′‐deoxyuridine (EdU) (10 µM/mL; Life Technologies) and with or without Galectins (50 µg/mL). At 6 div, cells were fixed and stained (Supp. Info. M5).
Statistical Analysis
Confocal laser scanning (Zeiss LSM5; Zeiss LSM710) microscope was used to quantify immunopositive cells in sections or cell culture. For each of the quantifications, at least three animals or experimental culture batches were examined. All quantifications of immunohistochemistry are based on analysis of at least five sections per animal. The incidence of immunopositive cells in cell culture was expressed either relative to the total cell number (500–1,000 DAPI‐stained nuclei per coverslip) or the number of co‐stained cells. Statistical analyses of data were performed using the GraphPrism 5.0 software. Data were tested for the normal distribution using the Kolmogorov–Smirnov test, and significance between means from two experimental groups was analyzed using of two‐tailed unpaired Student's t‐test and between multiple groups by one‐way analysis of variance (ANOVA) analysis. All values were plotted as mean ± standard error of the mean (SEM), unless otherwise stated. Significance indicated as *(P < 0.05), **(P < 0.01), ***(P < 0.001).
Results
Labeling Parenchymal Astrocytes in the Intact and Injured GM of the Adult Cerebral Cortex
NSCs have been previously isolated by FACS using two criteria, their GFAP expression using hGFAP‐eGFP mice (Nolte et al., 2001) and their epithelial properties with Prominin1 located on their apical surface (Beckervordersandforth et al., 2010; Pinto et al., 2008). When examining the expression of eGFP driven by the hGFAP promoter in the normal cerebral cortex GM (Fig. 1A), only a minority of GM astrocytes at subpial positions (Nolte et al., 2001) were labeled by GFP or GFAP (Fig. 1A′, A″), in contrast to the widespread GFAP labeling in the SEZ at the lateral ventricle (lower row in Fig. 1C). In our search for a mouse line labeling more astrocytes in the GM, we examined Aldh1L1‐eGFP mice (Fig. 1C, D). Aldh1L1 is expressed in astrocytes in young rodent tissue (Cahoy et al., 2008), but has been suggested to decrease during ageing (Yang et al., 2011). In the 2–3‐months old Aldh1L1‐eGFP mice, we found virtually all astrocytes labeled by S100β (Fig. 1C) to be GFP+, and no GFP signal was observed in NeuN+ neurons, NG2+ glia, or Iba1+ microglia (data not shown). Thus, the Aldh1L1‐eGFP animals were deemed best suited for isolating astrocytes from the uninjured cerebral cortex GM.
Figure 1.
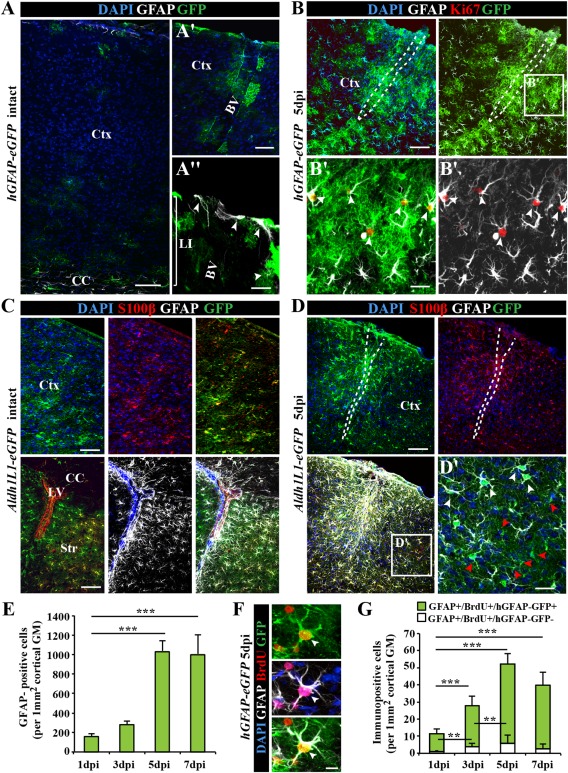
Characterization of astrocytes in the intact and injured somatosensory cortex. (A) Fluorescence micrographs of GFP immunoreactivity in the intact somatosensory cortex of hGFAP‐eGFP mice at postnatal day (P65). Note that GFP is limited to few subpial astrocytes in the cortical layer I (LI) or parenchymal astrocytes in the vicinity of blood vessels (BV) in the intact cerebral cortex (Ctx). (B) After stab wound injury of hGFAP‐eGFP mice of the same age, GFP was found in virtually all GFAP+ reactive astrocytes including proliferating (Ki67+) reactive astrocytes (white arrowheads in B′). (C) Fluorescence micrographs depict representative confocal images of GFP immunostaining in the intact cerebral cortex (upper panel) of Aldh1L1‐eGFP transgenic mice. Double‐immunopositive cells for the calcium binding protein S100β in the intact gray matter (GM) and GFAP in the intact corpus callosum (CC), striatum (Str) and subependymal layer of lateral ventricle (LV) (lower panel). (D) Stab wound injury of Aldh1L1‐eGFP somatosensory GM induces a broad expression of GFAP in many (white arrowhead), but not all GM astrocytes (red arrowheads) at the injury site (D′). (E) Histogram depicting the number of the hGFAP‐GFP+ astrocytes in the penumbra of the injury increasing significantly over the first 7 dpi. (F) Fluorescence micrograph shows labeling for the DNA base analog BrdU (given ×1 i.p. 2 h before immunohistological processing) in GFP+ and GFAP+ astrocytes 5 dpi in hGFAP‐eGFP mouse cerebral cortex GM. (G) Quantification of the number of such cells is shown in the histogram with a significant increase within the first 5 dpi. All data are plotted as mean ± SEM from n = 5 experiments. Significance between means was analyzed using of one‐way ANOVA test and indicated as *(P < 0.05), **(P < 0.01), ***(P < 0.001). Scale bars: (A–D) 100 μm, (A′) 50 μm, (B′, D′) 25 μm, (A″, F) 10 μm.
After stab wound, many but not all GM astrocytes upregulate GFAP (Fig. 1B, D; see also Bardehle et al., 2013; Buffo et al., 2008; Sirko et al., 2013). Accordingly, only about 60% of Aldh1L1‐GFP+ cells were GFAP+ within 500 µm of the lesion (Fig. 1D). Thus, GFP+ cells in this mouse line comprise reactive (GFAP+) and nonreactive (GFAP−) astrocytes (Fig. 1D′). Conversely, eGFP driven by the hGFAP promoter labeled virtually all GFAP+ astrocytes that were also hypertrophic in the injured GM (Fig. 1B). The hGFAP‐GFP+ astrocytes also comprised almost all cycling (Ki67+ or BrdU+ astrocytes (Fig. 1B′, F). The increase in hGFAP‐GFP+/GFAP+ astrocytes over the first days after injury (Fig. 1E) occurred in parallel with the increase of reactive astrocytes proliferation, which reached the peak at 5 dpi (Fig. 1F, G). Thus, expression of GFP in the injured GM of hGFAP‐eGFP mice is specifically restricted to reactive astrocytes and faithfully reflects the progression of astrogliosis, including the proliferative astrocyte reaction. Besides being best suited for monitoring reactive astrocytes, the hGFAP‐GFP line was also used to isolate SEZ NSCs (Beckervordersandforth et al., 2010) and hence allows for best comparison between reactive astrocytes and NSCs.
Selective Enrichment of Reactive and Nonreactive Cortical GM Astrocytes via FACS
To isolate GFP+ astrocytes (Fig. 2A), we prepared single cell suspensions from biopsy punches of the intact somatosensory GM of 2–3‐months old Aldh1L1‐eGFP mice. GFP+ cells were sorted by gating out cell debris, duplets, and non‐ or autofluorescent cells by setting the gate for GFP in comparison to nonfluorescent cells prepared from wild‐type (wt) mice (Fig. 2B). Cells plated before and after sorting documented the high enrichment of sorted cells from less than 20% in the starting population to 95% GFP+ cells after sorting (Fig. 2C, D). The astroglial identity of the GFP+ sorted cells was confirmed by co‐staining for S100β (91% of GFP+ cells isolated from Aldh1L1‐eGFP mice were S100β+, Supp. Info. Fig. 1A). For sorting of reactive astrocytes (Fig. 2A), we isolated GFP+ cells from hGFAP‐GFP mice at 5 dpi, the peak of astrocyte proliferation. This resulted in a similar enrichment (93% of sorted cells were GFP+, Fig. 2C, D) with 97% of the sorted GFP+ cells GFAP‐immunoreactive (Supp. Info. Fig. 1A). Thus, this reproducible sorting protocol allowed isolation of nearly pure astrocytes from intact and injured adult cerebral cortex GM.
Figure 2.
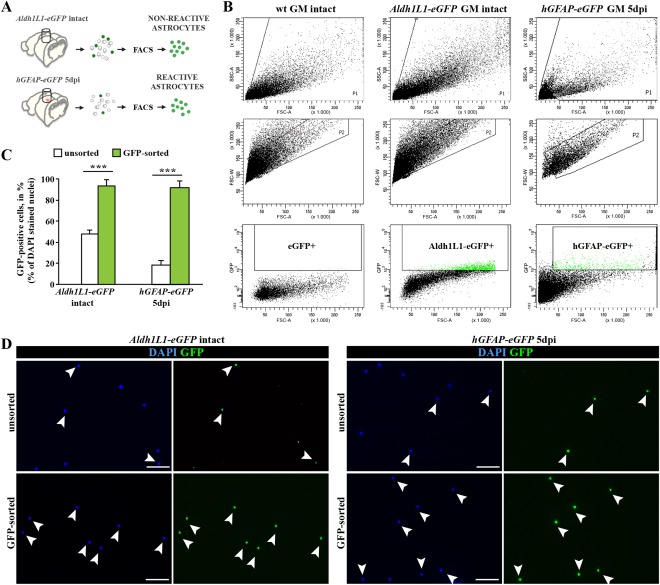
Purification of astrocytes from the intact and injured somatosensory cortex GM using fluorescence‐activated cell sorting (FACS). (A) Schematic representation of experimental design used for the tissue dissection and purification of astrocytes from intact or stab wound‐injured cerebral cortex GM. (B) FACS plots show the sorting gates that were used in hierarchical way for isolation of astrocytes, based on cell size (forward scatter, FSC) and granularity (side scatter, SSC) (upper layer), discrimination of doublets (FSC‐area vs. FSC‐width; middle layer) and positive selection for GFP+ cells (lower layer). (C) Bar graphs depict proportions of GFP+ astrocytes before (unsorted) and after FACS sorting (GFP‐sorted), indicating high efficiency of the sorting procedure. (D) Purity of sorting cell populations was confirmed by immunostaining of cells before (unsorted) and after FACS sorting (sorted). All data are plotted as mean ± SEM from n ≥ 5 experiments. Significance between means from two experimental groups was analyzed using of two‐tailed unpaired Student's t‐test and indicated as *(P < 0.05), **(P < 0.01), ***(P < 0.001). Scale bars: (D) 75 μm
Injury‐Induced Changes of Gene Expression in the Cerebral Cortex GM Astrocytes
RNA was extracted from the sorted astrocyte populations (see also Supp. Info. M1), and amplified for hybridization on Affymetrix Mouse Genome 430 2.0 arrays. As expected, a large number of common genes were found to be expressed in reactive and nonreactive astrocytes (Fig. 3A, B; see also Beckervordersandforth et al., 2010; Cahoy et al., 2008; Zamanian et al., 2012; Zhang et al., 2014). Conversely, genes specific for neurons, microglia, or oligodendroglia were consistently very low, confirming the selective sorting of astrocytes (Fig. 3B; see also Cahoy et al., 2008; Zhang et al., 2014). Notably, reactive astrocytes showed significantly lower expression of most astrocyte‐specific genes (selected as astrocyte‐specific from Cahoy et al. 2008, Zhang et al. 2014), except for Aldh1l1 and GFAP (Fig. 3B). However, this was not accompanied by upregulation of neuronal or other glia‐specific genes (Fig. 3B), suggesting that astrocytes do not acquire other lineage characteristics, while they apparently reduce astrocyte‐specific gene expression, consistent with partial dedifferentiation.
