

Abstract
Adrenocortical cancer (ACC) is a rare, but agressive malignancy with poor prognosis. Histopathological diagnosis is challenging and pharmacological options for treatment are limited. By the comparative reanalysis of the transcriptional malignancy signature with the cell cycle dependent transcriptional program of ACC, we aimed to identify novel biomarkers which may be used in the histopathological diagnosis and for the prediction of therapeutical response of ACC. Comparative reanalysis of publicly available microarray datasets included three earlier studies comparing transcriptional differences between ACC and benign adrenocortical adenoma (ACA) and one study presenting the cell cycle dependent gene expressional program of human ACC cell line NCI-H295R. Immunohistochemical analysis was performed on ACC samples. In vitro effects of antineoplastic drugs including gemcitabine, mitotane and 9-cis-retinoic acid alone and in combination were tested in the NCI-H295R adrenocortical cell line. Upon the comparative reanalysis, ribonucleotide reductase subunit 2 (RRM2), responsible for the ribonucleotide dezoxyribonucleotide conversion during the S phase of the cell cycle has been validated as cell cycle dependently expressed. Moreover, its expression was associated with the malignancy signature, as well. Immunohistochemical analysis of RRM2 revealed a strong correlation with Ki67 index in ACC. Among the antiproliferative effects of the investigated compounds, gemcitabine showed a strong inhibition of proliferation and an increase of apoptotic events. Additionally, RRM2 has been upregulated upon gemcitabine treatment. Upon our results, RRM2 might be used as a proliferation marker in ACC. RRM2 upregulation upon gemcitabine treatment might contribute to an emerging chemoresistance against gemcitabine, which is in line with its limited therapeutical efficacy in ACC, and which should be overcome for successful clinical applications.
Keywords: Fluorescence activated cell sorting (FACS), adrenocortical cancer (ACC), ribonucleotide reductase, ribonucleotide reductase subunit 2 (RRM2), cell cycle, cell cycle-dependent, malignancy signature, proliferation marker, gemcitabine-resistance, chemoresistance
Introduction
Adrenocortical cancer (ACC) is a rare, but highly agressive malignancy with poor prognosis [1], with annual incidence varying between 0.7 to 2.0 cases per million population [1,2]. Although surgical removal in the case of strictly localized tumors might result in favourable outcome, overall 5-year survival rate remains poor at only 22-37% [3,4]. Pharmacological options are limited mostly to the use of the adrenolytic agent mitotane [1], therefore, the applicability of various other drugs frequently used in advanced stage is under investigations [1,5-9]. Novel pharmacological targets are needed, although, due to the rarity of the disease, prospective, controlled trials of novel drugs are challenging [1].
The two most frequently altered molecular pathways in adrenocortical tumorigenesis are Wnt/β-catenin and IGF-2 signaling pathways [1]. Pharmacological targeting of IGF signaling resulted in a marked decrease in proliferation [10] and it potentiated the effect of simultaneously administered mitotane [10]. Additionally, blocking of intrinsically produced IGF-2 increased the antiproliferative effect of mTOR inhibitor sirolimus on ACC cell line [11]. In contrast with these promising in vitro data, the small molecular IGF1-receptor inhibitor linsitinib turned out to be ineffective in a recent clinical trial [12].
In search of potential new targets in ACC, gene expression comparison between ACCs and benign adrenocortical adenomas (ACA) is of the utmost importance. Hundreds of genes are upregulated in ACC’s malignancy signature, the gene expressional hallmark differentiating malignant neoplasms (ACC) from benign lesions (ACA) [13,15]. Among these, several genes may represent promising targets for further therapies. Upon a complex bioinformatics analysis, we previously identified (i) cell cycle, (ii) retinoic acid signaling and (iii) complement system and antigen presentation as the three major pathogenic pathways of adrenocortical tumorigenesis [16]. In addition, our group confirmed the antiproliferative effect of 9-cis retinoic acid in ACC in vitro and in vivo [7,9].
During the cell cycle, several key agents display cell cycle dependent expression on mRNA and protein level [17]. Additionally, post-transcriptional modifications of certain proteins harbor specific role for the proper maintanence of cell cycle progression [17]. Inhibiting key regulators of the cell cycle machinery might decelerate cell cycle progression. Therefore, large emphasis was dedicated to create novel drugs targeting specific cell cycle regulators being tested in different clinical trials [18].
Detecting cell cycle dependent expression in unperturbed cells is challenging [19,20]. Synchronization based techniques, followed by time-course gene expression profiling have been widely used to detect the cell cycle dependent transcription program [20,22]. However, synchronization procedures result in growth imbalance and alteration of the cell cycle machinery [23,25]. We previously showed that DNA content based fluorescence activated cell sorting (cell cycle sort) is able to discriminate cell cycle phases without perturbating the cell cycle machinery [25]. High-throughput profiling of mRNA and miRNA transcripts revealed the cell cycle dependent transcriptional profile in the ACC cell line NCI-H295R [25].
Here, by the reanalysis of the previously described gene expression profiles measured in ACC and ACA samples and comparing with the cell cycle dependent transcription profile of NCI-H295R cell line, we identified ribonucleotide reductase subunit M2 (RRM2) as a cell cycle dependently expressed gene overexpressed in ACC compared to ACA. Moreover, expression level of the RRM2 protein correlated well with Ki67 labeling index in ACC samples, therefore, its expression may be used as a novel proliferation marker in ACC with a possibility of contributing to prognostic information upon histopathological diagnosis. Additionally, antiproliferative effect, in particular with regard to RRM2 expression, of gemcitabine (G), mitotane (M), 9-cis-retinoic acid (R) (the former targeting cell cycle progression specifically on RRM2) and their combinations on ACC cell line NCI-H295R were studied. Although significant reduction in proliferative activity of various drugs have been shown, RRM2 has been upregulated upon gemcitabine treatment. This upregulation implies an emerging chemoresistance against gemcitabine [26,27], resulting in limited antiproliferative effect in patients with ACC observed earlier [5,8].
Materials and methods
Reanalysis of publicly available microarray data
For the comparison of the cell cycle dependent transcripts with the malignancy signature of ACC, data from three previous microarray studies [13,15] examining gene expression alterations between ACC and ACA tissues were downloaded from the European Bioinformatics Institute Array Express database (accession number: E-TABM-311) and from National Center for Biotechnology Information’s Gene Expression Omnibus (accession numbers: GSE10927 and GSE14922), and were reanalyzed. In total, one hundred fifty-four microarray data (ACA = 87; ACC = 67) were studied. Cell cycle dependent expression profile of ACC cell line NCI-H295R, previously established by our research group [25], was downloaded from National Center for Biotechnology Information’s Gene Expression Omnibus (accession number: GSE73256). Significant gene expressional alterations between ACC vs. ACA and S vs. G1 phase exceeding two-fold change were compared.
