

Abstract
Intratumoral hypoxia is a common feature of solid tumors. Recent advances in cancer biology indicate that hypoxia is not only a consequence of unrestrained tumor growth, but also plays an active role in promoting tumor progression, malignancy, and resistance to therapy. Hypoxia signaling is mediated by the hypoxia-inducible factors (HIFs), which are not only stabilized under hypoxia, but also by activated oncogenes or inactivated tumor suppressors under normoxia. Hypoxia is a prominent feature of the tumor microenvironment of pancreatic tumors, also characterized by the presence of a fibrotic reaction that promotes, and is also modulated by, hypoxia. As the mechanisms by which hypoxia signaling impacts invasion and metastasis in pancreatic cancer are being elucidated, hypoxia is emerging as a key determinant of pancreatic cancer malignancy as well as an important target for therapy. Herein we present an overview of recent advances in the understanding of the impact that hypoxia has in pancreatic cancer invasion and metastasis.
Keywords: PanIN, EMT, HIF-1, invadopodia, PDAC, invasion
Introduction
Pancreatic cancer is a disease with very poor prognosis. The median survival rate, which currently remains as low as 5% over a period of 5 years after diagnosis, has not improved over recent decades.1 Unfortunately, diagnosis frequently occurs after pancreatic cancer has already spread beyond the pancreas, a point after which therapy remains largely palliative. Reasons for late diagnosis include frequent clinical presentation with only mild symptoms and the lack of early detection markers. Furthermore, current treatments for pancreatic cancer are seldom curative because relapse is very common. Early detection tools and improved therapies are, therefore, needed to manage patients at risk as well as those already diagnosed. A fundamental step toward the development of better diagnostic and therapeutic approaches is a deep understanding of the biology of pancreatic cancer. In this regard, very important contributions have been made in past decades, including the identification of driver mutations and precursor lesions, the generation of relevant genetically engineered mouse (GEM) models, the isolation of cancer-initiating cells, and the identification of the pancreatic cancer stroma as a driver of malignancy that also promotes drug resistance. A number of reviews have addressed these important advances in pancreatic cancer biology.2–8 An emerging player in pancreatic cancer progression is hypoxia, a condition of low oxygen availability, to which cancer and stromal cells are often exposed as tumors evolve. Intratumoral hypoxia is a common feature of cancers and affects many aspects of tumor biology, as summarized in several review articles.9–13 Pancreatic cancer is highly hypoxic and, in this review, we focus on the impact that hypoxia has on the ability of pancreatic cancer cells to invade and metastasize. Hypoxia activates signaling pathways in both cell-autonomous and non-cell-autonomous manners in cancer and stromal cells, which directly affect the activation of pro-invasive programs in cancer cells, facilitate the maintenance of invasiveness by favoring a hypoxic environment, or aid pro-invasive interactions between cancer and stromal cells. In the first part of this article we present an overview of the cellular components of the pancreas, pancreatic tumor precursor lesions, and tumors, as well as the main features of pancreatic cancer invasion. In the second part, we summarize the current understanding of the cellular and molecular mechanisms by which hypoxic signaling promotes invasion and metastasis in pancreatic cancer.
Pancreatic cancer biology
The tissue architecture of the exocrine pancreas is composed of epithelial and mesenchymal tissues, which undergo important changes upon malignant transformation. Epithelial changes follow some established patterns that have helped clinicians and researchers to identify precursor lesions and understand some of the events that lead to tumor initiation. Recent discoveries indicate that epithelial plasticity is an important event in pancreatic tumor initiation and suggest that invasion and metastatic spreading may be initiated earlier than anticipated during pancreatic cancer progression. Furthermore, changes associated with the glandular mesenchyme are also prominent during pancreatic cancer progression, as the stroma associated with the tumor contains tumor-associated stromal cells that display important phenotypic modifications with respect to the stroma associated to the normal tissue. Epithelial and mesenchymal cells within the tumor generate a pathological microenvironment that changes dynamically as the tumor progresses, and in turn, promotes invasion and malignancy of the cancer cells.
The pancreas
Pancreatic ductal adenocarcinoma (PDAC), the most common type of pancreatic cancer, arises from the malignant transformation of the exocrine pancreas, a gland whose normal function is the secretion of digestive enzymes to the intestinal tract. Histologically, the exocrine compartment of the pancreas is an epithelial tubuloalveolar gland. The functional unit of this gland is referred to as an alveolus or acinus, and it is comprised of three main epithelial cell types: acinar, ductal, and centroacinar cells (Figure 1). Acinar cells group into small clusters to form the acini and secrete the components of the pancreatic juice to the central alveolar lumen. Ductal cells line the conducts that collect and transport the secretions of the acinar cells. The morphology of the ductal epithelial cells changes progressively, from almost squamous at the smaller intercalated ducts, to cuboidal in inter- and intralobular ducts, and finally to columnar in the main pancreatic duct draining the pancreatic juice into the duodenum. The cells at the terminal end of the ductal tree, in contact with the acinar lumen, are known as centroacinar cells (Figure 1). This third pancreatic exocrine cell population expresses markers of pancreatic embryonic progenitors and exhibits tissue stem cell properties in vitro.14 Also within the exocrine pancreas is an additional subset of cells expressing markers of pancreatic embryonic progenitors, which are found in close proximity to the larger pancreatic ducts. These cells, which are not well studied, reside within structures known as pancreatic duct glands.15 The pancreatic stroma that surrounds acini and ducts contains a noncellular component formed by extracellular matrix proteins, and a cellular component including neurons, endothelial cells, resident macrophages, adipocytes, fibroblasts, and pancreatic stellate cells (PSCs). This rare cell population was discovered in close proximity to pancreatic acini by the presence of vitamin A-containing cytoplasmic lipid droplets.16,17 Pancreatic fibroblasts secrete the interstitial collagen and other structural components of the extracellular matrix, but the normal function of PSCs in the healthy organ is still a matter of investigation. Another noncellular component, the basement membrane, surrounds acini and ducts, as well as local blood vessels separating them from the interstitial stroma (Figure 1). All these cellular and noncellular components of the exocrine pancreas will be subjected to substantial histological and functional changes upon the initiation and progression of PDAC.