Figure 3.
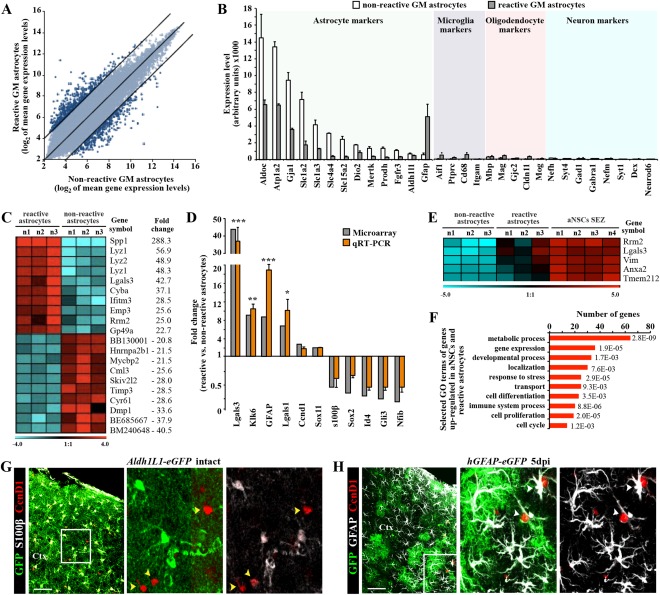
Genomewide expression analysis of astrocytes from the intact or injured cerebral cortex GM. (A) Scatter plot of whole genome expression profile in nonreactive (Aldh1L1‐eGFP) and reactive (hGFAP‐eGFP) astrocytes depicted at normalized expression levels of all probe sets from the Affymetrix GeneChip Mouse Genome 430 2.0 arrays presented on log2 scale. Black lines indicated boundaries of twofold difference in gene expression levels. (B) Both isolated astrocyte populations express high level of astroglia‐specific genes. The y‐axis represents normalized expression of probe sets for cell‐type‐specific markers from the array. (C) Heat map showing the top 20 genes that significantly differ between astrocytes from intact or injured cerebral cortex (the normalized values are plotted on a log2 color scale, and color indicates up‐ (red) and downregulation (blue), as compared with the mean and linear fold changes (reactive vs. nonreactive astrocytes) are provided). (D) Transcript levels of several genes were confirmed by quantitative RT‐PCR and are shown in comparison to GeneChip expression levels. Data are plotted as mean ± SEM from independent biological/technical replicates. Significance between experimental groups was analyzed using of two‐tailed unpaired Student's t‐test and indicated as *(P < 0.05), **( P < 0.01), ***( P < 0.001). (E) The top five genes significantly enriched in both reactive astrocyte and adult neural stem cells (aNSC SEZ; see Beckervordersandforth et al., 2010) in comparison to the non‐reactive astrocytes are shown as a heat map as described above (for complete list, see Table 2). (F) Bars show 10 selected major GO classes associated with genes significantly enriched in both reactive astrocytes and aNSCs. (G, H) Immunofluorescence for cyclin D1 (in red) in frontal sections of intact (G) or injured (5 dpi, H) cerebral cortex confirm the injury‐induced expression of cyclin D1 (Ccnd1) in reactive astrocytes (white arrowheads), while astrocytes in the intact GM do not express cyclin D1 (yellow arrowheads). The cell nuclei were counterstained with DAPI. Scale bars: (G, H) 100 μm.
Statistical analysis revealed 916 significantly regulated transcripts between astrocytes isolated from the injured versus intact GM (599 downregulated and 317 upregulated in reactive astrocytes; top 50 up‐ or downregulated genes in Table 1). Consistent with previous microarray analyses (Beckervordersandforth et al., 2010; Johansson et al., 2013; Ninkovic et al., 2013; Pinto et al., 2009, 2008; Stahl et al., 2013; Walcher et al., 2013), quantitative reverse transcription polymerase chain reaction (qRT‐PCR) confirmed the changes observed in the microarray (Fig. 3D), including the strongly upregulated genes Lgals1 (lectin, galactoside‐binding, soluble, 1) and Lgals3 (lectin, galactoside‐binding, soluble, 3) (40× and 10×, respectively).
Table 1.
Genes regulated in cortical astrocytes in response to stab wound injury
| Top 50 genes upregulated in reactive astrocytes | Top 50 genes downregulated in reactive astrocytes | ||||
|---|---|---|---|---|---|
| Probe set ID | Gene symbol | Fold induction | Probe set ID | Gene symbol | Fold induction |
| 1449254_at | Spp1 | 288.25 | 1443745_s_at | Dmp1 | −33.61 |
| 1436996_x_at | Lyz1 | 56.94 | 1442340_x_at | Cyr61 | −28.61 |
| 1423547_at | Lyz2 | 48.88 | 1449335_at | Timp3 | −28.46 |
| 1426808_at | Lgals3 | 42.71 | 1447517_at | Skiv2l2 | −27.99 |
| 1454268_a_at | Cyba | 37.11 | 1430966_at | Cml3 | −25.64 |
| 1423754_at | Ifitm3 | 28.54 | 1447150_at | Mycbp2 | −21.53 |
| 1417104_at | Emp3 | 25.58 | 1433830_at | Hnrnpa2b1 | −21.47 |
| 1434437_x_at | Rrm2 | 25.03 | 1453514_at | Gpm6b | −20.53 |
| 1420394_s_at | Gp49a | 22.66 | 1457410_at | Arhgap5 | −18.32 |
| 1452352_at | Ctla2b | 21.56 | 1440037_at | Pbx1 | −17.65 |
| 1420804_s_at | Clec4d | 18.76 | 1440770_at | Bcl2 | −16.03 |
| 1449164_at | Cd68 | 18.52 | 1452378_at | Malat1 | −15.61 |
| 1426278_at | Ifi27l2a | 17.70 | 1439675_at | Ppara | −15.43 |
| 1456292_a_at | Vim | 17.19 | 1433413_at | Nrxn1 | −12.90 |
| 1453773_at | Rnf220 | 16.46 | 1458469_at | Cblb | −12.80 |
| 1417268_at | Cd14 | 15.42 | 1439556_at | Ncam1 | −12.77 |
| 1425140_at | Lactb2 | 14.42 | 1424393_s_at | Adhfe1 | −12.37 |
| 1449401_at | C1qc | 13.98 | 1454904_at | Mtm1 | −12.33 |
| 1417381_at | C1qa | 13.32 | 1441185_at | Msi2 | −11.65 |
| 1417876_at | Fcgr1 | 12.99 | 1438103_at | Pcdhgc4 | −11.49 |
| 1417063_at | C1qb | 12.91 | 1440789_at | Neo1 | −10.98 |
| 1455660_at | Csf2rb | 12.42 | 1457445_at | Trps1 | −10.84 |
| 1417346_at | Pycard | 12.40 | 1438089_a_at | Bclaf1 | −10.21 |
| 1460351_at | S100a11 | 11.85 | 1455799_at | Rorb | −10.04 |
| 1460218_at | Cd52 | 11.67 | 1456923_at | Trpm3 | −10.00 |
| 1434447_at | Met | 11.44 | 1421180_at | Lix1 | −9.53 |
| 1460330_at | Anxa3 | 11.01 | 1417408_at | F3 | −9.50 |
| 1419004_s_at | Bcl2a1a | 10.77 | 1427143_at | Kdm5b | −9.46 |
| 1448982_at | Klk6 | 9.94 | 1429515_at | Ubr2 | −9.39 |
| 1451335_at | Plac8 | 9.90 | 1433757_a_at | Nisch | −9.29 |
| 1450826_a_at | Saa3 | 9.55 | 1439940_at | Slc1a2 | −9.23 |
| 1451931_x_at | H2−L | 9.17 | 1419748_at | Abcd2 | −9.06 |
| 1424754_at | Ms4a7 | 8.99 | 1425362_at | Agfg2 | −8.89 |
| 1419482_at | C3ar1 | 8.63 | 1427293_a_at | Auts2 | −8.82 |
| 1440142_s_at | Gfap | 8.61 | 1422955_at | Syt17 | −8.73 |
| 1438948_x_at | Tspo | 8.54 | 1421093_at | Slc7a10 | −8.64 |
| 1457644_s_at | Cxcl1 | 8.44 | 1460567_at | Rfx7 | −8.62 |
| 1435290_x_at | H2−Aa | 8.37 | 1442223_at | Enah | −8.53 |
| 1415828_a_at | Serp1 | 8.32 | 1454070_a_at | Ddhd1 | −8.41 |
| 1421578_at | Ccl4 | 8.20 | 1417962_s_at | Ghr | −8.31 |
| 1424164_at | Mrpl50 | 8.10 | 1438751_at | Slc30a10 | −8.24 |
| 1452428_a_at | B2m | 8.07 | 1423594_a_at | Ednrb | −8.18 |
| 1449227_at | Ch25h | 7.68 | 1444468_at | Paqr8 | −8.09 |
| 1449773_s_at | Gadd45b | 7.52 | 1451257_at | Acsl6 | −8.02 |
| 1419091_a_at | Anxa2 | 7.45 | 1439108_at | Mll5 | −7.96 |
| 1418204_s_at | Aif1 | 7.43 | 1439582_at | Macf1 | −7.95 |
| 1416326_at | Crip1 | 7.21 | 1429427_s_at | Tcf7l2 | −7.92 |
| 1427736_a_at | Ccrl2 | 7.18 | 1429461_at | Ints2 | −7.76 |
| 1419573_a_at | Lgals1 | 7.01 | 1416130_at | Prnp | −7.75 |
| 1418340_at | Fcer1g | 7.00 | 1418024_at | Naa15 | −7.68 |
Shown are top 50 genes selected by stringent filters for statistical significance (FDR < 10%), fold change > 2×, and an average expression level >50 in at least one cell population. Genes are sorted according to fold induction/reduction in reactive relative to nonreactive astrocytes.
To further identify genes regulated in reactive astrocytes with relevance for proliferation or regulation of NSC hallmarks, we then compared the genomewide expression pattern of reactive and nonreactive astrocytes with the one of NSCs from the SEZ of healthy and age‐matched hGFAP‐eGFP mice, which were previously analyzed in our lab under identical conditions (Beckervordersandforth et al., 2010). Thereby, we focused on genes (147 transcripts) expressed at significantly higher levels in both reactive astrocytes and NSCs compared with nonreactive astrocytes from the intact adult GM (Fig. 3E, Supp. Info. Table 1). Remarkably, beside the proliferation gene ribonucleotide reductase M2 (Rrm2), Lgals3 was the gene with the second highest upregulation (65×) in both reactive astrocytes and adult NSCs (Fig. 3E). As also Lgals1 is significantly upregulated in reactive astrocytes and both have been implicated in regulating adult NSCs (Beckervordersandforth et al., 2010; Di Lella et al., 2011; Kajitani et al., 2009; Sakaguchi et al., 2006; Yan et al., 2009), this prompted us to focus on the role of Lgals1 and Lgals3 in the injured cerebral cortex.