Cell culture and treatments
Human adrenocortical cancer cell line NCI-H295R was obtained from the American Type Culture Collection (ATCC). NCI-H295R cells were cultured in Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 Ham (DMEM: F12) supplemented with 6.25 ng/ml insulin, 6.25 ng/ml transferrin, 6.25 ng/ml sodium selenite, 1.25 mg/ml bovine serum albumine, 5.35 ng/ml linoleic acid, 1% HEPES, 1% Penicillin-Streptomycin, 2.5% L-glutamine (Sigma-Aldrich Chemical Co.) and 2.5% Nu-Serum (BD Biosciences), as previously reported [25]. Cells were cultured at 37°C in a humidified 5% CO2 atmosphere.
For different treatments, 5×105 cells were plated in each well of a 6-well-plate (for subsequent flow cytometry, gene and protein expression studies) or 1×104 cells were plated in each well of a 96-well plate (for subsequent proliferation assay). After 24 h, eight treatment regimens of gemcitabine (G, G6423, Sigma-Aldrich Chemical Co.), mitotane (M, N12706, Sigma-Aldrich Chemical Co.) and 9-cis-retinoic acid (R, sc-205589A, Santa Cruz Biotechnology) were introduced: control, G, M, R, G+M, G+R, M+R, G+M+R. Drugs were resolved in DMSO and were added in the optimal concentrations of 5×10-6 mol/dm3 (G and M) or 5×10-5 mol/dm3 (R), as previously proposed [6,7,28]. Equal volume of 4 μL of DMSO vehicle was added to control samples as well. Thereafter, samples were collected 24 h, 48 h and 72 h after treatments begun.
Flow cytometry analysis
DNA content based fluorescence activated cell sorting (cell cycle sort) was performed as described previously [25]. Sorted cells to G1, S and G2 phases were reanalyzed, centrifuged, washed with ice-cold PBS and resuspended in QIAzol lysis reagent (Qiagen) or Western blot lysis buffer for subsequent RNA and protein isolation. Until RNA isolation or Western blot, samples were stored at -80°C [25].
For apoptosis and cell cycle distribution detection upon different treatments, cells were trypsinized, washed and centrifuged for 5 minutes at 1000 rpm. The supernatant was discarded, cells were resuspended in ice-cold 70% ethanol and incubated at room temperature for 20 minutes. Thereafter, cells were stored at -20°C for 30 minutes and cells were centrifuged for 5 minutes at 1000 rpm. The supernatant was discarded and cells were resuspended in a 100 ng/ml RNase containing extraction buffer (200 mmol/l Na2HPO4, pH = 7.8) and incubated at room temperature for 15 minutes. Then, propidium iodide was added to a final concentration of 10 ng/ml and cells were incubated for another 15 minutes. Thereafter, samples were measured by a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). At least 10,000 events were collected. Data were analyzed by Cell Quest Pro and Winlist softwares.
Cortisol measurements in NCI-H295R cell supernatants after various treatments
After treatments, cell culture medium was aspirated, centrifuged for 10 minutes at 1000 rpm and the supernatants were stored at -80°C until steroid measurements.
For the liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, the external (cortisol solution 1 mg/ml in methanol) and the internal standard (cortisol-9, 11, 12, 12-D4 solution 100 µg/ml in methanol), both certified reference materials, were purchased from Sigma-Aldrich Hungary Ltd (Budapest, Hungary). LC-MS grade water, LC-MS grade methanol and LC-MS grade formic acid 85% were procured from VWR International Ltd (Debrecen, Hungary).
Sample preparation was based on protein precipitation. 5 µl internal standard (concentration 1 µg/ml) was added to 245 µl sample. 750 µl acetonitrile was then added to this mix and after vortexing it was centrifuged 5 minutes at 13500 rpm. The supernatant was diluted in 1:1 proportion with MS grade water.
LC-MS/MS assays were performed on a Perkin-Elmer Flexar FX10 ultra-performance liquid chromatograph coupled to a Sciex 5500QTRAP mass spectrometer (Per-Form Hungária Kft, Budapest, Hungary). For chromatographic separation we used a Phenomenex Kinetex XB-C18 stationary phase (50×2.1 mm, 1.7 µm) attached to a 2×4.6 mm Phenomenex SecurityGuard Ultra C18 guard column (Gen-Lab Kft, Budapest, Hungary) and kept at 35°C. The mobile phase consisted of water containing 0.1% formic acid (A) and methanol containing 0.1% formic acid (B). The following gradient program was used: 10% B, hold 1.5 minutes, ramp B to 90% in 2 minutes, hold 1.5 minutes, ramp B to 10% in 0.5 minutes, hold 1.5 minutes (total run time: 7 minutes). The eluent flow rate was 200 µl/min and the injection volume was 20 µl.
The mass spectrometer was operated in negative electrospray ionization mode with the following settings. Source temperature: 350°C, ionization voltage: -4500 V, curtain gas: 35 psi, gas 1: 35 psi, gas 2: 35 psi, entrance potential: -10 V.
Quantitative analysis was performed in multiple reaction monitoring (MRM) mode, with the following compound specific settings: cortisol quantifier ion 407.215→331 (declustering potential (DP): -55 V, collision energy (CE): -22 V, collision cell exit potential (CXP): -21 V), cortisol qualifier ion 407.215→282 (DP: -55 V, CE: -31 V, CXP: -27 V), internal standard quantifier ion 411.2→335.2 (DP: -60 V, CE: -31 V, CXP: -27 V) and internal standard qualifier ion 411.2→301.2 (DP: -60 V, CE: -45 V, CXP: -24 V).
Proliferation assay
The effect on cell proliferation of certain drug combinations was measured using alamarBlue cell proliferation reagent (DAL1025, Thermo Fischer Scientific). At time points 0 h, 24 h, 48 h and 72 h, 10 μL of AlamarBlue reagent was added to the culture medium. After 180 minutes incubation at 37°C in a humidified 5% CO2 atmosphere, fluorescence measurement was performed on Varioskan Flash spectral scanning reader (Thermo Fisher Scientific). Excitation and emission maxima were 560 and 590 nms, respectively. Acquired data at each time point were normalized to the fluorescence detected at 0 h at the same well.
RNA isolation and quantitative real-time PCR
For RNA isolation, samples were collected after successful cell cycle sort or at 24 h, 48 h, 72 h of the drug treatments. Cells were washed with ice-cold PBS, resuspended in QIAzol lysis reagent (Qiagen) and stored at -80°C until RNA isolation. Total RNA was isolated with miRNeasy Mini Kit (Qiagen), according to the manufacturer’s instructions. RNA concentration was determined by NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific). Total RNA was reverse transcribed with SuperScript VILO cDNA synthesis kit according to the manufacturer’s instructions (Thermo Fisher Scientific). Gene expression was quantified using predesigned TaqMan gene expression assays (probe IDs: RRM2: Hs00357247_g1, ACTB: Hs99999903_m1) on a 7500 Fast Real-time PCR system (Thermo Fisher Scientific). ACTB was used as internal control.
All measurements were performed in triplicate (three biological, two technical replicates). Expression level was calculated by the ΔCt (S-phase) ΔCt (G1-phase), the ΔCt (G2-phase) ΔCt (G1-phase) or the ΔCt (treated) ΔCt (control) (ΔΔCt) methods. Fold change was calculated by the FC = 2-ΔΔCt method.