Figure 1.

Diagram of the main anatomic features of the exocrine pancreas.
Notes: The exocrine pancreas, which secretes pancreatic juice to the duodenum, is a glandular structure formed by a number of functional units or acini (1). The secretion of each acinus drains into small intercalated ducts (2), which then drain into interlobular ducts (3). Each acinus is formed by the secretory acinar cells (4), which have basally located nuclei and contain abundant zymogen granules in the cytoplasm, whose content is secreted to the center of the acinus through their apical membranes. Acini also contain ductal cells (5) and centroacinar cells in contact with the acinar lumen. Each acinus is surrounded by a basement membrane (clear dotted sheet) that separates the epithelial cells from the stromal component of the gland. The stroma is composed of interstitial fibroblasts (6), pancreatic stellate cells (7) – characterized by the presence of cytoplasmic vitamin A-containing droplets – and a loose interstitial extracellular matrix (8). Sparse blood vessels, also surrounded by a basement membrane, can be found within the interstitial stroma (9) of the normal gland.
Pancreatic cancer and precursor lesions
PDAC arises from the successive accumulation of genetic mutations in the epithelium of the exocrine pancreas. These mutations frequently include activating mutations in the oncogene k-Ras and inactivating mutations in the tumor suppressor genes CDKN2A, TP53, and SMAD4.7,18,19 Although activating mutations in k-Ras are almost universally present in PDAC, the mutational landscape of PDAC is complex, and several additional genetic lesions have been described that affect a variety of signaling pathways, such as DNA damage control, Hedgehog signaling, integrin signaling, and apoptosis.20
Histological and molecular analysis of the human pancreas has led to the proposal of a progression model in which PDAC originates from pancreatic preneoplastic lesions such as pancreatic intraepithelial neoplasia (PanIN) through a series of successive mutational events.21–23 Histologically, PanINs have the appearance of an abnormal ductal epithelium and are often associated with the atrophy of the normal acini and the presence of inflammation and fibrosis – excessive deposition of extracellular matrix proteins (Figure 2). Human PanIN lesions (classified from 1 to 3 [see Figure 2]) are considered nonmalignant, but frequently contain mutations in k-Ras,24,25 indicating that k-Ras mutation is an early oncogenic event in the progression of PDAC. The development of GEM models of pancreatic cancer that recapitulate the histological features of PDAC has provided great insight into the initiation of this disease. The expression of a constitutively active allele of k-Ras in the mouse pancreas leads to the formation of PanIN lesions with high penetrance, some of which spontaneously progressed to PDAC,26 suggesting that PanIN lesions are precursors of PDAC. The hypothesis that PDAC progresses through a series of mutational events following the activation of k-Ras was further supported by different GEM models of PDAC that combined mutations in k-Ras with other known driver mutations found in human PDAC. For instance, the combination of an activating mutation in k-Ras with an inactivating mutation in the tumor suppressor TP53 led to the rapid onset of PDAC lesions with almost full penetrance.27 A number of GEM models of PDAC that combine k-Ras mutations with mutations in additional driver genes have helped to dissect the molecular pathways regulating PDAC initiation and progression, and provided tools for finding early markers of the disease and exploring novel therapies.5
Figure 2.

Diagram of the histology of precursor lesions and PDAC.
Notes: The progression model from normal exocrine pancreas to PDAC is depicted as diagrams of representative tissue sections (left column) and their corresponding magnifications (right column). The normal exocrine pancreas is formed by pancreatic acini surrounded by stromal tissue containing fibroblasts and pancreatic stellate cells. Each acinus contains ductal (not depicted), centroacinar, and acinar cells that contain zymogen granules and are separated from the stroma by a basement membrane (black). Recent studies indicate that acinar cells may be the epithelial cell type targeted by oncogenic transformation during PDAC initiation, although a centroacinar or ductal origin for PDAC cannot be completely ruled out (see text for details). PanIN lesions already contain mutations in K-RAS and are characterized by a columnar epithelium resembling the ductal epithelium. Cells in PanIN-1 often contain mucin in the cytoplasm and present normal nuclei located basally. PanIN-2 lesions are characterized by hyperplasia (excessive cell proliferation), which leads to papillary protrusions into the lumen. The epithelium often becomes pseudostratified, and not all nuclei are basally located, indicating initial loss of epithelial polarity. Nuclei are abnormally shaped. PanIN-3 is the most advanced precursor lesion and displays extensive hyperplasia with papillary protrusions into the lumen, loss of epithelial polarity, and abnormally shaped nuclei that pile up. In PDAC, cells become mesenchymal and invasive, cross the basement membrane, and migrate through the stroma. The integrity of the basement membrane is the main histological feature that differentiates PanIN-3 and PDAC, although recent studies indicate that cells may cross the basement membrane earlier during the progression of the disease (see text for details). Note that the stroma surrounding the lesions and the cancer cells also evolves during the progression of PDAC, becoming more dense and fibrotic. Fibrosis starts in PanIN-1 lesions and progressively increases through PanIN-2 and -3 until the formation of the intense desmoplastic reaction that characterizes PDAC (see text for details).
Abbreviations: PanIN, pancreatic intraepithelial neoplasia; PDAC, pancreatic ductal adenocarcinoma.