Expression of Galectins 1 and 3 Identifies a Subset of Reactive Astrocytes in the Injured Cerebral Cortex GM
By qRT‐PCR and ISH, Lgals1 and Lgals3 expression was very low in the intact cerebral cortex while much higher in the NSC niche, the SEZ (Fig. 4A, E). At 5 days after stab wound injury, however, expression was also much higher in the GM surrounding the injury site (Fig. 4D, E). Notably, Lgals mRNA co‐localized with GFAP, supporting its expression in reactive astrocytes (Fig. 4F). Immunohistochemistry also revealed low numbers of Galectin 1 (Gal‐1) or Galectin 3 (Gal‐3) immunopositive cells in the intact cortex, most of which were neurons and not astrocytes (Fig. 4B and data not shown; note that antibody specificity was assessed using brain sections of Lgals1− / −Lgals3− / − animals, see Supp. Info. Fig. 2). Conversely, many Gal‐1+ or Gal‐3+ cells were detected in the cerebral cortex GM after stab wound injury. These were hGFAP‐GFP+ and/or GFAP+ reactive astrocytes (Fig. 4D). We noted that immunostainings for both lectins correlate well with the perimeter of GFAP‐immunoreactivity in the injured parenchyma at 5 dpi, and appeared more intense within the lesion core or in the close vicinity to the wound site (Fig. 5A–C). At this time point, GFAP+ reactive astrocytes comprised only 23% of all Gal‐1+ and 42% of all Gal‐3+ cells around the lesion (Fig. 5B, C) with the remainder of Gal‐1+/Gal‐3+ cells identified as Iba1+ microglia or NG2+ glia (data not shown), consistent with previous data (Bischoff et al., 2012; Liu et al., 1995; Qu et al., 2011; Walther et al., 2000). Importantly, however, both Gal‐1 and Gal‐3 labeled only a subset of reactive astrocytes (16 and 22%, respectively; Fig. 5E), consistent with the concept of astrocyte heterogeneity after injury (Dimou and Götz, 2014). Strikingly, virtually all Ki67+ reactive astrocytes were Gal‐3+, while only half of the mitotic astrocytes were positive for Gal‐1 (Fig. 5D, F). Thus, especially Gal‐3 is a novel marker for cycling reactive astrocytes in the murine cerebral cortex GM.
Figure 4.
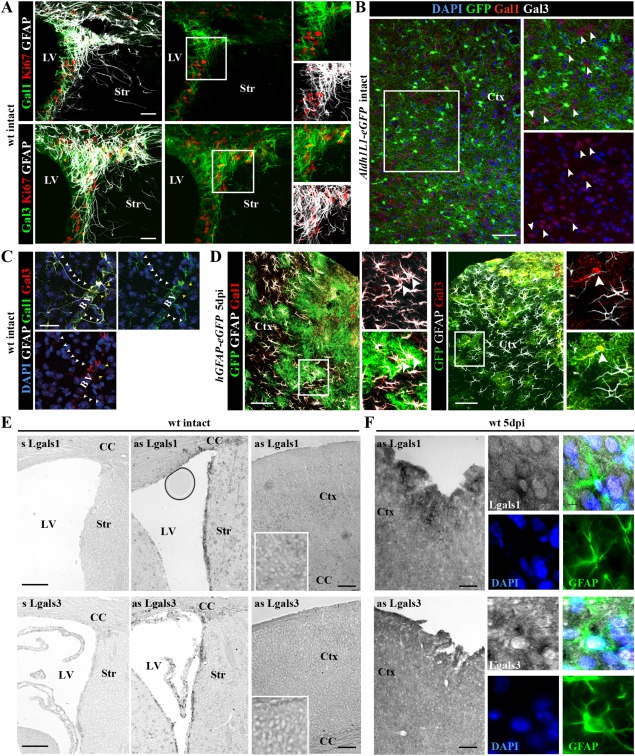
Injury‐induced expression of Galectins 1 and 3 in parenchymal astrocytes of the adult cortical GM. (A) In the intact forebrain, expression of Galectin‐1 (Gal‐1) (upper panels) or Galectin‐3 (Gal‐3) (lower panels) is restricted to the adult SEZ at the lateral ventricle (LV), where these lectins labeled also proliferating GFAP+ cells shown in the inserts to the right. (B) In the intact cerebral cortex, Gal‐3‐immunostaining is virtually not detectable, while Gal‐1 is occasionally associated with Aldh1L1‐eGFP‐negative neurons (white arrowheads). (C) Only few GFAP‐positive astrocytes were Gal‐1‐ and/or Gal‐3‐immunoreactive in the intact cerebral cortex and these were located in the perivascular surroundings (BV, blood vessels). (D) In the stab wound‐injured cerebral cortex, many hGFAP‐eGFP/GFAP‐positive astrocytes are Gal‐1+ or Gal‐3+ (white arrowheads) at 5 dpi. (E, F) Combined ISH and immunohistochemistry on frontal sections from intact (E) or injured (right panels) forebrains using an anti‐GFAP antibody and RNA probes for Lgals1 and Lgals3 transcripts show increased Lgals1/3 expression after injury (F) and co‐localization with GFAP (panels at higher magnification in F). The cell nuclei were counterstained with DAPI. Scale bars: (B, D, E, F) 100 μm, (A, C) 50 μm.
Figure 5.
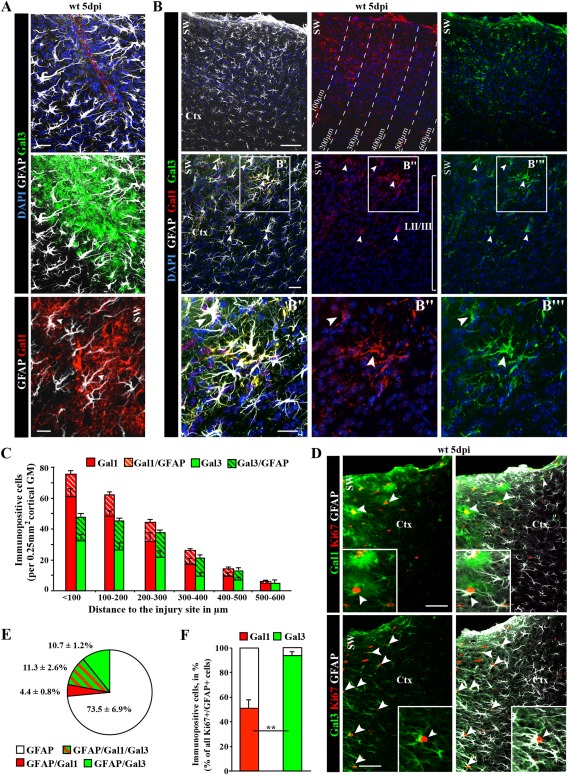
Galectin‐1/‐3 labels subsets of reactive astrocytes in the injured cortical GM. (A, B) Fluorescence micrographs depicting the stainings indicated on the left side of the panels show localization of Gal‐1 and Gal‐3 immunoreactivity in many cells, including a subset of reactive astrocytes (GFAP‐co‐labelling in A, B). (C) Histogram depicting the number of Gal‐1+ and Gal‐3+ cells or astrocytes at different distance to the injury site as indicated in B. Data are plotted as mean ± SEM from n ≥ 5 experiments. (D) Co‐localization with Ki67 and GFAP (white arrowheads) reveals that the subset of astrocytes double‐positive for Gal‐1 or Gal‐3 (for quantification, see pie chart in E) are all (Gal‐3; arrowheads) or most (Gal‐1; arrowheads) Ki67+ (for quantification in histogram see F). Data are plotted as mean ± SEM from n = 5 experiments. Significance was analyzed using of two‐tailed unpaired Student's t‐test and indicated as *(P < 0.05), **(P < 0.01), ***(P < 0.001). The cell nuclei were counterstained with DAPI. Scale bars: (B) 100 μm, (D) 75 μm, (B′–B″′) 50 μm, (A) 25 μm.
Lack of Lgals1/Lgals3 Results in Reduced GFAP Expression and Proliferation of Astrocytes in the Injured Cerebral Cortex GM
The prominent and highly specific upregulation of both lectins in astrocyte subsets after injury prompts the question of their functional role in this context. As Gal‐1 and Gal‐3 are also secreted and can hence be provided by several cell types in the cerebral cortex (e.g., neurons, microglia, and NG2 glia, see above), we took advantage of the double knockout mice Lgals1− / −Lgals3− / − (Colnot et al., 1998) that lack both galectins in all cells. After stab wound injury, we first noted that the region covered by GFAP+ cells was smaller (Fig. 6A) and the number of GFAP+ astrocytes was reduced in the Lgals1− / −Lgals3− / − cerebral cortex compared with control littermates (Fig. 6B). This effect occurred also at the transcriptional level, as shown by qRT‐PCR of mRNA from injured GM of the cerebral cortex from wt or mutant mice (Fig. 6B). As the number of astrocytes (S100β+) in the noninjured as well as in injured GM was well comparable between Lgals1 −/− Lgals3 −/− and wt mice (Supp. Info. Fig. 3A, B), this reduction is specific to the injury condition and seems to affect GFAP expression levels rather than astrocyte numbers. Indeed, when S100β+ astrocytes were double‐stained for GFAP, we noted a profound reduction in the GFAP+ proportion in the injured Lgals1 −/− Lgals3 −/− GM (55%) compared with wt GM (90%; Supp. Info. Fig. 3B). Note that cells were counted here within 300 µm of the lesion site, where we find a higher proportion of GFAP+ astrocytes than in the larger area of 500 µm as quantified above using the Aldh1L1‐eGFP mice. Thus, both GFAP and Gal‐1 and Gal‐3 are highest upregulated close to the injury site (Fig. 5B), and fewer astrocytes become GFAP‐immunoreactive in the injured Lgals1 −/− Lgals3 −/− GM.
Figure 6.
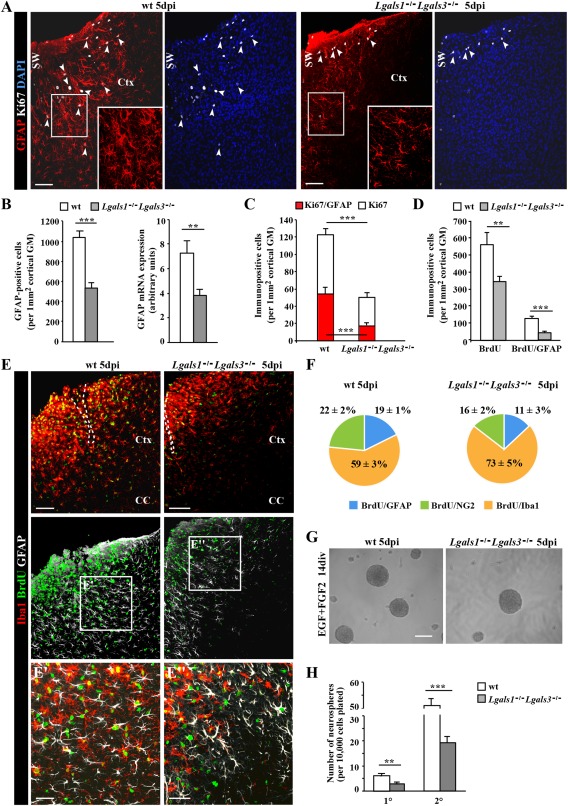
Reduced number and proliferation of reactive astrocytes in the stab wound‐injured cortical GM of Lgals1 −/− Lgals3 −/− mice. (A) Representative overview images shown distribution of proliferating reactive astrocytes (GFAP+/Ki67+, arrowheads) in the penumbra of the stab wound injury in the cerebral cortex of wild type (wt) and Lgals1 −/− Lgals3 −/− mice. The boxed regions are shown at higher magnification. (B) Quantification of the number of GFAP+ astrocytes (left histogram) and quantitative RT‐PCR analysis of GFAP mRNA (right histogram) showed significant reduction in the number of GFAP+ cells and GFAP mRNA levels in the cerebral cortex GM at 5 days after stab wound injury in Lgals1 −/− Lgals3 −/− mice compared with wt. (C) Quantification of cells (white bars) and astrocytes (red bars) proliferating (Ki67+) at 5 dpi revealed a significant reduction of both in Lgals1 −/− Lgals3 −/− compared with wt mice. (D) Quantification of cells (left bars) or astrocytes (GFAP/BrdU, right bars) proliferating at any time after stab wound injury labeled by BrdU given during these 5 dpi also revealed a significant decrease in proliferating cells and astrocytes (to less than 20% of the control) at the injury site of Lgals1 −/− Lgals3 −/− compared with wt cerebral cortex GM. (E) Micrograph showing reduced proliferative activity of microglial cells (BrdU+/Iba+) after stab wound injury in Lgals1 −/− Lgals3 −/− compared with wt cerebral cortex GM. (F) Pie chart depicting the composition of all cells proliferating within 5 dpi labeled by BrdU in wt (left) and Lgals1 −/− Lgals3 −/− (right) cerebral cortex GM. Note that macroglial cells are affected most. (G, H) Neurosphere formation (micrographs in G) of cells isolated at 5 dpi from the GM surrounding the injury site was significantly reduced for both primary (1°) and secondary (2°) neurospheres from Lgals1 −/− Lgals3 −/− compared with wt mice. Data are plotted as mean ± SEM from n ≥ 5 experiments. Significance between experimental groups was analyzed using of two‐tailed unpaired Student's t‐test and indicated as *(P < 0.05), **(P < 0.01), ***(P < 0.001). The cell nuclei were counterstained with DAPI. Scale bars: (A, E, G) 100 μm, (E′, E″) 50 μm.