Protein isolation and immunoblotting
Protein isolation, concentration measurements and Western blot analysis was performed as earlier reported [25]. Primary antibody dilutions were 1:200 for anti-RRM2 antibody (cat. No.: sc-10846, Santa Cruz Biotechnology) and 1:2000 for anti-β-actin antibody (cat. No.: 4967, Cell Signaling Technology). β-actin was used as loading control.
Immunohistochemistry
Formalin-fixed paraffin-embedded tissue sections of twelve ACCs were formerly diagnosed histopathologically at the 1st Department of Pathology and Experimental Cancer Research, Semmelweis University and were chosen for immunohistochemical analysis. The main demographic data of patients studied is summarized in Table 1. 4 µm-thick sections were cut, then mounted on SuperFrost/Plus slides and stored at 4°C until the staining. Slides were deparaffinized, rehydrated, and endogenous peroxidases were inhibited with 10% H2O2 for 20 minutes in methanol. Antigen retrieval was achieved by incubating slides at 100°C in TRS (10 mM Tris, 1 mM EDTA, 0.05% Tween 20, pH = 9) for 3 minutes. Ki67 staining was performed using the Novolink Polymer Detection System (Novocastra Laboratories, Peroxidase/DAB+, Rabbit). Non-specific binding was blocked (10 minutes, room temperature RT) with Novocastra™ Protein Block. Rabbit monoclonal anti-Ki67 (clone SP6, RM-9106, ThermoScientific) was used at 1:100 and incubated overnight at 4°C. After washing, incubation (30 minutes; RT) was performed with NovoLink Polymer. Slides were developed with DAB for 10 minutes and counterstained with haematoxylin. For RRM2 staining the non-specific binding was blocked with 5% bovine serum albumin (BSA, 20 minutes, RT). The endogenous biotin, biotin receptors and avidin binding sites in tissues were blocked (15-15 minutes, RT) with the avidin/biotin blocking kit (Vector laboratories, SP2001). Goat polyclonal anti-RRM2 (sc-10846, Santa Cruz Biotechnology) was used at 1:100 (overnight, 4°C). Biotinylated polyclonal rabbit anti-goat immunoglobulin (Dako Cytomation, E0466) was applied at 1:200 dilution for 1 h at RT. Sensitivity was enhanced with VECTASTAIN Elite ABC Kit (Standard, PK-6100, Vector Lab.) and staining was visualized with DAB Chromogen Kit (Biocare Medical, DB801R) for 10 minutes. Slides were counterstained with haematoxylin. The primary antibody was omitted in negative controls.
Table 1.
Baseline characteristics of tumors involved in the immunohistochemical analysis
| Pateint No. | Sex | Age (years) | Largest diameter (mm) | Modified Weiss score | Ki67 index (%) | RRM2 score |
|---|---|---|---|---|---|---|
| 1 | F | 62 | 85 | 5 | 14.8 | 23.3 |
| 2 | M | 79 | 120 | 5 | 7.5 | 18.2 |
| 3 | F | 46 | 200 | 6 | 36.9 | 56.7 |
| 4 | M | 25 | 100 | 6 | 14.1 | 17.5 |
| 5 | F | 50 | 120 | 5 | 14.6 | 18.6 |
| 6 | F | 55 | 90 | 6 | 21.2 | 28.0 |
| 7 | F | 69 | 110 | 6 | 21.8 | 44.8 |
| 8 | F | 61 | 80 | 5 | 16.8 | 26.8 |
| 9 | F | 54 | 80 | 5 | 18.5 | 30.2 |
| 10 | F | 62 | 130 | 6 | 4.2 | 15.7 |
| 11 | F | 61 | 120 | 5 | 18.6 | 27.6 |
| 12 | F | 47 | 140 | 5 | 37.1 | 86.2 |
F female, M male.
Evaluation of immunostainings
The stained slides were digitally scanned with a high-resolution scanner (Pannoramic Scan, 3DHISTECH Ltd.), and used for virtual microscopic evaluation with the Pannoramic Viewer 1.15 (3DHISTECH Ltd.). Ki67 and RRM2 stainings were scored independently in a blinded manner by 3 observers (VKG, KK and TM). Ki67 index was determined as the portion of positive cells upon examining 1000 cells. RRM2 score was determined as described earlier [29]. It was based on staining intensity of individual cells (0 = none, 1 = weak, 2 = moderate, 3 = strong) and the portion (0-100%) of cells staining at each intensity level [29]. RRM2 score was calculated by the RRM2 score = 1x+2y+3z method, where x, y, z are the percentage of cells with weak (x), moderate (y) and strong (z) immunoreactivity [29]. Modified Weiss score [30] was determined by a pathologist specialized in adrenal pathology (TM).
Statistical analysis
For detecting differently expressed mRNA transcripts, Students’ two sided independent samples T-test was used. Upon ΔCt values, differences were analyzed between G1 vs. S, S vs. G2 and G1 vs. G2 phases (cell cycle experiment) and treated vs. control groups (drug treatment experiment).
For detecting correlation between Ki67 index and RRM2 score, Spearman’s nonparametric correlation was used.
For detecting differences between drug-treated compared to untreated control groups at each time point in relative proliferation rate, cortisol concentration, apoptosis and cell cycle distribution, Students’ two sided independent samples T-test was used.
In all comparisons p-value <0.05 was considered statistically significant.
Results
Overlap between cell cycle dependent transcription program and the malignancy signature of ACC
Upon the average of the three microarray studies, 1752 transcripts had a minimum of two-fold change alteration between ACC and ACA, while 29 transcripts showed a minimum of two-fold change alteration between S and G1 phase in ACC cell line NCI-H295R (Figure 1A). 18 transcripts were altered in both the malignancy signature and in the cell cycle (Table 2). Among them, RRM2 had the largest upregulation in S compared to G1 phase. Additionally, RRM2 was significantly upregulated in ACC compared to ACA in all three microarray studies (ASPM was the other gene universally upregulated in all three microarray studies comparing ACCs with ACAs and upregulated in S phase compared to G1 phase as well). Upon these results, RRM2 was chosen for further examination.
Figure 1.