Despite the fact that cells within PanIN lesions are histologically very similar to ductal epithelial cells, some studies have questioned the ductal origin of PanIN and PDAC, favoring instead the hypothesis that PanIN and PDAC originate in the acinar/centroacinar compartment,28,29 an idea also suggested after histological analysis of human PDAC samples.30 Recently, lineage tracing experiments in mice have shown that the expression of mutant k-Ras in cells expressing Sox9 (a marker of ductal and centroacinar cells) rarely led to PanIN formation, whereas expression of mutant k-Ras in cells expressing Ptf1a (a marker of acinar cells) led to the formation of PanIN lesions with high penetrance.31 These results indicate that acinar cells in the exocrine pancreas of mice are likely the main cell type targeted by mutant k-Ras to initiate PanIN and PDAC.
But, why do PanIN cells resemble ductal cells? The initiation of PDAC by mutant k-Ras in mice is associated with the phenomenon of acinar-to-ductal metaplasia (ADM),28,29,32 which leads to the phenotypic conversion of acinar cells into ductal-like cells. This conversion likely occurs through persistent activation of a dedifferentiation program leading to the acquisition of ductal-like characteristics by acinar cells.32 During pancreatitis, a risk factor for PDAC,33,34 tissue injury may lead to the aberrant activation of this acinar regeneration program leading to ADM acceleration of oncogene-induced PDAC.29 Acinar cells reprogrammed to ductal-like cells during ADM might be more susceptible to oncogenic transformation and PanIN formation, although a direct cell lineage relationship between ADM and PanIN remains to be proven. Nevertheless, it seems clear that acinar cell plasticity is a novel contributor to the development of PanIN and PDAC.31
In summary, current studies support a model in which PDAC often arise from the successive accumulation of mutations in PanIN lesions, which are preneoplastic lesions initiated by early mutation events in the pancreatic acinar cells. This extremely simplified model obviously does not account for the complexity of this disease. For instance, different premalignant lesions in addition to PanIN give rise to PDAC.35,36 Furthermore, under different oncogenic insults, centroacinar or distal ductal cells may also initiate PanIN and/or PDAC in mice.37,38 Understanding the role of cellular plasticity and ADM in the origin of PDAC, the possible different cellular origins of PDAC, and how that may relate to PDAC subtypes and/or other pancreatic cancers, along with a better understanding of the crosstalk between the cancer cells and their microenvironment, should help researchers to tackle the complexity of this disease and improve therapy and early detection.
Invasion and metastasis in pancreatic cancer
Cancer malignancy is associated with the ability of cancer cells to spread outside their tissue of origin, forming secondary tumors or metastasis. Invasive migration is a prerequisite for metastasis and a hallmark of cancer.39 PDAC is highly invasive, extending to regional areas that include peritoneum, portal vein, and regional nerves,7 and also metastasizes to regional lymph nodes and distant organs, such as liver and lungs. Metastatic disease is the main cause of mortality by PDAC, whereas local and regional invasion may impede tumor resection and are important causes of morbidity, as they may lead to jaundice, duodenal obstruction, and severe pain derived from perineural invasion.40 As in other cancer types, metastasis to regional lymph nodes is a negative prognostic factor in PDAC.41,42
In order to invade, cancer cells have to cross the epithelial basement membrane, a specialized region of the extracellular matrix that separates epithelium from mesenchyme. Indeed, the presence of an intact basement membrane is the main histological feature used to differentiate premalignant lesions such as carcinoma in situ (PanIN-3 in the pancreas [see Figure 2]) from malignant carcinoma.43 A recent study, however, suggests that invasion may start earlier than understood during the progression of PDAC, as preneoplastic cells from premalignant PanIN-3 lesions delaminated from the epithelia, crossed the basement membrane, entered the circulation, and seeded the liver in a mouse model of PDAC.44 Whether these disseminated cells actually form metastasis is not known, but early precancerous cell dissemination followed by metastasis has also been described in a mouse model of breast cancer.45 The possibility of early dissemination indicates that pancreatic primary tumor and metastasis may form and evolve simultaneously44 rather than sequentially,46 which would be consistent with a previously proposed model of early dissemination.47,48 It would be very important to address whether cells disseminated from PanIN lesions are capable of forming metastatic lesions because of the impact in clinical and preclinical studies.
After the epithelial basement membrane is crossed, invasive ability is successively required at different steps of the metastatic cascade to migrate through the surrounding stroma, breach endothelial basement membranes, and reach the circulatory systems.49 Once in the capillary bed of distant organs, invasive traits facilitate extravasation (exit from the circulatory systems), which precedes dissemination. Disseminated cells may remain dormant in distant organs, either as solitary cells or as micrometastasis, before acquiring unrestricted proliferation and forming clinically detectable metastatic disease.49 The mechanisms that regulate dormancy after dissemination are a subject of intense investigation due to their clinical implications. Maintaining disseminated cells in a dormant state would restrain the formation of metastasis and perhaps shift cancer from a life-threatening condition to a chronic disease. Whether the early onset of metastatic disease in PDAC is a result of an inherently short period of dormancy of pancreatic cancer cells or to some other factor(s) is unknown.