As we had also seen a close connection between the Gal‐1/‐3+ subset of reactive astrocytes and the cycling reactive astrocytes, we labeled cells in the cell cycle by immunostaining for Ki67. Indeed, double+ astrocytes (Ki67+/GFAP+) were significantly reduced to less than half in the mutant mice (Fig. 6A, C). However, their preferential position close to the blood vessels, as described previously (Bardehle et al., 2013), was not altered in the absence of Gal‐1 and Gal‐3 (82.8 ± 8.2% in wt and 88.6 ± 9.3% in Lgals1 −/− Lgals3 −/−) (Supp. Info. Fig. 3C).
Notably, also other glial cells were less often Ki67+ and the total number of Ki67+ cells was significantly reduced to less than half in the injured Lgals1 −/− Lgals3 −/− GM (Fig. 6C). To examine whether reduced proliferation was restricted to 5 dpi or also occurred throughout the entire time after the injury, BrdU was continuously supplied after injury (Fig. 6D, E). This revealed an even larger decrease in cells entering S‐phase, with less than 20% of BrdU‐labeled reactive astrocytes in the Lgals1 −/− Lgals3 −/− compared with control cerebral cortex injured GM (Fig. 6D, F). While microglia proliferation was also reduced, it was least affected compared with astrocytes and NG2 glia (Fig. 6F). Taken together, these data show that macroglial cells are most deficient in eliciting proliferation after stab wound injury in the absence of Gal‐1 and Gal‐3.
A further measure of astrocyte reaction correlated so far to their proliferative reaction in vivo (Sirko et al., 2013) is the acquisition of neurosphere‐forming potential. As Gal‐1 and Gal‐3 were also high in adult SEZ NSCs, we would predict that in the absence of these proteins, the neurosphere‐forming capacity of astrocytes reacting to stab wound injury should be reduced. As expected, the number of primary neurospheres emerging from Lgals1 −/− Lgals3 −/− cells dissociated at 5 dpi was significantly diminished to less than half of the control (Fig. 6H). This indicated that the number of cells with neurosphere‐forming potential is reduced in the cerebral cortex GM lacking both Gal‐1 and Gal‐3. To determine whether these lectins also play a role in the neurosphere self‐renewal spheres were passaged to determine the number of secondary neurospheres emerging from the same number of plated cells. As also the number of secondary neurospheres was reduced (Fig. 6H), this indicates that either Gal‐1 and/or Gal‐3 are also implicated in regulating self‐renewal of neurosphere cells in vitro. The remaining neurospheres, however, were still multipotent even in the absence of Gal‐1 or Gal‐3 to the same extent as in controls (data not shown). Thus, Gal‐1/Gal‐3 signaling affects GFAP activation and proliferation in reactive astrocytes as well as their stem cell potential.
Distinct Effects of Extrinsic Galectins 1 and 3 on Reactive Glia Proliferation
Next we aimed to determine whether the observed defects in astrocyte proliferation and neurosphere formation could be rescued by acute supply with either Gal‐1 or Gal‐3 to dissect which of these lectins may be responsible for the observed effects. To do so, we examined reactive glia in adherent cultures of cells isolated from the injured cerebral cortex GM of either wt or Lgals1 −/− Lgals3 −/− animals. In line with our in vivo observations, the number of GFAP+ cells obtained at 3 or 5 dpi from Lgals1 −/− Lgals3 −/− was reduced at 6 div (Fig. 7A, B). To monitor the proliferation of Lgals1 −/− Lgals3 −/− glial cells, EdU was supplied to the culture medium at 2 div and the cells incorporating EdU during S‐phase were examined 4 days later. While more than 65% of wt cells had incorporated EdU, only 25% of Lgals1 −/− Lgals3 −/− cells did so (Fig. 7A, C), confirming the decline in reactive glia proliferation also in vitro.
Figure 7.
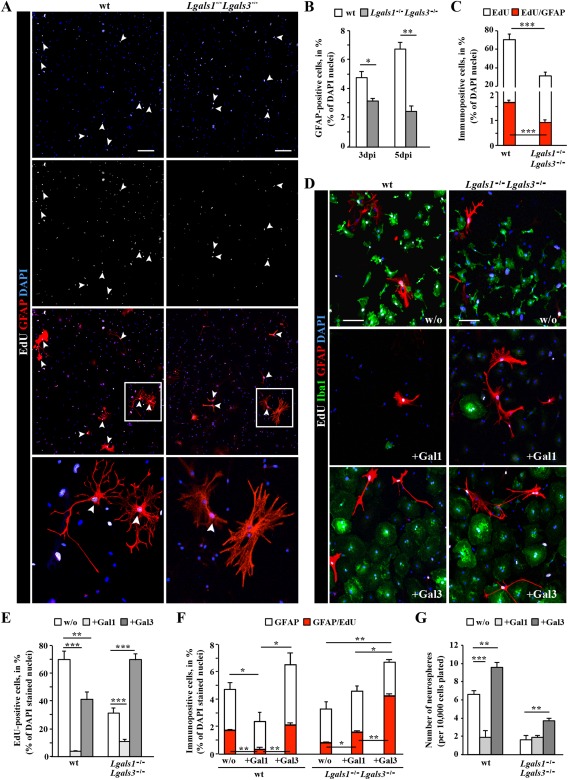
In vitro addition of Galectin 3, but not Galectin 1 promotes adult reactive glia proliferation, neurosphere formation and rescues the Lgals1 −/− Lgals3 −/− reactive glia phenotype. (A) Fluorescence micrographs of cells stained for EdU (added at 2 div) and GFAP in adherent cultures derived at 3 dpi from the injured cerebral cortex GM of wt or Lgals1 −/− Lgals3 −/− mice. Note the double‐labeled reactive astrocytes proliferating during the 4 days in culture after EdU addition (white arrowheads). The boxed regions are shown at higher magnification. (B, C) Histograms depicting the percent of GFAP+ cells (B, percent of DAPI+ cells), proliferating (EdU+, white in C) or proliferating GFAP+ cells (red in C) in the above cultures isolated at 3 or 5 dpi (as indicated on the x‐axis) from wt and Lgals1 −/− Lgals3 −/− cerebral cortex GM. Note that approximately twice as many astrocytes are proliferating among wt cells compared with Lgals1 −/− Lgals3 −/− cells, indicating a cell autonomous effect. Data are plotted as mean ± SEM from n = 5 experiments. (D–G) In vitro addition of Gal‐1 or Gal‐3 as indicated in the fluorescence micrograph panels (D) or the histograms (E‐G). For experiments shown in D‐F, Gal‐1 (+Gal‐1) or Gal‐3 (+Gal‐3) were added 1× and cells were fixed without further medium change 4 days later, stained for GFAP and Iba1 in combination with EdU labeling, as shown in the micrograph in (D) and quantified as indicated in (E, F). Note the differential effect of Gal‐1 addition, which largely mediated cell death versus Gal‐3 addition, which promoted proliferation of cells (E) and fully rescued the defective proliferation of Lgals1 −/− Lgals3 −/− cells (E, F). (G) The total number of primary neurospheres generated from wt or Lgals1 −/− Lgals3 −/− reactive glia, plated in the growth factors‐containing medium for 14 days with addition of Gal‐1 or Gal‐3. Note, that the number of spheres obtained from the Gal‐3‐treated wt or Lgals1 −/− Lgals3 −/− reactive glia was significantly increased in comparison to control cultures (w/o). All data are plotted as mean ± SEM from n = 5 experiments. Significance between experimental groups was analyzed using of two‐tailed unpaired Student's t‐test (in A, C, F) or one‐way ANOVA test (in E, G) and indicated as *(P < 0.05), **(P < 0.01), ***(P < 0.001). The cell nuclei were counterstained with DAPI. Scale bars: (A) 100 μm, (D) 50 μm.
We then added Gal‐1 or Gal‐3 (their activity was monitored in parallel in a bioassay, see Supp. Info. M6) first to cultures at 2 div from wt injured cerebral cortex GM and observed a strong difference between the effects of Gal‐1 and Gal‐3 addition. Gal‐1 addition strongly reduced the total pool of proliferating EdU+ cells from about 70 to 5% in Gal‐1‐treated cultures (Fig. 7E). This effect was much weaker for Gal‐3 addition reducing EdU+ cells only by about half (40%, Fig. 7E). In this regard, it is interesting to note that the majority of proliferating cells in these cultures are microglia (93.5 ± 2.4% and 76.6 ± 2.4% of EdU+ cells are Iba1+ in wt and Lgals1 −/− Lgals3 −/−, respectively), and Gal‐1 has been previously shown to inhibit microglia activation (Bischoff et al., 2012; Starossom et al., 2012). Thus the reduced number of cells in the reactive glia cultures upon Gal‐1 addition is likely due to reduced microglia activation and proliferation. In addition, however, Gal‐1 has been shown to elicit cell death via integrin ligands (see Discussion section), such that both cell death and reduced proliferation may lead to the rather cell‐depleted cultures upon Gal‐1 treatment.
Next, we added Gal‐1 and Gal‐3 to the Lgals1 −/− Lgals3 −/− cells. Remarkably, addition of Gal‐3 alone was sufficient to rescue the proliferation of Lgals1 −/− Lgals3 −/− cells (Fig. 7D, E), including astrocytes, to a level close to untreated cultures of wt cells (Fig. 7F). Gal‐3 also significantly augmented the number of GFAP+ astrocytes, as well as their proliferation, even beyond wt levels (Fig. 7F). Notably, also in the Lgals1 −/− Lgals3 −/− cell cultures, Gal‐1 addition led to reduced cell numbers (Fig. 7D), but nevertheless increased proliferation of remaining Lgals1 −/− Lgals3 −/− astrocytes (Fig. 7F). Taken together, these results indicate that both lectins positively regulate proliferation of reactive Lgals1 −/− Lgals3 −/− astrocytes.