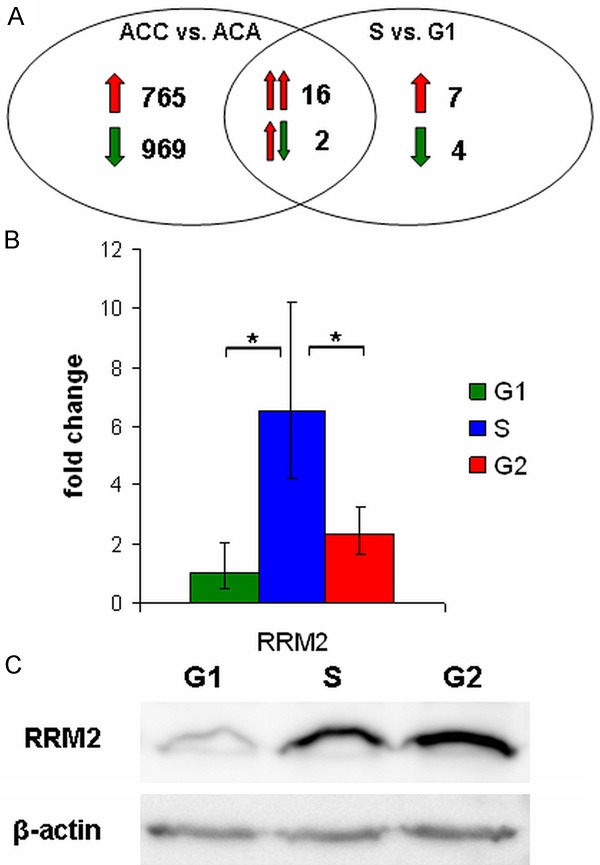
Cell cycle dependent expression of RRM2. A: Venn diagram displaying the overlap of the malignancy signature and the cell cycle regulated transcriptional program of ACC. Red arrows represent up-regulation, green arrows represent down-regulation in ACC vs. ACA and S vs. G1 comparisons. Double arrows represent expression alterations between ACC vs. ACA (left arrow) and S vs. G1 phase (right arrow) in the intersection. For gene list of the overlap genes see Table 2. B: qRT-PCR validation of cell cycle regulated RRM2. ΔCt value was normalized to G1 phase (ΔΔCt) and was subjected to FC = 2-ΔΔCt transformation. Error bars show standard deviation. Asterisks mark statistical significance (P<0.05). C: Western blot analysis of cell cycle regulated RRM2. β-actin was used as loading control.
Table 2.
List of genes displayed in the overlap of the Venn diagram (Figure 1A)
| Gene Symbol | Fold change (ACC vs. ACA) | Fold change S vs. G1 phase | |||
|---|---|---|---|---|---|
|
| |||||
| de Reynies et al. | Giordano et al. | Tombol et al. | Average | ||
| RRM2 | 5.245 | 12.662 | 9.586 | 9.164 | 5.365 |
| HJURP | 2.709 | 7.373 | 5.041 | 5.120 | |
| SPC24 | 8.543 | 8.543 | 5.022 | ||
| WDR62 | 2.597 | 2.597 | 4.414 | ||
| RTKN2 | 7.382 | 7.382 | 4.410 | ||
| CDCA2 | 3.084 | 13.485 | 8.285 | 4.068 | |
| SKA1 | 7.462 | 7.462 | 3.638 | ||
| GTSE1 | 3.949 | 6.678 | 5.314 | 3.614 | |
| STIL | 2.671 | 3.114 | 2.893 | 3.415 | |
| ASPM | 3.466 | 11.213 | 28.779 | 14.486 | 3.307 |
| IQGAP3 | 2.169 | 2.169 | 3.296 | ||
| ARHGAP11A | 8.580 | 8.580 | 3.283 | ||
| PLK4 | 2.201 | 2.279 | 2.240 | 3.086 | |
| CDKN2D | 4.593 | 4.593 | 2.733 | ||
| SMC2 | 2.571 | 2.571 | 2.676 | ||
| KIF14 | 4.031 | 9.100 | 6.566 | 2.472 | |
| COL1A1 | 2.680 | 2.680 | -3.805 | ||
| HOXB13 | 10.458 | 10.458 | -3.615 | ||
List of genes altered in ACC vs. ACA and S vs. G1 phase as well. In all comparisons significant (p-value <0.05) alterations of at least two-fold change in expression are presented. Lacking values represent nonsignificant alterations or significant alterations below two-fold change in expression.
Expression of RRM2 is cell cycle dependent in ACC cell line NCI-H295R
Successful validation of cell cycle dependent expression of RRM2 in ACC cell line NCI-H295R was performed on cell cycle sorted samples (Figure 1B and 1C). As reported previously in other cells [31], RRM2 expression rises in S phase, acquiring an important role in the creation of the ribonucleotide reductase complex.
RRM2 correlates well with Ki67 index in ACC
Upon immunohistochemical analysis of RRM2 and Ki67 in 12 selected ACC samples of various proliferative activity (Table 1), RRM2 expression was found to be tightly correlated with the widely used proliferation marker Ki67 index (Figure 2).
Figure 2.
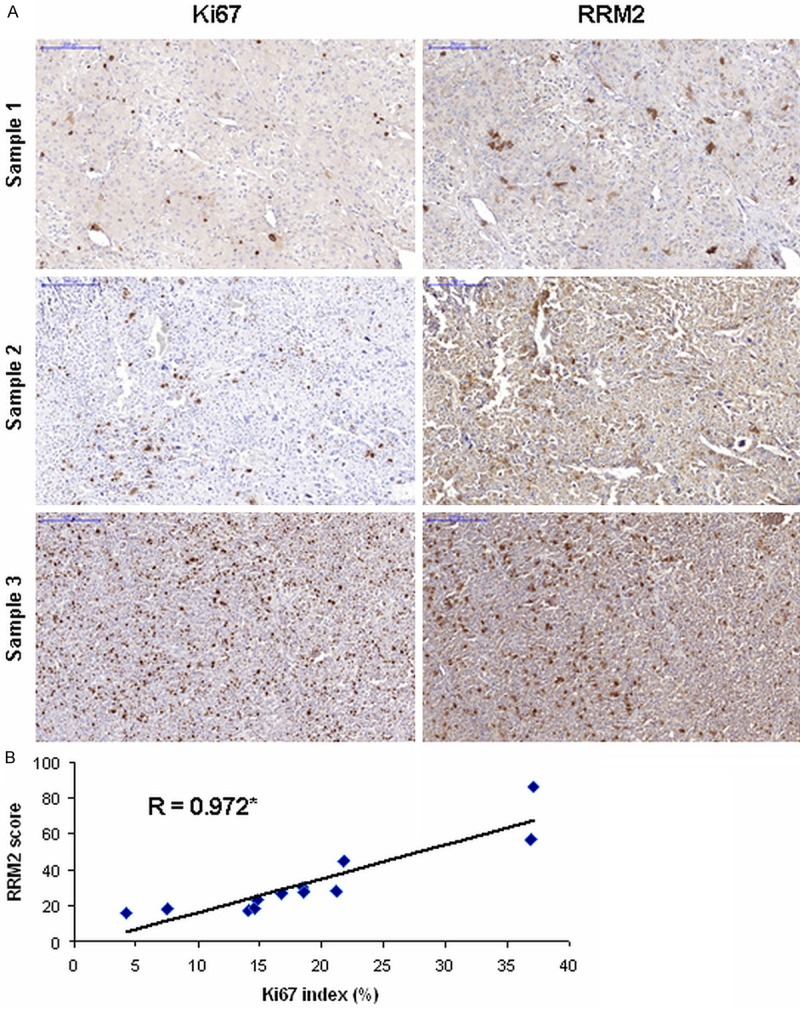
Immunohistochemical analysis of Ki67 and RRM2. A: Representative slides of immunohistochemical analysis of Ki67 (left column) and RRM2 (right column) on the same area of three different ACCs. Blue scale corresponds to 200 μm, magnification is ×100. B: Correlation of Ki67 index and RRM2 score. Spearman’s correlation coefficient is shown. Asterisk marks statistical significance (P<0.005).