PDAC stroma and microenvironment
The tumor stroma surrounds and infiltrates the tumor parenchyma (cancer cells) and influences cancer progression.50,51 The stroma of PDAC is characterized by an extensive fibrotic stroma (or desmoplastic reaction) with a strong immune infiltration and low vascularization.4 The stroma of PDAC often represents the majority of the tumor volume.52 More importantly, stroma of pancreatic tumors is a major driver of tumor malignancy53 and a physical barrier for therapy.54 Cancer-associated fibroblasts (CAFs), activated PSCs, immune cells, endothelial cells, adipocytes, nerve cells, extracellular matrix proteins, cytokines, and growth factors are the main components of the PDAC stroma. The tumor stroma contributes to the generation of the conditions of acidosis, low abundance of nutrients, and reduced oxygen availability (hypoxia) that characterize the tumor microenvironment. CAFs and activated PSCs drive fibrosis in PDAC. CAFs secrete type-I collagen and proteoglycans to the extracellular matrix, and a distinct subpopulation of these cells participates in immune suppression.55 Pancreatic cancer cells induce the transdifferentiation (or “activation”) of PSCs, which leads to phenotypic and functional changes including exit from quiescence and increased secretion of type-I collagen, fibronectin, osteopontin, periostin,53 and other extracellular matrix components of the fibrotic stroma.56,57 Furthermore, interactions between activated PSCs and cancer cells are established to support disease progression. For instance, activated PSCs increase tumor size and metastasis after orthotopic co-injection with human pancreatic cell lines in mice,58,59 and support the self-renewal of human pancreatic cancer stem cell-like populations in vitro.60,61 Immune cells are also recruited to the stroma of mouse K-Ras-driven PDAC62 and human tumors.63,64 Immunosuppressive M2 macrophages at the invasive front of human PDAC tumors may promote lymphatic metastasis through the induction of lymphangiogenesis.63 Indeed, the “activated stroma index” – a measure of PSC activity – and the abundance of tumor-infiltrating macrophages are negative prognostic values in human PDAC.53,63,64 The effects of the stroma in PDAC seem to extend beyond the primary tumor, as PSCs may disseminate along with PDAC cells, perhaps to establish the metastatic stroma.65
The formation of a tumor stroma may initially represent a defense mechanism, but the stroma eventually evolves to support and promote malignant progression.66 In fact, components of the wound healing response, such as inflammation and fibrosis, are already present in PanIN lesions and become exacerbated in PDAC.4 Moreover, several findings indicate that a pathological stroma might promote cancer initiation. For instance, pancreatitis induces stromal changes, such as activation of PSCs and fibrosis,67 or increased immune infiltration,68 which may contribute to the initiation of PDAC. Along these lines, infiltrating macrophages have recently been shown to contribute to ADM during pancreatitis.69
As tumors progress, stroma and parenchyma change and adapt to each other in a dynamic fashion. In a constantly evolving tumor microenvironment, the availability of oxygen depends on tumor size, perfusion patterns, and stromal reaction. This generates a dynamic pattern of intratumoral hypoxia, which has clinical relevance due to its effects in cancer progression, metastasis, and resistance to therapy.70 Hypoxia, and its implication in invasion and metastasis in PDAC, will be the main focus of the next section.
Hypoxia in pancreatic cancer
A study reporting direct measurement of oxygenation in human PDAC tumors71 analyzed the oxygen partial pressure (pO2) of PDAC and adjacent normal pancreatic tissue immediately before surgical resection by inserting a probe into the exposed organ. Despite the small number of patients enrolled in this study, the result was striking, as the average oxygenation of PDACs was highly decreased when compared to normal tissue (median pO2 of tumors was between 0 and 5.3 mmHg, whereas median pO2 of normal adjacent tissue was between 9.3 and 92.7 mmHg). These results indicate that human PDACs are highly hypoxic. In mouse models of PDAC, measurement of hypoxia after systemic administration of pimonidazole (a chemical that becomes reduced in low oxygen environments and binds to thiol-containing molecules inside cells, forming adducts that can be detected by antibodies) revealed the presence of frequent intratumoral hypoxic areas.72 A similar result was observed in orthotopic implants of human PDAC samples into mice,73 supporting the idea that intratumoral hypoxia is an important component of the PDAC microenvironment.
Sensing and responding to hypoxia
The main molecular mechanism by which oxygen homeostasis is maintained in tissues is regulated by the hypoxia-inducible factors (HIFs).74 This transcriptional regulator is a heterodimer consisting of a constitutively expressed HIF-1β subunit and an oxygen-regulated HIF-α subunit. There are three isoforms of HIF-α (HIF-1α, HIF-2α, and HIF-3α), and each may heterodimerize with HIF-1β to form the HIF-1, HIF-2, and HIF-3 transcriptional regulators. The HIF-1/2α proteins are made unstable in normoxia by a mechanism that includes the hydroxylation of proline residues by specific prolyl hydroxylases.75 Proline hydroxylation of HIF-1/2α facilitates their binding to the von Hippel–Lindau protein (pVHL), which targets them for degradation by the proteasomal pathway. The activity of prolyl hydroxylases, which require oxygen for their catalytic activity, is decreased in hypoxia, thereby preventing the binding of HIF-α to pVHL and promoting oxygen-dependent HIF-α accumulation. HIF-1 and HIF-2 transcriptional regulators activate partially overlapping sets of genes that regulate glycolysis, erythropoiesis, angiogenesis, and pH homeostasis, with an overall contribution to the cellular and organismal adaptation to hypoxia.11 Furthermore, HIFs interact with other transcriptional regulators such as Notch,76 p53, and myc11 to regulate additional downstream pathways. Also, HIF-dependent signaling may be activated in normoxia, generally by signals that increase cell proliferation, which will likely result in an overall increased oxygen demand. Examples include signaling through growth factors,77 activated oncogenes,78 and inactivated tumor suppressors.79 For instance, pancreatic cancer cell lines in the absence of hypoxia have detectable HIF-1α protein, which is probably stabilized through the PI3K/Akt pathway.80 In human PDAC samples, HIF-1α expression correlates with tumor size,81 bad prognosis,82 advanced stage, presence of lymph node metastasis,83 and a higher incidence of hepatic metastasis.84 HIF-2α expression was elevated in human and mouse PanIN lesions when compared to normal exocrine pancreas or PDAC.85 Lack of HIF-2α in the pancreatic epithelial compartment of a Ras-induced GEM model of PanIN led to increased formation of early PanIN lesions and decreased progression of these to high-grade PanIN.85 It would be very interesting to determine the relative contributions of hypoxia, HIF-1, and HIF-2 to ADM, PanIN formation, and progression to PDAC.