Galectin 3, But Not Galectin 1 Increases Neurosphere Formation from Reactive Glia
Next, we examined whether Gal‐1 and Gal‐3 would affect the NSC potential of reactive glia and rescue the severely reduced neurosphere formation of Lgals1 −/− Lgals3 −/− cells obtained from the injured cerebral cortex GM. Gal‐3, but not Gal‐1 was found to significantly increase the number of neurospheres, both from wt as well as Lgals1 −/− Lgals3 −/− cells, although the rescue was incomplete in the latter case (Fig. 7G). By contrast, Gal‐1 addition rather inhibited the number of neurospheres formed in wt cells, and could not improve this low level of neurosphere formation in cultures obtained from Lgals1 −/− Lgals3 −/−‐injured cortex (Fig. 7G). These findings thus reveal a selective effect of Gal‐3 in promoting neurosphere formation from reactive astrocytes. These findings support the notion that elevated Gal‐3 expression in the reactive cortical astrocytes is not only a predictor of their reactive status and proliferative activity in vivo, but also correlates with their NSC potential in vitro.
Discussion
Insights from Genomewide Expression Analysis
Here we described the first genomewide expression analysis of astrocytes reacting to stab wound in the adult cerebral cortex GM. Previous expression analyses had either focused on noninvasive injury, such as amyloid plaque deposition (Orre et al., 2014), or were performed with young postnatal (P30) astrocytes pooled from white matter (WM) and GM of the cerebral cortex, striatum and hippocampus after stroke or lipopolysaccharide (LPS) treatment (Zamanian et al., 2012). Our transcriptome data, therefore, provides relevant, as well as novel information about similarities and differences in astrocyte gene expression in the adult cerebral cortex GM following stab wound injury.
Interestingly, several of the top‐regulated genes were also regulated in young postnatal astrocytes upon other injury condition (Zamanian et al., 2012), such as osteopontin (SPP1). Indeed, SPP1 is also upregulated in various other CNS region after injury, such as astrocytes and Müller glia in the retina (Chan et al., 2014; Plantman, 2012; Wahl et al., 2013; Zamanian et al., 2012). Likewise Lgals3, Vim, p65/RelA, GFAP, and Lgals1 have been observed at increased levels after different brain injury conditions (Engelmann et al., 2014; Qu et al. 2011; Robel et al., 2011; Yan et al., 2009; Zamanian et al., 2012; Zhang et al., 1998).
When analyzing GO terms and pathways beyond single regulated genes, the top‐significant GO term is “metabolic process” (Supp. Info. Fig. 1B). Astrocytes play key roles in brain metabolism (Bouzier‐Sore and Pellerin, 2013; Pellerin, 2008), and the switch from a resting to a reactive phenotype can trigger a change in metabolic phenotype (Steele and Robinson, 2012). Interestingly, we found that many genes whose expression was significantly increased in both SEZ NSCs and reactive astrocytes compared with nonreactive astrocytes are also related to metabolic processes (Fig. 3F, Supp. Info. Table 2), demonstrating similarities in metabolic programs between reactive astrocytes and NSCs different from the ones of normal parenchymal astrocytes. This is particularly intriguing, as metabolic pathways have been shown to be key in regulating fate decisions and changes (see, e.g., Maryanovich and Gross, 2013) and specific aspects of the lipid metabolism have been identified as a key pathway regulating adult NSC activity (Bracko et al., 2012; Codega et al., 2014; Knobloch et al., 2013). Thus, the metabolic signatures shared between SEZ NSCs and reactive astrocytes are likely involved in regulating the transition of astrocytes to a more plastic progenitor/NSC‐like phenotype after injury.
Notably, consistent with recent observations that FGF signaling counteracts the reactive state of astrocytes (Kang et al., 2014), FGF signaling was the strongest downregulated pathway, after injury. Ultimately, reduced FGF signaling in astrocytes reduces formation of the GFAP+ scar, implying that reduced expression of members of this signaling pathway helps to limit gliotic scar formation. In contrast, nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) signaling pathways mediating response to inflammatory signals was the top upregulated GO term highlighting significant gene expression changes in reactive astrocytes related to immune or inflammatory signaling (Table 2, including interleukin [IL] 1β, IL‐6, and IL‐10 pathways, tumor necrosis factor α [TNFα] and toll‐like receptor (TLR); see also Falsig et al., 2004; Hamby et al., 2012; Henn et al., 2011; Zamanian et al., 2012; Zimmer et al., 2011). These are the mediators released by reactive astrocytes modulating immune cell invasion after injury (Burda and Sofroniew, 2014). Specifically NF‐κB signaling has been implicated in gene expression events that impact on cell survival, differentiation, and proliferation (Napetschnig and Wu, 2013; Oeckinghaus et al., 2011). Interestingly, cyclin D1 (Ccnd1), a known NF‐κB target (Guttridge et al., 1999; Hinz et al., 1999), is also significantly upregulated in reactive astrocytes (Fig. 3E, F) and might thus be one of the positive regulators of reactive astrocyte proliferation, integrating various damage and inflammatory signals to instruct astrocyte proliferation.
Table 2.
Gene expression pattern analysis of regulated signal transduction pathways
| Signal transduction pathway associations | P value | Genes |
|---|---|---|
| Pathways upregulated in reactive astrocytes at 5 days after stab wound injury | ||
| NF kappa B | 6,54 E −06 | Cd14, Cd83, Aif1, Litaf, Nfkbia, Cebpb, Nfkbid, Cxcl1, Ier3, Cd74, Bcl3, Il1b, Tnfrsf1b, Spp1, Nfkbib, Bcl2a1a, S100a4, Ccl4, Siva1, Saa3, Malt1, Pycard |
| Tumor necrosis factor (TNF superfamily, member 2) | 2,77 E −05 | Cd14, Cd83, Aif1, Litaf, S100a6, Nfkbia, H2‐Ab1, Cxcl1, Ctsd, Il1b, Tnfrsf1b, Nfkbib, Ccl4, Saa3, Socs3, Erg |
| Spleen tyrosine kinase | 1,11 E −04 | Tyrobp, Sla, Lgals1, Fcgr1, Cmtm7, Ptprc, Fcer1g, Trem2, Egr2 |
| Caspase | 1,84 E −03 | Lgals1, Plp1, Ctsd, Afp, Ctsc, B2m, Siva1, Malt1, Pycard, Creg1 |
| Chemokine (CC MOTIF) ligand 2 | 2,52 E −03 | Cd14, Nfkbia, Cxcl1, Il1b, Vim, Ccl4 |
| Interleukin 10 | 3,07 E −03 | Cd83, Lgals1, Il1b, Etv3, Fcgr1, Trem2, Socs3 |
| Interleukin 1 | 3,95 E −03 | Cd14, Nfkbia, Cxcl1, Il1b, Nfkbib, Ctsc, Ptgds, Socs3, Pycard |
| Interleukin 6 | 4,21 E −03 | Grn, Cebpb, Cxcl1, Lgals3, Il1b, Gfap, Junb, Socs3 |
| Toll‐like receptor | 6,94 E −03 | Cd14, Cxcl1, Grk5, Anxa2, Hspe1, Ly86, Saa3, Trem2 |
| Protein tyrosine phosphatase, receptor type | 8,19 E −03 | Tyrobp, Lgals1, Ptpre, Fcgr1, Cd52, Ptprc |
| Pathways downregulated in reactive astrocytes at 5 days after stab wound injury | ||
| Fibroblast growth factor | 8,99 E −05 | Ccnd2, Syt1, Serpine2, Fgfr3, Pax6, Sdc4, Sox2, Nedd4, Gpc5, Dmp1, Ncam1, Vegfa, Gja1, Spry2, Hip1, Hs2st1, Gli3, Sox9, Etv4, Cyr61 |
| Rhoa RAS homolog | 4,78 E −04 | Neo1, Arhgap5, Ednrb, Rock1, Gna13, Pias1, Sdc4, Ophn1, S1pr1, Plxnb1, Rapgef3, Sox9, Sdc2, Cyr61 |
| BETA catenin | 1,75 E −03 | Faf1, Tle1, Fzd4, Ppap2b, Sfrp1, Fbxw11, Rora, Sox2, Chd8, Prdm5, Tcf7l2, Trrap, Enah, Ptn, Sox9, Sdc2, Apc, Cyr61, Zeb1, Ptprz1 |
| Lymphoid enhancer binding factor 1 (TCF/LEF) | 1,85 E −03 | Tle1, Ppap2b, Nrarp, Sfrp1, Tcf7l2, Hipk2 |
| Paxillin | 2,00 E −03 | Pld2, Gna13, Ptpn11, Adcyap1r1, Nek3, Flii, Ptprt, Hgf |
| Homeodomain interacting protein kinase | 4,01 E −03 | Daxx, Pax6, Tcf7l2, Zbtb4, Hipk2 |
| Wingless type | 4,44 E −03 | Faf1, Tnik, Tle1, Fzd4, Ppap2b, Fermt2, Znrf3, Nrarp, Lrp4, Sfrp1, Rora, Pax6, Sox2, Chd8, Prdm5, Tcf7l2, Rhou, Trps1, Trrap, Enah, Gli3, Sox9, Sdc2, Apc, Macf1 |
| Progestin and ADIPOQ receptor | 7,64 E −03 | Srebf1, Ppara, Paqr8, Appl2 |
| Nuclear receptor subfamily 2, group F, member 2 | 8,13 E −03 | Srebf1, Ppara, Paqr8, Appl2 |
| Pathways upregulated in both the reactive astrocytes at 5 days after stab wound injury and the adult NSCs | ||
| Aurora kinase | 3,03 E −03 | Cdc20, Ncaph, Vim, Ccna2, Cdk1 |
| Mixed lineage kinase | 3,22 E −03 | Pim1, Txn1, Gspt1, Nfkbia, Gadd45b, Anxa2 |
| Cyclin B1 | 3,61 E −03 | Cdc20, Pbk, Ccna2, Cdk1 |
| Cell division cycle 2, (G1 to S and G2 to M) | 4,14 E −03 | Cdc20, Pbk, Ncaph, Vim, Ccna2, Cdk1 |
| Minichromosome maintenance complex | 4,49 E −03 | Ppp2r3c, Ccna2, Cdk1 |
| Cell division cycle 25C | 4,78 E −03 | Pim1, Ccna2, Cdk1 |
| BUB1 mitotic checkpoint serine/threonine kinase | 5,64 E −03 | Cdc20, Cdk1 |
Shown are the significantly (P < 0.01) enriched signal transduction pathways identified using GePS for genes significantly (FDR < 10%) regulated in reactive relative to nonreactive astrocytes or upregulated (P < 0.01, fold change > ×2) in both the adult NSCs and reactive astrocytes in comparison to nonreactive astrocytes.
Ccnd1 is also a target of Shh pathway (Kenney and Rowitch, 2000) and the injury‐induced activation of Shh signaling promotes proliferation of reactive astrocytes (Sirko et al., 2013). Notably, at 5 dpi, the peak of proliferation of reactive astrocytes, we detected no significant upregulation of the genes encoding for Patched, Smo, and Gli family proteins, suggesting either that their upregulation may have taken place prior to reaching the peak of proliferation, or these are not regulated at the transcriptional level in this context. Interestingly, Patched 1 regulates cell cycle progression by affecting the subcellular localization of the M‐phase promoting factor that consists of two proteins: the kinase Cdk1 and the cyclin B1 (Barnes et al., 2001; Doree and Hunt, 2002). Although the level of Ptch1 expression in reactive astrocytes was not significantly changed, it is possible that binding of extracellular Shh can disrupt interaction between Ptch1 and phosphorylated cyclin B1, thus allowing the complex to localize to the nucleus where it stimulates the G2 to M‐phase transition, as has been shown for Shh‐treated cells in vitro (Barnes et al., 2001; Ruiz i Altaba et al., 2002). As transcriptional regulation during G1‐to‐S transition is dependent on E2F transcription factors family (for review, see Bertoli et al., 2013), it is relevant to note that E2F2 and E2F8 were also strongly induced in reactive astrocytes. Thus, proliferation of reactive astrocytes is regulated at the transcriptional level by increasing the transcription of several proproliferative components, while their regulation by the Shh pathway is not reflected at the transcriptome level at least at this time point.