Effects of different drug treatments on the proliferation, apoptosis, cortisol production and cell cycle distribution of NCI-H295R cells
Although all treatment regimens decreased proliferation of ACC cells after 72 h, cells treated with G alone or in combination showed the largest decrease in proliferation compared to other treatments (Figure 3A). Additionally, G-mediated decrease in proliferation was detectable at shorter treatment times as well. Higher rates of apoptotic cells, in particular after 72 h of treatment with G alone or in combination contributed to the observed decrease in proliferation (Figure 3B). Although M treatment inhibited cortisol production as expected, G treatment had no effect on cortisol production (Figure 3C). Moreover, effects on cell cycle distribution showed a larger portion of G1 phase cells upon G treatment. However, 24 h M and R treatments and especially their combination resulted in a larger portion of G2 phase cells (Figure 3D-F).
Figure 3.
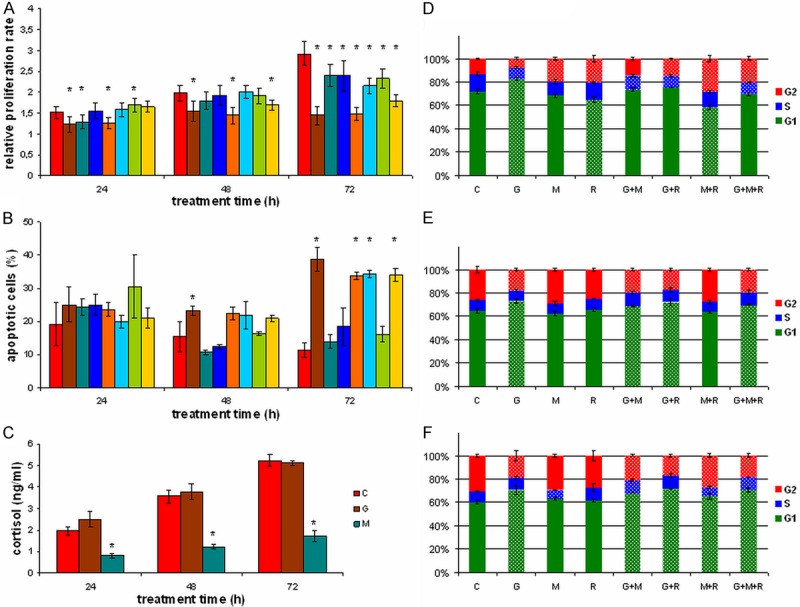
Effects of different treatments on proliferation, apoptosis, cortisol production and cell cycle in NCI-H295R cells. Effects of different treatments on proliferation (A), apoptosis (B), cortisol production (C), and cell cycle distribution ((D) 24 h treatment, (E) 48 h treatment, (F) 72 h treatment). C control, G gemcitabine, M mitotane, R 9-cis-retinoic acid. Error bars show standard deviation. Asterisks (A-C) or white dotted boxes (D-F) mark statistical significant (P<0.05) alterations from control samples.
RRM2 expression is upregulated upon gemcitabine treatment in NCI-H295R cells
Independently from treatment duration, RRM2 was found to be upregulated upon G treatment by three-fold. Combination with other drugs did not alter G’s effect. Conversely, M and R had no effect on RRM2 expression (Figure 4A). Western blot analysis confirmed the G-mediated alterations on protein level (Figure 4B).
Figure 4.
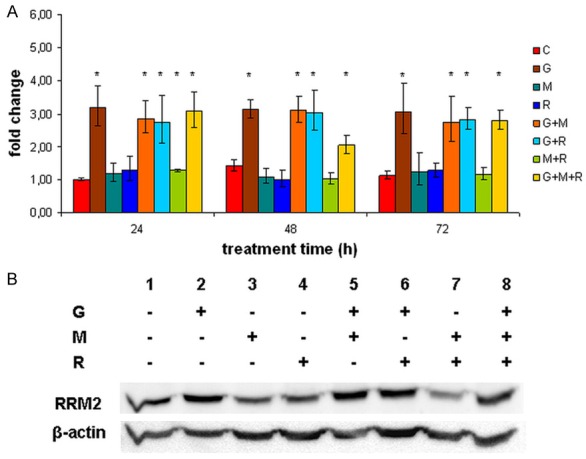
RRM2 expression upon different treatments in NCI-H295R cells. A: qRT-PCR measurements of RRM2 upon different treatments. ΔCt value was normalized to RRM2 expression in 24 h control samples (ΔΔCt) and was subjected to FC = 2-ΔΔCt transformation. Error bars show standard deviation. Asterisks mark statistical significance (P<0.05). B: Western blot analysis of RRM2 expression after 48 h treatment. β-actin was used as loading control. C control, G gemcitabine, M mitotane, R 9-cis-retinoic acid.
Discussion
Targeting the altered cell cycle machinery in cancers resulted in novel pharmacological agents with confirmed antiproliferative impact [18]. However, different, cell type-specific driver genes contribute to the accelerated cell cycle in various cancers. Therefore, novel cancer-specific target genes are needed for the development of novel, effective and targeted chemotherapeutic approaches.
The only adrenal cortex specific drug is mitotane [1], whereas several other cytostatic compounds are administered mostly in advanced stages of ACC with moderate efficacy [1,5-8]. Due to its rarity, clinical trials that might result in evidence-based recommendations in ACC are limited [1].
In this study, by comparing the cell cycle dependent transcription program with the malignancy signature of ACC, we aimed to detect novel proliferation markers, which may facilitate the rather challenging histopathological diagnosis of ACC [32]. Additionally, by detecting cell cycle dependently expressed genes overexpressed in the malignancy signature as well, new pharmacological targets of ACC had been identified.
More than 60% of cell cycle regulated genes in NCI-H295R cells were dysregulated in the malignancy signature as well. This significant overlap is the consequence of phase-dependent and phase-independent overexpression of cell cycle genes [22,25]. Phase-dependent overexpression arise from the larger portion of cells being in S and G2 phases in cancer vs. benign cells [22], while phase-independent overexpression is the consequence of basally higher expression of cell cycle genes in cancer cells independently from cell cycle phase [25]. Among genes overexpressed in S vs. G1 phase and in the malignancy signature as well, ribonucleotide subunit M2 (RRM2) had the largest upregulation in S vs. G1 and was also detected as a part of the malignancy signature by all three microarray studies [13,15]. ASPM was the other gene with cell cycle dependent expression manner being present in the malignancy signature by all three micrroarray studies [13,15].
While abnormal forms of ASPM (abnormal spindle-like microcephaly-associated) gene have been associated with autosomal recessive primary microcephaly [33], overexpression of this proliferative gene has been detected in various cancers [13-15,34,35]. Rapid ASPM expression changes upon adjuvant aromatase-inhibitor therapy were found to have important predictive value for response to endocrine therapy in breast cancer [34]. Additionally, by the analysis of oncogenic signaling networks in glioblastoma, ASPM was identified as a key molecular target downstream from mutant epidermal growth factor receptor, which might be inhibited by the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib [35]. Erlotinib, however, only had limited clinical effect in the salvage therapy of ACC [8], although the salvage therapy was initiated only in patients with progressive ACC after two to four previous systemic therapeutical approaches [8].