Causes and consequences of hypoxia
Poor perfusion and/or high oxygen demand cause intratumoral hypoxia, mostly due to an accelerated rate of tumor growth. Hypoxic cancer cells promote angiogenesis in a process known as the “angiogenic switch”.86 However, PDAC is a poorly vascularized tumor, displaying even less vascularization than the normal surrounding pancreatic tissue.54,87,88 Consequently, the existence of a robust angiogenic switch in PDAC might even be questioned.4 Indeed, microvessel density was not correlated with hypoxia in patient-derived PDAC tumors implanted in mouse pancreas after pimonidazole administration,73 suggesting that hypoxia may not be sufficient to increase angiogenesis in PDAC tumors. Why PDAC is poorly vascularized and how pancreatic cells thrive in a highly hypoxic and hypovascular environment, seemly without robust angiogenesis, is not entirely understood. The specific microenvironment of the desmoplastic reaction, as well as the activation of a metabolic switch in PDAC cancer cells, is likely to hold some answers to that question. For instance, PSCs increase the production of angiostatic factors by pancreatic cancer cells, which overcome the proangiogenic effects of vascular endothelial growth factor (VEGF) secreted by PSCs inside the tumor stroma.87 Hypoxic tumor areas in mouse models of PDAC are highly glycolytic and release lactate, which is metabolized by nearby normoxic cancer cells to sustain proliferation.72 Additionally, pancreatic cancer cells catabolize glutamine in addition to glucose,89 an ability that increases pancreatic cancer cell survival under hypoxia.72 The combination of a Notch inhibitor with gemcitabine (the standard of care in PDAC) in a mouse model of PDAC has shown a survival benefit by disrupting the tumor vasculature.90 This indicates that, despite being hypovascular, PDAC relies on the tumor vasculature for its maintenance. The apparently poor angiogenic response of PDAC tumors to hypoxia may contribute to further sustain the conditions of low oxygenation.
Although robust angiogenesis does not seem to occur in response to hypoxia in PDAC, hypoxia has a profound effect in the tumor stroma because it increases the desmoplastic reaction.91 Furthermore, migration of cancer cells away from the suboptimal conditions of hypoxic areas toward blood vessels is considered a trigger of invasiveness and metastasis;92 along these lines, a correlation between hypoxia, cancer cell proliferation, and spontaneous metastasis is found in human PDAC samples growing orthotopically in mice.73 The prognostic value of hypoxia in human PDAC is currently being analyzed in a clinical trial (NCT01248637). A recent study monitored HIF-1α activity in vivo during tumor progression by using a human pancreatic cancer line engineered to express a reporter of HIF-1α activity.93 Interestingly, the authors observed that the activity of HIF-1α accompanied local invasion, peritoneal dissemination, and liver metastasis of pancreatic cells.93
Hypoxia and pancreatic cancer invasion and metastasis
Cellular invasiveness is required at different steps of the metastatic cascade, and it is also needed for regional invasion. Several cellular mechanisms are associated with cancer cell invasion, including epithelial-to-mesenchymal transition (EMT) and invasive migration (ability to migrate while remodeling the extracellular matrix). Both mechanisms are neither mutually exclusive nor completely dependent, and are the subject of intense investigation due to their importance in cancer malignancy. EMT and invasive migration are regulated through homotypic interactions (between cancer cells themselves), or through heterotypic interactions (between cancer and stromal cells), and also by tumor microenvironmental cues such as hypoxia. Next, we will discuss recent advances in the regulatory role of hypoxia-dependent signaling in cancer cell invasion and metastasis and how hypoxia mediates the interaction between cancer and stromal cells to induce invasion and metastasis in PDAC.
Hypoxia and EMT in pancreatic cancer
The acquisition of invasiveness by epithelial cancers, such as PDAC, is associated with aberrant epithelial plasticity, which is evidenced by the pathological (ectopic and often incomplete) reactivation of an EMT program.94 This program, controlled by a network of transcription factors,95 promotes cellular changes that allow epithelial cancer cells to acquire mesenchymal characteristics, become migratory, and invade (Figure 3). These changes affect cell polarity, cell–cell junctions, and the cellular cytoskeleton.96 Although EMT- independent programs may also facilitate cancer cell invasion,97 the importance of activating the EMT program for tumor progression extends beyond increasing invasiveness, because EMT in cancer is associated with the acquisition of cancer stem cell-like properties98 as well as drug resistance in different cancer cells, including PDAC.99
Figure 3.
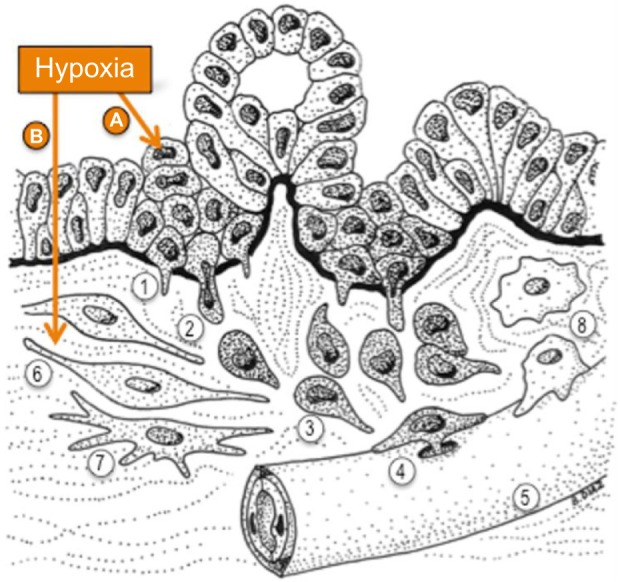
Hypoxia potentiates invasive steps in PDAC.