Our analysis was performed at the peak of reactive astrocyte proliferation and accordingly, we found that many upregulated genes are involved in the generation of precursor metabolites including RNA metabolism and cell cycle promotion. However, only about 10–20% of astrocytes divide in this injury model (Bardehle et al., 2013; Simon et al., 2011), suggesting that only some may reach levels of expression sufficient to complete the cell cycle. Indeed, some genes, for example, Ccnd1 appear clearly in a subset of astrocytes (Fig. 3G, H and Supp. Info. Fig. 3D–F; Zamanian et al., 2012), supporting the concept that our analysis allows detection of genes expressed in the proliferative subset of reactive astrocytes.
Subtypes of astrocytes also exist in regard to GFAP expression as only a subset of Aldh1L1‐GFP+ have GFAP after stab wound injury. As our transcriptome analysis examined the GFAP+ cells using the hGFAP‐eGFP mice, this prompts the question to which extent altered gene expression in our transcriptome would also be detectable in the GFAP‐negative astrocytes. To get a first idea on this we examined five of the upregulated genes by co‐immunostaining for GFAP and GFP in Aldh1L1‐eGFP mice (Supp. Info. Fig. 4; Spp1, Gal‐1, Gal‐3, cyclin D1, and FGFR3). All of these were typically restricted to the vicinity of the injury site and to GFAP‐immunopositive cells, but virtually not detectable in GFAP‐negative, Aldh1L1‐GFP+ cells. Note that some of these nonreactive astrocytes are rather close to the injury site. Indeed, also live in vivo imaging of astrocytes after stab wound injury revealed a subset of astrocytes, sometimes in close vicinity to the injury site that showed no signs of reactivity (no proliferation, polarization, or detectable hypertrophy; Bardehle et al., 2013), consistent with the concept of reactive astrocyte heterogeneity with a subset showing little to no reaction. To further scrutinize this concept of a subset of nonreactive astrocytes, it will, however, be important to perform genomewide expression analysis by, for example, isolating the GFP+ and RFP+/GFP+ astrocytes from crosses of Aldh1L1‐eGFP and hGFAP‐RFP mice.
For the GFAP+ reactive astrocytes analyzed here, it is also important to note that not only proliferation genes are upregulated, but also the GO term analysis also indicates significant enrichment of genes involved in developmental processes and cell differentiation (Supp. Info. Fig. 1B, C and Supp. Info. Table 3), indicating the more immature nature of reactive astrocytes compared with astrocytes in the intact brain. Notably, some neurogenic factors, such as Sox11 (sex‐determining region y‐box, 11) (Mu et al., 2012; Ninkovic et al. 2013) are upregulated in reactive astrocytes, but do not reach the levels of the neurogenic priming observed in SEZ NSCs (Beckervordersandforth et al., 2010; Bracko et al, 2012; Götz et al., 2015). Consistent with some degree of cell fate change, the gliogenic genes, Nfib (nuclear factor I/B) and Sox9 are downregulated in reactive astrocytes compared with astrocytes from the intact brain. Likewise some genes also expressed in SEZ NSCs, such as Sox2, Musashi1 and its up‐stream regulator Rfx7 (regulatory factor, ×7), are rather downregulated in reactive astrocytes (Supp. Info. Table 2), possibly counteracting the further dedifferentiation. Taken together, genomewide expression analysis not only reveals a significant number of genes expressed by endogenous adult SEZ NSCs, which are upregulated by reactive astrocytes, but also identifies genes involved in limiting their self‐renewal capacity in vivo and neurogenic progeny (see, e.g., Shimada et al., 2012).
We note that previous studies have indicated that NSCs activate similar pathways when exposed to injury, such as production of chemokines, cytokines, and TLRs (Kokaia et al., 2012; Lindvall and Kokaia, 2010; Martino and Pluchino, 2006). This is often accompanied by a switch from neurogenesis to gliogenesis after injury (Sierra et al., 2015), suggesting that some of the activated pathways may also contribute to favor the glial lineage. Intriguingly, some of this glial progeny derived from SEZ NSCs after injury becomes migratory toward the injury site (Benner et al., 2013), while local reactive astrocytes in the cerebral cortex GM do not migrate (Bardehle et al., 2013). NSC‐derived reactive astrocytes may exert beneficial functions in the injury site (Sabelström et al. 2013), further emphasizing the significance of comparing the properties and molecular signature of endogenous adult NSCs with those of reactive astrocytes (for review, see Götz et al., 2015; Grégoire et al., 2015; Robel et al., 2011; Silver and Steindler, 2009).
Galectins 1 and 3 as Necessary and Sufficient Regulators of Reactive Astrocyte Proliferation and Their NSC Potential
Both Galectins 1 and 3 are prominently expressed in adult NSCs and reactive astrocytes (Beckervordersandforth et al., 2010; Di Lella et al., 2011; Kajitani et al., 2009; Sakaguchi et al., 2006; Yan et al., 2009), and their expression levels also increase after other injuries, including middle cerebral artery occlusion (MCAo) (Qu et al., 2011; Young et al., 2014; Zamanian et al., 2012) and cancer (for review, see Camby et al., 2006; Griffioen and Thijssen, 2014; LeMercier et al., 2010; Newlaczyl and Yu, 2011). Interestingly, Lgals3 expression is not increased upon LPS stimulation, an injury condition with limited proliferative response of reactive astrocytes (Zamanian et al., 2012).
Our data now show that Gal‐1 and Gal‐3 label a specific subset of reactive astrocytes (∼25%) with Gal‐3 selectively labeling the set of proliferating astrocytes. This correlation was further substantiated at the functional level using both loss‐ and gain‐of‐function experiments. Reactive astrocyte proliferation was significantly reduced in Lgals1 −/− Lgals3 −/− mice, while their proliferation was increased after treatment with Gal‐1 or Gal‐3 in vitro. Moreover, these experiments demonstrate that defects in proliferation of Lgals1 −/− Lgals3 −/− astrocytes may not reflect developmental defects, since they can be fully rescued by Gal‐3 addition to astrocytes from the adult cerebral cortex. The absence of developmental contributions to the phenotype observed in reactive astrocytes is further corroborated by normal numbers of S100β+ astrocytes without and with injury in the Lgals1 −/− Lgals3 −/− mice. In contrast to Gal‐3 addition in vitro, Gal‐1 addition markedly reduces the number and proliferation of microglia and astrocytes, consistent with previous in vivo application after the focal ischemia (Qu et al., 2011). The antiproliferative effect of Gal‐1 was also observed in neuroblastoma cells (Kopitz et al., 2001) and a panel of carcinoma cells of different origin (Fischer et al., 2005). Gal‐1 selectively increases p27 protein stability by inhibition of the Ras‐MEK‐ERK cascade and induces p21 transcription via interaction with α5β1 integrin (Fischer et al., 2005). It is thus conceivable that Gal‐1‐mediated accumulation of these two potent cyclin‐dependent kinase 2 inhibitors may operate also in reactive glial cells, ultimately arresting them in G1 phase of cell cycle. Besides regulation of cell cycle, exogenously added Gal‐1 also exerts effects through interaction with integrins (Astorgues‐Xerri et al., 2014; Moiseeva et al., 2003), such as activating proapoptotic α5β1‐integrin signaling (Sanchez‐Ruderisch et al., 2011). Gal‐1‐induced apoptosis involves several intracellular mediators including the transcription factor AP1 and the downregulation of Bcl2 protein production (Hahn et al., 2004; Rabinovich et al., 2000; Walzel et al., 2000). Taking into account that α5β1 integrin is an important mediator of microglial activation (Milner et al., 2007), cell surface presentation of Gal‐1 may be implicated in the α5β1‐integrin signaling also leading to death of microglia, consistent with our in vitro observations. Thus, Gal‐1 addition reduces the number of microglia, the vast majority of glial cells in the cultures of reactive glia isolated from the stab wound injured cerebral cortex, likely by both reducing their activation and proliferation as well as their survival. This provides the intriguing perspective to selectively regulate specific types of glial cells after injury by taking advantage of the selective effects of Gal‐1 reducing microglia numbers, while Gal‐3 promotes astrocyte number and proliferation.
Most importantly, our experiments in vitro reveal functional divergence of Galectins, as extracellular Gal‐3 can exert a proproliferative effect on reactive astrocyte proliferation in vitro. One mode of Gal‐3 function may be to bind and/or crosslink cell‐surface glycoconjugates, influencing thereby their activity (for review, see Liu and Rabinovich, 2005; Priglinger et al., 2013; Yang et al., 2008). This highlights that Gal‐1 and Gal‐3 can also exert effects in a noncell‐autonomous manner on reactive astrocytes, as also microglia, some neurons and NG2 glia express Gal‐1 and Gal‐3 in the cerebral cortex GM. This is why it is important to examine in this case mice with Galectin depletion in all cells, rather than cell type‐specific gene deletion. Gal‐3 has been shown to affect various cell cycle regulators also by intracellular function in other cell types, including upregulation of cyclin D1 through direct activation of its promoter or by its binding to β‐catenin, a known mediator of the canonical Wnt signaling that regulates cyclin D1 and cell cycle progression (Kim et al., 1999; Lin et al., 2002; Shimura et al., 2004). Cyclin D1 is required for the injury‐induced proliferation of glial cells, including astrocytes (Nobs et al., 2013). Indeed, beyond downregulation of cyclin D1 also Ki67 was reduced in Lgals1 −/− Lgals3 −/− reactive astrocytes (Supp. Info. Fig. 3D–F), suggesting that Galectins may regulate cell cycle progression at the G1‐S‐phase transition, as shown also in other cell types (Kim et al., 1999; Lin et al., 2002; Shimura et al., 2004). We note that previous studies of Lgals3 −/− mice did not report reductions in reactive astrocyte proliferation in striatum after MCAo (Young et al., 2014), suggesting that other Galectins or co‐factors may compensate loss of Lgals3, but not the loss of both Lgals1 and Lgals3, as our results indicate.
We found that Galectins also regulate the neurosphere‐forming capacity of reactive astrocytes: it is reduced in the Lgals1 −/− Lgals3 −/− mice after injury and increased upon Gal‐3, but not Gal‐1 addition. Thus, Gal‐3 expression is not only shared between adult NSCs and reactive astrocytes, but also seems to be a key factor regulating NSC properties as reflected in the multipotency and self‐renewal of the neurosphere assay. Given the role of Galectins in promoting the NSC potential of reactive astrocytes observed in our study, future regenerative therapies may use such tools to enhance the local stem cell pool. This is particularly promising as we could show that extracellular addition of Gal‐3 is able to promote NSC potential.
In sum, our comparative transcriptome analyses identified two Galectins not only as markers in the lesioned adult cerebral cortex, but also as regulators of the proliferating and neurosphere‐forming capacity of a subset of reactive astrocytes, thereby providing a rich data source toward a better understanding of the key factors regulating astrocyte heterogeneity after injury.
Supporting information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgment
The authors thank Gabi Jäger, Carmen Meyer, Andrea Steiner‐Mezzadri, Tatiana Simon‐Ebert and Detlef Franzen for excellent technical assistance, Dr. Corinna Haupt for great help in the initial phase of the Galectin mutant project and Dr. Ruth Beckervordersandforth for great help in sorting of reactive astrocytes.