We evaluated the expression of RRM2 in ACC samples and performed an in vitro study in the human adrenocortical cell line (NCI-H295R) in order to analyze whether the expression of RRM2 might predict the therapeutical response of different antineoplastic drugs used in the treatment of ACC. RRM2 is a subunit of the ribonucleotide reductase complex (RR), which catalyzes the formation of deoxyribonucleotides from ribonucleotides [36]. The enzyme acquires an important role in regulating the total rate of DNA synthesis [37] and its’ activity is strictly regulated during the cell cycle [31,38]. The short half life of RRM2 is an important factor contributing to the dynamic upregulation of the enzyme activity during S phase [38].
As we identified RRM2 a cell cycle dependently expressed gene in ACC cell line NCI-H295R, also being a contributor to the malignancy signature of ACC, we aimed to analyze its expression in ACC samples, in particular with regard to Ki67 index. Our hypothesis was that, due to its cell cycle-specific expression, it may correlate well with Ki67 index and, therefore, it may be an additional molecular biomarker of proliferation in the histopathological analysis of ACC. Although in a limited sample size, RRM2 score [29] showed strong correlation with Ki67 index, therefore, upon a larger scale validation, it may be used in the determination of proliferative status of ACCs, as well. RRM2’s role as a biomarker of proliferation has been confirmed in colorectal [39,40], breast [41,42], non-small cell lung [29] and hepatocellular [43] cancer predicting poor survival [29,40,42]. Moreover, it has been identified as an important molecular target in anticancer therapy [41,42].
As RRM2 expression is a hallmark of ACC’s proliferative nature, we aimed to analyze whether its expression decreases upon different treatment regimens in ACC cell line NCI-H295R. Due to their proven antiproliferative effects, mitotane (M) [28], 9-cis-retinoic-acid (R) [7,9], gemcitabine (G) [6] and their simultaneous combinations were chosen in our in vitro study. Additionally, G has earlier been shown to specifically target RR complex [44], therefore, it became especially important for us to assess RRM2 expression upon G treatment. Although all treatment regimens resulted in a decrease of proliferation, G was the most efficient reaching significant and more robust decrease at an earlier timepoint. However, this correlated with the observed cytotoxic, in particular apoptotic events upon G treatment [6]. Interestingly, G did not alter steroid hormone production, although the administration of M showed the well-known adrenolytic consequence [28]. Assessment of cell cycle distribution confirmed the early S phase block of G, resulting in a larger portion of cells residing in G1 phase. Additionally, results of M and R treatments are also in line with earlier studies characterizing G2 arrest upon M treatment [45,46] and alteration of the cell cycle upon R treatment [7]. It seems, that simultaneous delivery of M and R synergistically results in rapid G2 arrest, with nearly 30% of cells residing in G2/M phase.
Interestingly, however, G treatment alone as well as in combinations resulted in a time-independent three-fold increase in RRM2 expression, detectable also on protein level, while other treatments did not have any effect on RRM2 expression. Therefore, contrary to the expectations based on the changes in cell cycle distribution [22], a robust, phase-independent up-regulation of RRM2 occurred. This result is in line with the up-regulation of RR-subunit RRM1 upon G treatment in NCI-H295R cells [6].
As higher levels of RRM1 at the initiation of gemcitabine therapy predict poorer efficacy in biliary tract carcinoma [47], and siRNA- or miRNA-mediated silencing of RRM2 restores gemcitabine chemosensitivity in pancreatic cancer [48,49], we hypothesize, that RRM2 upregulation upon G treatment in ACC cells is the consequence of an emerging chemoresistance aginst G. This may contribute to the limited effect of G in the therapy of ACC [8]. SiRNA-mediated silencing or inhibition of de novo RRM2 production might as well optimize G teatment in ACC.
In conclusion, by the re-analysis of the malignancy signature and the cell cycle dependent transcription program of ACC, RRM2 had been identified as a key proliferative gene and a potential novel molecular target in ACC. As an immunohistochemical marker, RRM2 might be used to assess the proliferative nature of ACC, therefore, it may be helpful in the diagnosis of borderline lesions. Our in vitro study demonstrated that gemcitabine had a robust effect on proliferation and apoptosis, however, RRM2 upregulation upon gemcitabine treatment may contribute to an emerging chemoresistance against gemcitabine, which should be overcome for successful further clinical applications.
Acknowledgements
This work was supported by the Hungarian Academy of Sciences “Lendület” grant awarded to Attila Patocs (Lendület 2013), by the Hungarian Scientific Research Fund (OTKA, PD100648 (AP)) and by the Technology Innovation Fund, National Developmental Agency (KTIA-AIK-2012-12-1-0010).
Disclosure of conflict of interest
None.
References
- 1.Fassnacht M, Kroiss M, Allolio B. Update in adrenocortical carcinoma. J Clin Endocrinol Metab. 2013;98:4551–4564. doi: 10.1210/jc.2013-3020. [DOI] [PubMed] [Google Scholar]
- 2.Kerkhofs TM, Verhoeven RH, Van der Zwan JM, Dieleman J, Kerstens MN, Links TP, Van de Poll-Franse LV, Haak HR. Adrenocortical carcinoma: a population-based study on incidence and survival in the Netherlands since 1993. Eur J Cancer. 2013;49:2579–2586. doi: 10.1016/j.ejca.2013.02.034. [DOI] [PubMed] [Google Scholar]
- 3.Luton JP, Cerdas S, Billaud L, Thomas G, Guilhaume B, Bertagna X, Laudat MH, Louvel A, Chapuis Y, Blondeau P, et al. Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med. 1990;322:1195–1201. doi: 10.1056/NEJM199004263221705. [DOI] [PubMed] [Google Scholar]
- 4.Abiven G, Coste J, Groussin L, Anract P, Tissier F, Legmann P, Dousset B, Bertagna X, Bertherat J. Clinical and biological features in the prognosis of adrenocortical cancer: poor outcome of cortisol-secreting tumors in a series of 202 consecutive patients. J Clin Endocrinol Metab. 2006;91:2650–2655. doi: 10.1210/jc.2005-2730. [DOI] [PubMed] [Google Scholar]