Notes: Hypoxia in the tumor microenvironment potentiates PDAC cell invasiveness by inducing molecular pathways that affect (A) the cancer cells and (B) the tumor stroma. (A) Hypoxia promotes EMT and also facilitates the invasive migration of cancer cells through extracellular barriers. Upon EMT, epithelial cells lose the attachment to their neighboring cells within the epithelial layer and acquire a migratory phenotype. The process of invasion starts when PDAC cells breach the epithelial basement membrane that separates the epithelium from the stroma (black). Invasion is aided in vitro and possibly in vivo by the formation of proteolytic protrusions named invadopodia (1) that locally digest the basement membrane, generating pores that cancer cells use to cross it and reach the stroma (2). Once in the stroma, PDAC cells display invasive directed migration, aided by EMT and the proteolytic activity of invadopodia (3) toward blood (5) or lymphatic vessels to intravasate and disseminate through the circulation. PDAC cells are likely to use invadopodia to cross endothelial basement membranes (4) and reach the circulation. (B) Hypoxia affects the tumor stroma by promoting the profibrotic activity of cancer-associated fibroblasts (6) and pancreatic stellate cells (7), which further increases hypoxia in the tumor microenvironment, and also by promoting the crosstalk between cancer and stromal cells to increase cancer cells invasiveness (see text for details). Hypoxia may also affect the recruitment and activation of macrophages (8), which also interact with cancer cells to facilitate invasion, for instance, during intravasation. Hypoxia is a determinant of PDAC invasiveness through the activation of cell autonomous and non-cell-autonomous mechanisms that operate in both cancer and associated stroma.
Abbreviations: EMT, epithelial-to-mesenchymal transition; PDAC, pancreatic ductal adenocarcinoma.
Hypoxia triggers EMT in cancer through a number of mechanisms.100 Analysis of tumor sections from a mouse model of PDAC driven by mutant Ras and lack of Ink4a/Arf revealed that cells residing in hypoxic areas (marked after pimonidazole administration) exhibit decreased expression of E-cadherin (epithelial marker) and increased expression of N-cadherin (mesenchymal marker), indicating activation of EMT in hypoxic areas of pancreatic tumors.72 The existence of early events of EMT during PDAC initiation in a mouse model of PDAC driven by mutant Ras and lack of p53 has recently been described using lineage tracing of yellow fluorescent protein (YFP)-labeled pancreatic epithelial cells and immunodetection of the mesenchymal markers Zeb1, Slug, and Snail1.44 In this study, the authors observed EMT (detected as YFP-positive cells expressing mesenchymal markers) as early as in PanIN2 lesions. Furthermore, some of the cells engaged in EMT, delaminated from the PanIN lesions, and were found in the stroma, indicating that the epithelial basement membrane in PanIN lesions was already compromised, at least in mouse models.44 YFP-labeled cells expressing mesenchymal markers were detected as circulating tumor cells in the blood, displayed markers of cancer stem cells, and seeded the liver before any evidence of PDAC.44 It is not known whether PanIN lesions are hypoxic when compared to the normal acini, but this would be conceivable because PanIN lesions are already surrounded by a fibrotic stroma. If so, hypoxia in PanIN lesions could act as an early trigger of EMT and dissemination. Whereas a number of studies in vitro have revealed molecular pathways that regulate EMT in pancreatic cancer,101 the activation of EMT by hypoxia in pancreatic cancer is less studied. Some mechanisms include increased expression of Twist102 and the activation of additional signaling pathways such as Hedgehog and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), as summarized in Table 1.
Table 1.
Current studies on EMT induction by hypoxia in pancreatic cancer
| Hypoxic conditions | EMT markers | Mechanism | Increased migration | Increased invasion | Increased metastasis | Reference |
|---|---|---|---|---|---|---|
| 3% oxygen (48 h) | E-cadherin, Snail, vimentin | Hedgehog pathway | ND | Yes | ND | 147 |
| 1% oxygen (72 h) | Slug, Snail, Twist | ROS, HIF-1α, NFκB | Yes | Yes | Yes | 148 |
| 1% oxygen (48 h) | E-cadherin, Slug, Twist, vimentin, Zeb1 | ND | Yes | ND | ND | 149 |
| 3% oxygen (48 or 72 h) | E-cadherin, Snail, α-SMA | HIF-1α, ROS, VEGF | Yes | Yes | ND | 150 |
Abbreviations: α-SMA, α-smooth muscle actin; EMT, epithelial-to-mesenchymal transition; h, hours; HIF-1α, hypoxia-inducible factor 1α; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; ND, not determined; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor.
Hypoxia, invasion, and extracellular matrix remodeling in pancreatic cancer
During invasion, cancer cells cross a number of extracellular matrix barriers, including the epithelial basement membrane, the interstitial collagen, and the endothelial basement membranes (lymphatic and/or hematoid), which are approached from either the adluminal or the luminal side during intra- and extravasation, respectively. The detailed mechanisms that cancer cells employ to negotiate all these barriers is a matter of intense investigation, and both proteolysis-dependent103 and -independent104 mechanisms have been proposed. It is plausible that both types of invasion are used by cancer cells in vivo depending on the mode of migration (single-cell or collective), the composition of the extracellular barrier, the tissue of origin, the tumor microenvironment, the route of dissemination, and the site of metastasis. For instance, cancer cells that transit the interstitial matrix through tunnels created by CAFs,105 enter the blood vessels at sites where activated macrophages are present,106 or enter the circulation through the discontinuous basement membrane of lymphatic vessels,107 are likely less dependent on proteolysis, at least during those steps of metastasis, than cells that transit as single cells through the dense interstitial matrix of fibrotic tumors.