References
- Anderson MA, Ao Y, Sofroniew MV. 2014. Heterogeneity of reactive astrocytes. Neurosci Lett 565:23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astorgues‐Xerri L, Riveiro ME, Tijeras‐Raballand A, Serova M, Neuzillet C, Albert S, Raymond E, Faivre S. 2014. Unraveling galectin‐1 as a novel therapeutic target for cancer. Cancer Treat Rev 40:307–319. [DOI] [PubMed] [Google Scholar]
- Bardehle S, Krüger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, Snippert HJ, Theis FJ, Meyer‐Luehmann M, Bechmann I, Dimou L, Götz M. 2013. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 16:580–586. [DOI] [PubMed] [Google Scholar]
- Barnes EA, Kong M, Ollendorff V, Donoghue DJ. 2001. Patched1 interacts with cyclin B1 to regulate cell cycle progression. Embo J 20:2214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckervordersandforth R, Tripathi P, Ninkovic J, Bayam E, Lepier A, Stempfhuber B, Kirchhoff F, Hirrlinger J, Haslinger A, Lie DC, Beckers J, Yoder B, Irmler M, Götz M. 2010. In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell 7:744–758. [DOI] [PubMed] [Google Scholar]
- Behrendt G, Baer K, Buffo A, Curtis MA, Faull RL, Rees MI, Götz M, Dimou L. 2013. Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia 61:273–286. [DOI] [PubMed] [Google Scholar]
- Benner EJ, Luciano D, Jo R, Abdi K, Paez‐Gonzalez P, Sheng H, Warner DS, Liu C, Eroglu C, Kuo CT. 2013. Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature 497:369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Skotheim JM, de Bruin RA. 2013. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14:518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff V, Deogracias R, Poirier F, Barde YA, 2012. Seizure‐induced neuronal death is suppressed in the absence of the endogenous lectin Galectin‐1. J Neurosci 32:15590–15600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzier‐Sore AK, Pellerin L. 2013. Unraveling the complex metabolic nature of astrocytes. Front Cell Neurosci 7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracko O, Singer T, Aigner S, Knobloch M, Winner B, Ray J, Clemenson GD, Jr , Suh H, Couillard‐Despres S, Aigner L, Gage FH, Jessberger S. 2012. Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J Neurosci 32:3376–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill MS, Ninkovic J, Winpenny E, Hodge RD, Ozen I, Yang R, Lepier A, Gascón S, Erdelyi F, Szabo G, Parras C, Guillemot F, Frotscher M, Berninger B, Hevner RF, Raineteau O, Götz M. 2009. Adult generation of glutamatergic olfactory bulb interneurons. Nat Neurosci 12:1524–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Götz M. 2008. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci USA 105:3581–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. 2014. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81:229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. 2008. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camby I, Le Mercier M, Lefranc F, Kiss R. 2006. Galectin‐1: a small protein with major functions. Glycobiology 16:137–157. [DOI] [PubMed] [Google Scholar]
- Chan JL, Reeves TM, Phillips LL. 2014. Osteopontin expression in acute immune response mediates hippocampal synaptogenesis and adaptive outcome following cortical brain injury. Exp Neurol 261:757–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codega P, Silva‐Vargas V, Paul A, Maldonado‐Soto AR, Deleo AM, Pastrana E, Doetsch F. 2014. Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron 82:545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C, Fowlis D, Ripoche MA, Bouchaert I, Poirier F. 1998. Embryonic implantation in galectin 1/galectin 3 double mutant mice. Dev Dyn 211:306–313. [DOI] [PubMed] [Google Scholar]
- Di Lella S, Sundblad V, Cerliani JP, Guardia CM, Estrin DA, Vasta GR, Rabinovich GA. 2011. When lectins recognize glycans: from biochemistry to physiology and back again. Biochemistry 50:7842–7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimou L, Götz M. 2014. Glial cells as progenitors and stem cells: new roles in the healthy and diseased brain. Physiol Rev 94:709–737. [DOI] [PubMed] [Google Scholar]
- Doree M, Hunt T. 2002. From cdc2 to Cdk1: When did the cell cycle kinase join its cyclin partner?. J Cell Sci 115:2461–2464. [DOI] [PubMed] [Google Scholar]
- Engelmann C, Weih F, Haenold R. 2014. Role of nuclear factor kappa B in central nervous system regeneration. Neural Regen Res 9:707–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsig J, Pörzgen P, Lotharius J, Leist M. 2004. Specific modulation of astrocyte inflammation by inhibition of mixed lineage kinases with CEP‐1347. J Immunol 173:2762–2770. [DOI] [PubMed] [Google Scholar]
- Fischer C, Sanchez‐Ruderisch H, Welzel M, Wiedenmann B, Sakai T, André S, Gabius HJ, Khachigian L, Detjen KM, Rosewicz S. 2005. Galectin‐1 interacts with the α5β1 fibronectin receptor to restrict carcinoma cell growth via induction of p21 and p27. J Biol Chem 280:37266–37277. [DOI] [PubMed] [Google Scholar]
- Gadea A, Schinelli S, Gallo V. 2008. Endothelin‐1 regulates astrocyte proliferation and reactive gliosis via a JNK/c‐Jun signaling pathway. J Neurosci 28:2394–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz M, Sirko S. 2013. Potential of glial cells In: Stewart S, editor. Stem cells handbook. 2nd ed., New York: Springer, pp 347–361. [Google Scholar]
- Götz M, Sirko S, Beckers J, Irmler M. 2015. Reactive astrocytes as neural stem or progenitor cells: In vivo lineage, in vitro potential, and genome‐wide expression analysis. Glia 63:1452–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande A, Sumiyoshi K, López‐Juárez A, Howard J, Sakthivel B, Aronow B, Campbell K, Nakafuku M. 2013. Environmental impact on direct neuronal reprogramming in vivo in the adult brain. Nat Commun 4:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grégoire CA, Goldenstein BL, Floriddia EM, Barnabé‐Heider F, Fernandes KJL. 2015. Endogenous neural stem cell responses to stroke and spinal cord injury. Glia 63:1469–1482. [DOI] [PubMed] [Google Scholar]
- Griffioen AW, Thijssen VL. 2014. Galectins in tumor angiogenesis. Ann Transl Med 2:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr. 1999. NF‐kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 19:5785–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn HP, Pang M, He J, Hernandez JD, Yang RY, Li LY, Wang X, Liu FT, Baum LG. 2004. Galectin‐1 induces nuclear translocation of endonuclease G in caspase‐ and cytochrome c‐independent T cell death. Cell Death Differ 11:1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby ME, Coppola G, Ao Y, Geschwind DH, Khakh BS, Sofroniew MV. 2012. Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G‐protein‐coupled receptors. J Neurosci 32:14489–14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz N. 2004. Gene expression nervous system atlas (GENSAT). Nat Neurosci 7:483. [DOI] [PubMed] [Google Scholar]
- Henn A, Kirner S, Leist M. 2011. TLR2 hypersensitivity of astrocytes as functional consequence of previous inflammatory episodes. J Immunol 186:3237–3247. [DOI] [PubMed] [Google Scholar]
- Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. 1999. NF‐kappaB function in growth control: regulation of cyclin D1 expression and G0/G1‐to‐S‐phase transition. Mol Cell Biol 19:2690–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson PA, Irmler M, Acampora D, Beckers J, Simeone A, Götz M. 2013. The transcription factor Otx2 regulates choroid plexus development and function. Development 140:1055–1066. [DOI] [PubMed] [Google Scholar]
- Kajitani K, Nomaru H, Ifuku M, Yutsudo N, Dan Y, Miura T, Tsuchimoto D, Sakumi K, Kadoya T, Horie H, Poirier F, Noda M, Nakabeppu Y. 2009. Galectin‐1 promotes basal and kainate‐induced proliferation of neural progenitors in the dentate gyrus of adult mouse hippocampus. Cell Death Differ 16:4117–4127. [DOI] [PubMed] [Google Scholar]
- Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, Verveer M, de Groot LR, Smith VD, Rangarajan S, Rodríguez JJ, Orre M, Hol EM. 2012. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One 7:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, Balordi F, Su N, Chen L, Fishell G, Hébert JM. 2014. Astrocyte activation is suppressed in both normal and injured brain by FGF signaling. Proc Natl Acad Sci USA 111:2987–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney AM, Rowitch DH. 2000. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol 20:9055–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. 2011. Physiology of microglia. Physiol Rev 91:461–553. [DOI] [PubMed] [Google Scholar]
- Kim HR, Lin HM, Biliran H, Raz A. 1999. Cell cycle arrest and inhibition of anoikis by galectin‐3 in human breast epithelial cells. Cancer Res 59:4148–4154. [PubMed] [Google Scholar]
- Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Araúzo‐Bravo MJ, Kovacs WJ, Karalay O, Suter U, Machado RA, Roccio M, Lutolf MP, Semenkovich CF, Jessberger S. 2013. Metabolic control of adult neural stem cell activity by Fasn‐dependent lipogenesis. Nature 493:226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia Z, Martino G, Schwartz M, Lindvall O. 2012. Cross‐talk between neural stem cells and immune cells: the key to better brain repair? Nat Neurosci 15:1078–1087. [DOI] [PubMed] [Google Scholar]
- Kopitz J, von Reitzenstein C, André S, Kaltner H, Uhl J, Ehemann V, Cantz M, Gabius HJ. 2001. Negative regulation of neuroblastoma cell growth by carbohydrate‐dependent surface binding of galectin‐1 and functional divergence from galectin‐3. J Biol Chem 76:35917–35923. [DOI] [PubMed] [Google Scholar]
- Le Mercier M, Fortin S, Mathieu V, Kiss R, Lefranc F. 2010. Galectins and gliomas. Brain Pathol 20:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HM, Pestell RG, Raz A, Kim HR. 2002. Galectin‐3 enhances cyclin D1 promoter activity through SP1 and a cAMP‐responsive element in human breast epithelial cells. Oncogene 21:8001–8010. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Kokaia Z. 2010. Stem cells in human neurodegenerative disorders—time for clinical translation? Physiol J Clin Invest 120:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu FT, Hsu DK, Zuberi RI, Kuwabara I, Chi EY, Henderson Jr WR. 1995. Expression and function of galectin‐3, a beta‐galactosidebinding lectin, in human monocytes and macrophages. Am J Pathol 147:1016–1028. [PMC free article] [PubMed] [Google Scholar]
- Liu FT, Rabinovich GA. 2005. Lectins as modulators of tumour progression. Nat Rev Cancer 5:29–41. [DOI] [PubMed] [Google Scholar]
- Martín‐López E, García‐Marques J, Núñez‐Llaves R, López‐Mascaraque L. 2013. Clonal astrocytic response to cortical injury. PLoS One 8:e74039, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino G, Pluchino S. 2006. The therapeutic potential of neural stem cells. Nat Rev Neurosci 7:395–406. [DOI] [PubMed] [Google Scholar]