- 5.Sperone P, Ferrero A, Daffara F, Priola A, Zaggia B, Volante M, Santini D, Vincenzi B, Badalamenti G, Intrivici C, Del Buono S, De Francia S, Kalomirakis E, Ratti R, Angeli A, Dogliotti L, Papotti M, Terzolo M, Berruti A. Gemcitabine plus metronomic 5-fluorouracil or capecitabine as a second-/third-line chemotherapy in advanced adrenocortical carcinoma: a multicenter phase II study. Endocr Relat Cancer. 2010;17:445–453. doi: 10.1677/ERC-09-0281. [DOI] [PubMed] [Google Scholar]
- 6.Germano A, Rapa I, Volante M, Lo Buono N, Carturan S, Berruti A, Terzolo M, Papotti M. Cytotoxic activity of gemcitabine, alone or in combination with mitotane, in adrenocortical carcinoma cell lines. Mol Cell Endocrinol. 2014;382:1–7. doi: 10.1016/j.mce.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 7.Szabo DR, Baghy K, Szabo PM, Zsippai A, Marczell I, Nagy Z, Varga V, Eder K, Toth S, Buzas EI, Falus A, Kovalszky I, Patocs A, Racz K, Igaz P. Antitumoral effects of 9-cis retinoic acid in adrenocortical cancer. Cell Mol Life Sci. 2014;71:917–932. doi: 10.1007/s00018-013-1408-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quinkler M, Hahner S, Wortmann S, Johanssen S, Adam P, Ritter C, Strasburger C, Allolio B, Fassnacht M. Treatment of advanced adrenocortical carcinoma with erlotinib plus gemcitabine. J Clin Endocrinol Metab. 2008;93:2057–2062. doi: 10.1210/jc.2007-2564. [DOI] [PubMed] [Google Scholar]
- 9.Nagy Z, Baghy K, Hunyadi-Gulyas E, Micsik T, Nyiro G, Racz G, Butz H, Perge P, Kovalszky I, Medzihradszky KF, Racz K, Patocs A, Igaz P. Evaluation of 9-cis retinoic acid and mitotane as antitumoral agents in an adrenocortical xenograft model. Am J Cancer Res. 2015;5:3645–3658. [PMC free article] [PubMed] [Google Scholar]
- 10.Barlaskar FM, Spalding AC, Heaton JH, Kuick R, Kim AC, Thomas DG, Giordano TJ, Ben-Josef E, Hammer GD. Preclinical targeting of the type I insulin-like growth factor receptor in adrenocortical carcinoma. J Clin Endocrinol Metab. 2009;94:204–212. doi: 10.1210/jc.2008-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Martino MC, van Koetsveld PM, Feelders RA, Sprij-Mooij D, Waaijers M, Lamberts SW, de Herder WW, Colao A, Pivonello R, Hofland LJ. The role of mTOR inhibitors in the inhibition of growth and cortisol secretion in human adrenocortical carcinoma cells. Endocr Relat Cancer. 2012;19:351–364. doi: 10.1530/ERC-11-0270. [DOI] [PubMed] [Google Scholar]
- 12.Fassnacht M, Berruti A, Baudin E, Demeure MJ, Gilbert J, Haak H, Kroiss M, Quinn DI, Hesseltine E, Ronchi CL, Terzolo M, Choueiri TK, Poondru S, Fleege T, Rorig R, Chen J, Stephens AW, Worden F, Hammer GD. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol. 2015;16:426–435. doi: 10.1016/S1470-2045(15)70081-1. [DOI] [PubMed] [Google Scholar]
- 13.de Reynies A, Assie G, Rickman DS, Tissier F, Groussin L, Rene-Corail F, Dousset B, Bertagna X, Clauser E, Bertherat J. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J. Clin. Oncol. 2009;27:1108–1115. doi: 10.1200/JCO.2008.18.5678. [DOI] [PubMed] [Google Scholar]
- 14.Giordano TJ, Kuick R, Else T, Gauger PG, Vinco M, Bauersfeld J, Sanders D, Thomas DG, Doherty G, Hammer G. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res. 2009;15:668–676. doi: 10.1158/1078-0432.CCR-08-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tombol Z, Szabo PM, Molnar V, Wiener Z, Tolgyesi G, Horanyi J, Riesz P, Reismann P, Patocs A, Liko I, Gaillard RC, Falus A, Racz K, Igaz P. Integrative molecular bioinformatics study of human adrenocortical tumors: microRNA, tissue-specific target prediction, and pathway analysis. Endocr Relat Cancer. 2009;16:895–906. doi: 10.1677/ERC-09-0096. [DOI] [PubMed] [Google Scholar]
- 16.Szabo PM, Tamasi V, Molnar V, Andrasfalvy M, Tombol Z, Farkas R, Kovesdi K, Patocs A, Toth M, Szalai C, Falus A, Racz K, Igaz P. Meta-analysis of adrenocortical tumour genomics data: novel pathogenic pathways revealed. Oncogene. 2010;29:3163–3172. doi: 10.1038/onc.2010.80. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J. Clin. Oncol. 2005;23:9408–9421. doi: 10.1200/JCO.2005.01.5594. [DOI] [PubMed] [Google Scholar]
- 18.Pitts TM, Davis SL, Eckhardt SG, Bradshaw-Pierce EL. Targeting nuclear kinases in cancer: development of cell cycle kinase inhibitors. Pharmacol Ther. 2014;142:258–269. doi: 10.1016/j.pharmthera.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Shedden K, Cooper S. Analysis of cell-cycle-specific gene expression in human cells as determined by microarrays and double-thymidine block synchronization. Proc Natl Acad Sci U S A. 2002;99:4379–4384. doi: 10.1073/pnas.062569899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bar-Joseph Z, Siegfried Z, Brandeis M, Brors B, Lu Y, Eils R, Dynlacht BD, Simon I. Genome-wide transcriptional analysis of the human cell cycle identifies genes differentially regulated in normal and cancer cells. Proc Natl Acad Sci U S A. 2008;105:955–960. doi: 10.1073/pnas.0704723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho RJ, Huang M, Campbell MJ, Dong H, Steinmetz L, Sapinoso L, Hampton G, Elledge SJ, Davis RW, Lockhart DJ. Transcriptional regulation and function during the human cell cycle. Nat Genet. 2001;27:48–54. doi: 10.1038/83751. [DOI] [PubMed] [Google Scholar]
- 22.Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, Botstein D. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong J, Traganos F, Darzynkiewicz Z. Growth imbalance and altered expression of cyclins B1, A, E, and D3 in MOLT-4 cells synchronized in the cell cycle by inhibitors of DNA replication. Cell Growth Differ. 1995;6:1485–1493. [PubMed] [Google Scholar]
- 24.Darzynkiewicz Z, Halicka HD, Zhao H, Podhorecka M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: analysis by flow cytometry. Methods Mol Biol. 2011;761:85–96. doi: 10.1007/978-1-61779-182-6_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grolmusz VK, Toth EA, Baghy K, Liko I, Darvasi O, Kovalszky I, Matko J, Racz K, Patocs A. Fluorescence activated cell sorting followed by small RNA sequencing reveals stable microRNA expression during cell cycle progression. BMC Genomics. 2016;17:412. doi: 10.1186/s12864-016-2747-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano Y, Tanno S, Koizumi K, Nishikawa T, Nakamura K, Minoguchi M, Izawa T, Mizukami Y, Okumura T, Kohgo Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer. 2007;96:457–463. doi: 10.1038/sj.bjc.6603559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C, Zhang W, Fu M, Yang A, Huang H, Xie J. Establishment of human pancreatic cancer gemcitabineresistant cell line with ribonucleotide reductase overexpression. Oncol Rep. 2015;33:383–390. doi: 10.3892/or.2014.3599. [DOI] [PubMed] [Google Scholar]