Cancer malignancy begins when cancer cells cross the epithelial basement membrane, a specialized sheet-like extracellular matrix structure formed by a highly cross-linked mixture of laminin, type-IV collagen, nidogen, and proteoglycans.108,109 During embryonic development and immune surveillance in vivo, cells cross basement membranes by mechanisms that include breakage by physical force or sliding to generate temporary gaps.110 The exact mechanism by which cancer cells cross the epithelial basement membrane in vivo is not known, as the role of proteases has not been formally proven in vivo.110 Whereas defective turnover could compromise the integrity of basement membranes adjacent to tumors in the absence of proteolysis,111 a critical role for proteolysis in invasion through mesothelial basement membrane ex vivo is demonstrated by the presence of type-IV collagen degradation products adjacent to invading cells and by the need for the catalytic activity of the membrane-anchored membrane type 1 matrix metalloproteinase MT1-MMP in that process.112 Schoumacher et al used a similar ex vivo system, whereby cancer cells were plated on top of rat peritoneal basement membrane and imaged over time with confocal fluorescence microscopy.113 This system allowed the visualization of individual cancer cells during the process of crossing the intact basement membrane in vitro. The basement membrane was crossed within 5–7 days through a proteolytic-dependent mechanism mediated by the elaboration of invadopodia.113 Invadopodia are subcellular protrusive structures that contain cytoskeletal and signaling proteins as well as membrane-anchored metalloproteases that mediate focal pericellular proteolysis of extracellular matrix proteins.114–116 Whether invading through basement membranes or through interstitial tissue, cancer cells are likely to use invadopodia to perform regulated proteolysis during proteolytic-dependent invasion (Figure 3), although the existence of proteolytic-dependent invadopodia-independent invasion has not been formally ruled out. The need for proteolysis in order for cells to migrate through interstitial collagen is supported by the observation of collagen proteolytic degradation products adjacent to cells invading through the extracellular matrix in xenografted tumor sections.117 Recently, invadopodia-like structures have been observed in vivo in the Caenorhabditis elegans gonadal anchor cell118 and in xenografted breast cancer cells.117 A number of proteins necessary for invadopodia formation, including cortactin, Tks5, N-WASP, and MT1-MMP are necessary for tumor growth and/or metastasis in vivo,117,119–123 suggesting an important role for invadopodia during cancer progression and malignancy in vivo. A recent study indicated a relationship between EMT and invadopodia formation;121 however, it is unknown whether invadopodia formation precedes or follows EMT at the onset of cancer invasiveness in vivo.
Human pancreatic cancer cells elaborate invadopodia.124–126 Interestingly, recent reports indicate that hypoxia induces the formation of invadopodia in different cancer cells.125,127,128 Increased invadopodia formation under hypoxia (Figure 4) represents an additional mechanism by which hypoxia induces cancer cell invasiveness. In human pancreatic cancer cells, hypoxia increases the formation of invadopodia as well as their activity (measured by the local proteolytic degradation of gelatin) through HIF-1α-dependent activation of the Notch signaling pathway.125 Activation of Notch signaling under hypoxia led to increased protease-dependent shedding of the membrane-anchored epidermal growth factor (EGF) receptor ligand Heparin-binding EGF-like growth factor (HB-EGF). Soluble HB-EGF, in turn, induced the formation of invadopodia in a non-cell autonomous manner through the activation of the EGF receptor.125 These findings indicate that hypoxia elicits a complex signaling pathway that couples cell contact- dependent signaling (mediated by Notch) with paracrine signaling (mediated by protease-dependent release of soluble HB-EGF), as summarized in Figure 5. Although the contribution of Notch signaling to cancer progression in pancreatic cancer seems to be context-dependent,129 these data suggest that hypoxia-activated Notch potentiates pancreatic cancer cell invasion and metastasis. Likewise, the effect of HB-EGF-dependent activation of the EGF receptor in invadopodia formation and activity in pancreatic cancer cells125 indicates a function for the EGF receptor pathway in PDAC invasion and metastasis in addition to its recently reported roles in the induction of Ras-driven PDAC in mice.130–132 Furthermore, activation of paracrine signaling by hypoxia suggests that hypoxia might affect not only the behavior of cells that reside in hypoxic areas, but also that of cells that reside in nearby normoxic areas. Increased shedding of HB-EGF under hypoxia would allow paracrine signaling through the EGF receptor, which could be activated in nearby normoxic cells located within the diffusion limit of HB-EGF. The possibility of a crosstalk between hypoxic and normoxic areas of tumors has not been formally proven in vivo, but it is also suggested by the release of lactate under hypoxia72 and its possible effect in the proliferation of normoxic cells. Additional mechanisms by which hypoxia induces invasiveness in pancreatic cancer include the increased expression of urokinase-type plasminogen activator receptor, which promotes increased invasion and intravasation of pancreatic cells into the chicken chorioallantoic membrane model,133 as well as the HIF-1α-mediated induction of the actin-bundling protein fascin, which increased the expression of MMP-2 in human pancreatic cell lines.134 Based on knowledge from other cancer types, hypoxia could also promote invadopodia formation in pancreatic cancer cells through the generation of reactive oxygen species by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, which regulate invadopodia formation135 and facilitate glioblastoma invasion in vivo.136
Figure 4.
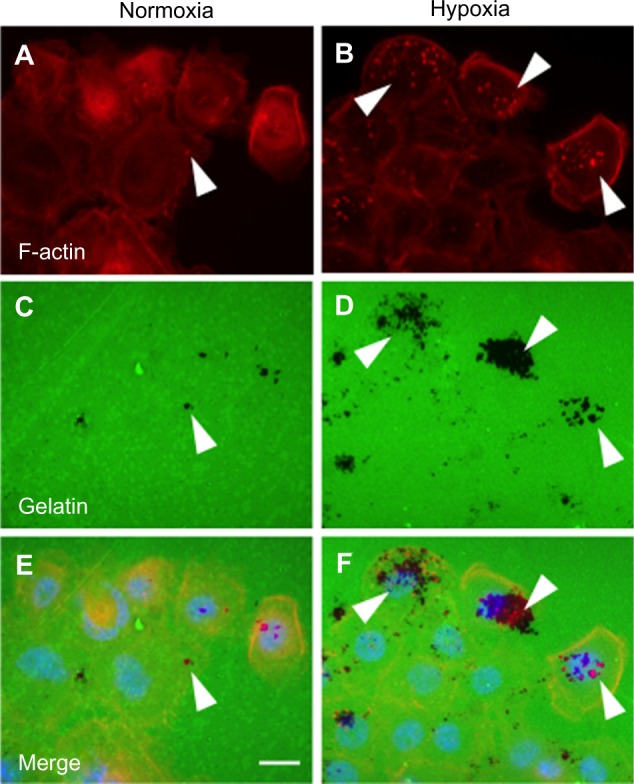
Hypoxia increases invadopodia formation in human PDAC cells.