- Maryanovich M, Gross A. 2013. A ROS rheostat for cell fate regulation. Trends Cell Biol. 23:129–134. [DOI] [PubMed] [Google Scholar]
- Milner R, Crocker SJ, Hung S, Wang X, Frausto RF, del Zoppo GJ. 2007. Fibronectin‐ and vitronectin‐induced microglial activation and matrix metalloproteinase‐9 expression is mediated by integrins alpha5beta1 and alphavbeta5. J Immunol 178:8158–8167. [DOI] [PubMed] [Google Scholar]
- Moiseeva EP, Williams B, Goodall AH, Samani NJ. 2003. Galectin‐1 interacts with beta‐1 subunit of integrin. Biochem Biophys Res Commun 310:1010–1016. [DOI] [PubMed] [Google Scholar]
- Mu L, Berti L, Masserdotti G, Covic M, Michaelidis TM, Doberauer K, Merz K, Rehfeld F, Haslinger A, Wegner M, Sock E, Lefebvre V, Couillard‐Despres S, Aigner L, Berninger B, Lie DC. 2012. SoxC transcription factors are required for neuronal differentiation in adult hippocampal neurogenesis. J Neurosci 32:3067–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napetschnig J, Wu H. 2013. Molecular basis of NF‐κB signaling. Annu Rev Biophys 42:443–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlaczyl AU, Yu LG. 2011. Galectin‐3: a jack‐of‐all‐trades in cancer. Cancer Lett 313:123–128. [DOI] [PubMed] [Google Scholar]
- Ninkovic J, Steiner‐Mezzadri A, Jawerka M, Akinci U, Masserdotti G, Petricca S, Fischer J, von Holst A, Beckers J, Lie CD, Petrik D, Miller E, Tang J, Wu J, Lefebvre V, Demmers J, Eisch A, Metzger D, Crabtree G, Irmler M, Poot R, Götz M. 2013. The BAF complex interacts with Pax6 in adult neural progenitors to establish a neurogenic cross‐regulatory transcriptional network. Cell Stem Cell 13:403–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobs L, Nestel S, Kulik A, Nitsch C, Atanasoski S. 2013. Cyclin D1 is required for proliferation of Olig2‐expressing progenitor cells in the injured cerebral cortex. Glia 61:1443–1455. [DOI] [PubMed] [Google Scholar]
- Nolte C, Matyash M, Pivneva T, Schipke CG, Ohlemeyer C, Hanisch UK, Kirchhoff F, Kettenmann H. 2001. GFAP promoter‐controlled EGFP‐expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia 33:72–86. [PubMed] [Google Scholar]
- Oeckinghaus A, Hayden MS, Ghosh S. 2011. Crosstalk in NF‐κB signaling pathways. Nat Immunol 12:695–708. [DOI] [PubMed] [Google Scholar]
- Orre M, Kamphuis W, Osborn LM, Jansen AH, Kooijman L, Bossers K, Hol EM. 2014. Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol Aging 35:2746–2760. [DOI] [PubMed] [Google Scholar]
- Pellerin L. 2008. Brain energetics (thought needs food). Curr Opin Clin Nutr Metab Care 11:701–705. [DOI] [PubMed] [Google Scholar]
- Pinto L, Drechsel D, Schmid MT, Ninkovic J, Irmler M, Brill MS, Restani L, Gianfranceschi L, Cerri C, Weber SN, Tarabykin V, Baer K, Guillemot F, Beckers J, Zecevic N, Dehay C, Caleo M, Schorle H, Götz M. 2009. AP2gamma regulates basal progenitor fate in a region‐ and layer‐specific manner in the developing cortex. Nat Neurosci 12:1229–1237. [DOI] [PubMed] [Google Scholar]
- Pinto L, Mader MT, Irmler M, Gentilini M, Santoni F, Drechsel D, Blum R, Stahl R, Bulfone A, Malatesta P, Beckers J, Götz M. 2008. Prospective isolation of functionally distinct radial glial subtypes–lineage and transcriptome analysis. Mol Cell Neurosci 38:15–42. [DOI] [PubMed] [Google Scholar]
- Plantman S. 2012. Osteopontin is upregulated after mechanical brain injury and stimulates neurite growth from hippocampal neurons through β1 integrin and CD44. Neuroreport 23:647–652. [DOI] [PubMed] [Google Scholar]
- Priglinger CS, Szober CM, Priglinger SG, Merl J, Euler KN, Kernt M, Gondi G, Behler J, Geerlof A, Kampik A, Ueffing M, Hauck SM. 2013. Galectin‐3 induces clustering of CD147 and integrin‐201transmembrane glycoprotein receptors on the RPE cell surface. PLoS One 8:e70011, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu WS, Wang YH, Ma JF, Tian DS, Zhang Q, Pan DJ, Yu ZY, Xie MJ, Wang JP, Wang W. 2011. Galectin‐1 attenuates astrogliosis‐associated injuries and improves recovery of rats following focal cerebral ischemia. J Neurochem 116:217–226. [DOI] [PubMed] [Google Scholar]
- Rainer J, Sanchez‐Cabo F, Stocker G, Sturn A, Trajanoski Z. 2006. CARMAweb: comprehensive R‐ and bioconductor‐based web service for microarray data analysis. Nucleic Acids Res 34:498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich GA, Alonso CR, Sotomayor CE, Durand S, Bocco JL, Riera CM. 2000. Molecular mechanisms implicated in galectin‐1‐induced apoptosis: activation of the AP‐1 transcription factor and downregulation of Bcl‐2. Cell Death Differ 7:747–753. [DOI] [PubMed] [Google Scholar]
- Robel S, Berninger B, Götz M. 2011. The stem cell potential of glia: lessons from reactive gliosis. Nat Rev Neurosci 12:88–104. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A, Sánchez P, Dahmane N. 2002. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer 2:361−372. [DOI] [PubMed] [Google Scholar]
- Sabelström H, Stenudd M, Réu P, Dias DO, Elfineh M, Zdunek S, Damberg P, Göritz C, Frisén J. 2013. Resident neural stem cells restrict tissue damage and neuronal loss after spinal cord injury in mice. Science 342:637–640. [DOI] [PubMed] [Google Scholar]
- Sakaguchi M, Shingo T, Shimazaki T, Okano HJ, Shiwa M, Ishibashi S, Oguro H, Ninomiya M, Kadoya T, Horie H, Shibuya A, Mizusawa H, Poirier F, Nakauchi H, Sawamoto K, Okano H. 2006. A carbohydrate‐binding protein, Galectin‐1, promotes proliferation of adult neural stem cells. Proc Natl Acad Sci USA 103:7112–7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Ruderisch H, Detjen KM, Welzel M, André S, Fischer C, Gabius HJ, Rosewicz S. 2011. Galectin‐1 sensitizes carcinoma cells to anoikis via the fibronectin receptor α5β1‐integrin. Cell Death Differ 18:806–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada IS, LeComte MD, Granger JC, Quinlan NJ, Spees JL. 2012. Self‐renewal and differentiation of reactive astrocyte‐derived neural stem/progenitor cells isolated from the cortical peri‐infarct area after stroke. J Neurosci 32:7926–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura T, Takenaka Y, Tsutsumi S, Hogan V, Kikuchi A, Raz A. 2004. Galectin‐3, a novel binding partner of β‐catenin. Cancer Res 64:6363–6367. [DOI] [PubMed] [Google Scholar]
- Silver DJ, Steindler DA. 2009. Common astrocytic programs during brain development, injury and cancer. Trends Neurosci 32:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Martín‐Suárez S, Valcárcel‐Martín R, Pascual‐Brazo J, Aelvoet SA, Abiega O, Deudero JJ, Brewster AL, Bernales I, Anderson AE, Baekelandt V, Maletić‐Savatić M, Encinas JM. 2015. Neuronal hyperactivity accelerates depletion of neural stem cells and impairs hippocampal neurogenesis. Cell Stem Cell 16:488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C, Götz M, Dimou L. 2011. Progenitors in the adult cerebral cortex: cell cycle properties and regulation by physiological stimuli and injury. Glia 59:869–881. [DOI] [PubMed] [Google Scholar]
- Sirko S, Behrendt G, Johansson PA, Tripathi P, Costa M, Bek S, Heinrich C, Tiedt S, Colak D, Dichgans M, Fischer IR, Plesnila N, Staufenbiel M, Haass C, Snapyan M, Saghatelyan A, Tsai LH, Fischer A, Grobe K, Dimou L, Götz M. 2013. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. Cell Stem Cell 12:426–439. [DOI] [PubMed] [Google Scholar]
- Sirko S, Neitz A, Mittmann T, Horvat‐Bröcker A, von Holst A, Eysel UT, Faissner A. 2009. Focal laser‐lesions activate an endogenous population of neural stem/progenitor cells in the adult visual cortex. Brain 132:2252–2264. [DOI] [PubMed] [Google Scholar]
- Stahl R, Walcher T, De Juan Romero C, Pilz GA, Cappello S, Irmler M, Sanz‐Aquela JM, Beckers J, Blum R, Borrell V, Götz M. 2013. Trnp1 regulates expansion and folding of the mammalian cerebral cortex by control of radial glial fate. Cell 153:535–549. [DOI] [PubMed] [Google Scholar]
- Starossom SC, Mascanfroni ID, Imitola J, Cao L, Raddassi K, Hernandez SF, Bassil R, Croci DO, Cerliani JP, Delacour D, Wang Y, Elyaman W, Khoury SJ, Rabinovich GA. 2012. Galectin‐1 deactivates classically activated microglia and protects from inflammation‐induced neurodegeneration. Immunity 37:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele ML, Robinson SR. 2012. Reactive astrocytes give neurons less support: implications for Alzheimer's disease. Neurobiol Aging 33:423.e1–e13. [DOI] [PubMed] [Google Scholar]
- Wahl V, Vogler S, Grosche A, Pannicke T, Ueffing M, Wiedemann P, Reichenbach A, Hauck SM, Bringmann A. 2013. Osteopontin inhibits osmotic swelling of retinal glial (Müller) cells by inducing release of VEGF. Neuroscience 246:59–72. [DOI] [PubMed] [Google Scholar]
- Walcher T, Xie Q, Sun J, Irmler M, Beckers J, Öztürk T, Niessing D, Stoykova A, Cvekl A, Ninkovic J, Götz M. 2013. Functional dissection of the paired domain of Pax6 reveals molecular mechanisms of coordinating neurogenesis and proliferation. Development 140:1123–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther M, Kuklinski S, Pesheva P, Guntinas‐Lichius O, Angelov DN, Neiss WF, Asou H, Probstmeier R. 2000. Galectin‐3 is upregulated in microglial cells in response to ischemic brain lesions, but not to facial nerve axotomy. J Neurosci Res 61:430–435. [DOI] [PubMed] [Google Scholar]
- Walzel H, Blach M, Hirabayashi J, Kasai K, Brock J. 2000. Involvement of CD2 and CD3 in galectin‐1 induced signaling in human Jurkat T‐cells. Glycobiology 10:131–140. [DOI] [PubMed] [Google Scholar]
- Yan YP, Lang BT, Vemuganti R, Dempsey RJ. 2009. Galectin‐3 mediates post‐ischemic tissue remodeling. Brain Res 1288:116–124. [DOI] [PubMed] [Google Scholar]
- Yang RY, Rabinovich GA, Liu FT. 2008. Galectins: structure, function and therapeutic potential. Expert Rev Mol Med 13:1–12. [DOI] [PubMed] [Google Scholar]
- Yang Y, Vidensky S, Jin L, Jie C, Lorenzini I, Frankl M, Rothstein JD. 2011. Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia 59:200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CC, Al‐Dalahmah O, Lewis NJ, Brooks KJ, Jenkins MM, Poirier F, Buchan AM, Szele FG. 2014. Blocked angiogenesis in Galectin‐3 null mice does not alter cellular and behavioral recovery after middle cerebral artery occlusion stroke. Neurobiol Dis 63:155–164. [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. 2012. Genomic analysis of reactive astrogliosis. J Neurosci 32:6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer B, Kuegler PB, Baudis B, Genewsky A, Tanavde V, Koh W, Tan B, Waldmann T, Kadereit S, Leist M. 2011. Coordinated waves of gene expression during neuronal differentiation of embryonic stem cells as basis for novel approaches to developmental neurotoxicity testing. Cell Death Differ 18:383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Tobaru T, Zivin JA, Shackelford DA. 1998. Activation of nuclear factor‐kappaB in the rabbit spinal cord following ischemia and reperfusion. Brain Res Mol Brain Res 63:121–132. [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. 2014. An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information