- 28.Zsippai A, Szabo DR, Tombol Z, Szabo PM, Eder K, Pallinger E, Gaillard RC, Patocs A, Toth S, Falus A, Racz K, Igaz P. Effects of mitotane on gene expression in the adrenocortical cell line NCI-H295R: a microarray study. Pharmacogenomics. 2012;13:1351–1361. doi: 10.2217/pgs.12.116. [DOI] [PubMed] [Google Scholar]
- 29.Mah V, Alavi M, Marquez-Garban DC, Maresh EL, Kim SR, Horvath S, Bagryanova L, Huerta-Yepez S, Chia D, Pietras R, Goodglick L. Ribonucleotide reductase subunit M2 predicts survival in subgroups of patients with non-small cell lung carcinoma: effects of gender and smoking status. PLoS One. 2015;10:e0127600. doi: 10.1371/journal.pone.0127600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aubert S, Wacrenier A, Leroy X, Devos P, Carnaille B, Proye C, Wemeau JL, Lecomte-Houcke M, Leteurtre E. Weiss system revisited: a clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol. 2002;26:1612–1619. doi: 10.1097/00000478-200212000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Eriksson S, Graslund A, Skog S, Thelander L, Tribukait B. Cell cycle-dependent regulation of mammalian ribonucleotide reductase. The S phase-correlated increase in subunit M2 is regulated by de novo protein synthesis. J Biol Chem. 1984;259:11695–11700. [PubMed] [Google Scholar]
- 32.Papotti M, Libe R, Duregon E, Volante M, Bertherat J, Tissier F. The Weiss score and beyond--histopathology for adrenocortical carcinoma. Horm Cancer. 2011;2:333–340. doi: 10.1007/s12672-011-0088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pattison L, Crow YJ, Deeble VJ, Jackson AP, Jafri H, Rashid Y, Roberts E, Woods CG. A fifth locus for primary autosomal recessive microcephaly maps to chromosome 1q31. Am J Hum Genet. 2000;67:1578–1580. doi: 10.1086/316910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turnbull AK, Arthur LM, Renshaw L, Larionov AA, Kay C, Dunbier AK, Thomas JS, Dowsett M, Sims AH, Dixon JM. Accurate Prediction and Validation of Response to Endocrine Therapy in Breast Cancer. J. Clin. Oncol. 2015;33:2270–2278. doi: 10.1200/JCO.2014.57.8963. [DOI] [PubMed] [Google Scholar]
- 35.Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, Lee Y, Scheck AC, Liau LM, Wu H, Geschwind DH, Febbo PG, Kornblum HI, Cloughesy TF, Nelson SF, Mischel PS. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A. 2006;103:17402–17407. doi: 10.1073/pnas.0608396103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elledge SJ, Zhou Z, Allen JB. Ribonucleotide reductase: regulation, regulation, regulation. Trends Biochem Sci. 1992;17:119–123. doi: 10.1016/0968-0004(92)90249-9. [DOI] [PubMed] [Google Scholar]
- 37.Herrick J, Sclavi B. Ribonucleotide reductase and the regulation of DNA replication: an old story and an ancient heritage. Mol Microbiol. 2007;63:22–34. doi: 10.1111/j.1365-2958.2006.05493.x. [DOI] [PubMed] [Google Scholar]
- 38.Engstrom Y, Eriksson S, Jildevik I, Skog S, Thelander L, Tribukait B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J Biol Chem. 1985;260:9114–9116. [PubMed] [Google Scholar]
- 39.Fang Z, Gong C, Liu H, Zhang X, Mei L, Song M, Qiu L, Luo S, Zhu Z, Zhang R, Gu H, Chen X. E2F1 promote the aggressiveness of human colorectal cancer by activating the ribonucleotide reductase small subunit M2. Biochem Biophys Res Commun. 2015;464:407–415. doi: 10.1016/j.bbrc.2015.06.103. [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Zhang H, Lai L, Wang X, Loera S, Xue L, He H, Zhang K, Hu S, Huang Y, Nelson RA, Zhou B, Zhou L, Chu P, Zhang S, Zheng S, Yen Y. Ribonucleotide reductase small subunit M2 serves as a prognostic biomarker and predicts poor survival of colorectal cancers. Clin Sci (Lond) 2013;124:567–578. doi: 10.1042/CS20120240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H, Liu X, Warden CD, Huang Y, Loera S, Xue L, Zhang S, Chu P, Zheng S, Yen Y. Prognostic and therapeutic significance of ribonucleotide reductase small subunit M2 in estrogen-negative breast cancers. BMC Cancer. 2014;14:664. doi: 10.1186/1471-2407-14-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Putluri N, Maity S, Kommagani R, Creighton CJ, Putluri V, Chen F, Nanda S, Bhowmik SK, Terunuma A, Dorsey T, Nardone A, Fu X, Shaw C, Sarkar TR, Schiff R, Lydon JP, O’Malley BW, Ambs S, Das GM, Michailidis G, Sreekumar A. Pathway-centric integrative analysis identifies RRM2 as a prognostic marker in breast cancer associated with poor survival and tamoxifen resistance. Neoplasia. 2014;16:390–402. doi: 10.1016/j.neo.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee B, Ha SY, Song DH, Lee HW, Cho SY, Park CK. High expression of ribonucleotide reductase subunit M2 correlates with poor prognosis of hepatocellular carcinoma. Gut Liver. 2014;8:662–668. doi: 10.5009/gnl13392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cerqueira NM, Fernandes PA, Ramos MJ. Understanding ribonucleotide reductase inactivation by gemcitabine. Chemistry. 2007;13:8507–8515. doi: 10.1002/chem.200700260. [DOI] [PubMed] [Google Scholar]
- 45.Cerquetti L, Bucci B, Marchese R, Misiti S, De Paula U, Miceli R, Muleti A, Amendola D, Piergrossi P, Brunetti E, Toscano V, Stigliano A. Mitotane increases the radiotherapy inhibitory effect and induces G2-arrest in combined treatment on both H295R and SW13 adrenocortical cell lines. Endocr Relat Cancer. 2008;15:623–634. doi: 10.1677/erc.1.1315. [DOI] [PubMed] [Google Scholar]
- 46.Cerquetti L, Sampaoli C, Amendola D, Bucci B, Misiti S, Raza G, De Paula U, Marchese R, Brunetti E, Toscano V, Stigliano A. Mitotane sensitizes adrenocortical cancer cells to ionizing radiations by involvement of the cyclin B1/CDK complex in G2 arrest and mismatch repair enzymes modulation. Int J Oncol. 2010;37:493–501. doi: 10.3892/ijo_00000698. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura J, Kohya N, Kai K, Ohtaka K, Hashiguchi K, Hiraki M, Kitajima Y, Tokunaga O, Noshiro H, Miyazaki K. Ribonucleotide reductase subunit M1 assessed by quantitative double-fluorescence immunohistochemistry predicts the efficacy of gemcitabine in biliary tract carcinoma. Int J Oncol. 2010;37:845–852. [PubMed] [Google Scholar]
- 48.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine. Oncogene. 2004;23:1539–1548. doi: 10.1038/sj.onc.1207272. [DOI] [PubMed] [Google Scholar]
- 49.Bhutia YD, Hung SW, Krentz M, Patel D, Lovin D, Manoharan R, Thomson JM, Govindarajan R. Differential processing of let-7a precursors influences RRM2 expression and chemosensitivity in pancreatic cancer: role of LIN-28 and SET oncoprotein. PLoS One. 2013;8:e53436. doi: 10.1371/journal.pone.0053436. [DOI] [PMC free article] [PubMed] [Google Scholar]