Notes: The human PDAC line BxPC3 was cultured under normoxia or hypoxia (1% oxygen) for 16 hours on coverslips covered by fluorescein isothiocyanate-conjugated gelatin (green). Cells were processed for detection of invadopodia by F-actin labeling with Alexa468-conjugated phalloidin (red). Nuclei were stained with DAPI (blue). Invadopodia are detected as F-actin-rich dots (arrowheads in A and B) that display local protease activity by digesting the gelatin (black areas indicate absence of gelatin upon invadopodia activity; arrowheads in C and D). Gelatin degradation often co-localizes with invadopodia (arrowheads in E and F). Size bar: 10 µm.
Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; PDAC, pancreatic ductal adenocarcinoma.
Figure 5.

Cell-contact-dependent signaling and paracrine activation of the EGF receptor are coupled under hypoxia to promote invadopodia formation in pancreatic cancer cells.
Notes: In pancreatic cancer cells, hypoxia promotes HIF-1α-dependent activation of the Notch signaling pathway, which mediates cell-contact-dependent signaling upon binding of a transmembrane ligand to a transmembrane receptor expressed in adjacent cells. Upon ligand binding, the receptor is cleaved by proteolysis to release a Notch intracellular domain (NIC) that travels to the nucleus (N) and partners with HIF-1α to promote gene transcription. Activation of Notch signaling under hypoxia promotes protease-dependent shedding of the EGF receptor ligand HB-EGF (red). Upon shedding, the transmembrane HB-EGF becomes soluble and signals in autocrine, juxtacrine, or paracrine fashions. Activation of the EGF receptor by soluble HB-EGF promotes invadopodia (depicted in orange) formation and increases cancer cell invasiveness. Adapted with permission from Díaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201(2):279–292.125
Abbreviations: EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; HB-EGF, heparin-binding epidermal growth factor; HIF-1α, hypoxia-inducible factor 1α.
Hypoxia, tumor stroma, and invasion in pancreatic cancer
The stroma of PDAC tumors is also often hypoxic, as indicated by the increased expression of HIF-1α in stromal cells from human tumor sections.81,137–139 The presence of hypoxia in the stroma of PDAC has also been observed after pimonidazole administration to mice bearing PDAC tumor patient xenografts.140 A number of studies indicate that hypoxia and the pancreatic cancer desmoplastic reaction are interdependent and potentiate each other. PSCs in culture respond to hypoxia through increased HIF-1α expression, increased motility,91 and elevated alpha-smooth muscle actin (SMA) expression (a marker of activation).87 Furthermore, PSCs under hypoxia display increased synthesis and secretion of type-I collagen,87,91 periostin, and fibronectin,87 which potentiate the desmoplastic reaction under hypoxia. Hypoxia also affects the hypovascularity of the pancreatic stroma, which further reinforces hypoxia in the tumor microenvironment. For instance, PSCs under hypoxia stimulate the production of the angiostatic factor endostatin by pancreatic cancer cells, which may reduce angiogenesis.87 Also in line with these findings, hypoxia induces the expression of the Sonic Hedgehog ligand in pancreatic cancer cells, which acts in a paracrine manner, binding to the Patched-1 receptor in fibroblasts and promoting the secretion of type-I collagen and fibronectin, which may contribute to the desmoplastic reaction.139 A recent report indicates that pancreatic CAFs elaborate invadopodia in response to phorbol esters.141 It would be interesting to determine if hypoxia directly or indirectly (for instance, through HB-EGF shed under hypoxia by cancer cells) affects invadopodia formation in CAFs as a means to increase the remodeling of the stroma and facilitate the invasion of pancreatic cancer cells.
Hypoxia, therefore, is an important determinant of the desmoplastic reaction through a process that implicates the activation of a signaling crosstalk between cancer and stromal cells. In addition, the desmoplastic reaction could be considered as an effector of hypoxia that activates a positive feedback loop to maintain and further increase hypoxia and desmoplasia, which, in turn, activates invasion and metastasis.
Hypoxia in additional steps of the metastatic cascade in pancreatic cancer
The effects of hypoxia in additional steps of the metastatic cascade are still to be defined, but some studies in other cancer types indicate that hypoxia or hypoxia signaling (upon non-hypoxic activation of HIFs) could affect all the steps of the metastatic cascade, including intravasation in blood or lymphatic vessels, survival in the circulation, margination, extravasation, formation of the pre-metastatic niche, angiogenesis in the secondary tumor, and, perhaps, regulation of dormancy.142,143 Secondary pancreatic tumors might also be hypoxic since pimonidazole staining is detected in small liver metastatic lesions in mice bearing patient-derived pancreatic tumor xenografts.73 Although hypoxia induces lymphangiogenesis in cancer,143,144 the formation of new lymphatics is not detected in PDAC,145 which leads to the suggestion that lymphatic metastasis in PDAC occurs at peritumoral rather than intratumoral lymphatic vessels.146 Lymphatic spreading is an important feature of PDAC, and hypoxia signaling may be an important contributor to lymph node metastasis, as these lesions show detectable levels of HIF-1α.81 Along these lines, implantation of a human pancreatic cancer line expressing an HIF-1α reporter into mouse pancreas showed elevated HIF-1α activity after spreading from the pancreas to the abdominal cavity, peritoneum, and liver.93 High levels of VEGF, a target of HIF-1α, were found in the ascites from these mice,93 suggesting that hypoxic signaling is active over the course of dissemination to different target tissues in PDAC. In addition to affecting lymphatic nodes, liver, and lungs, PDAC often displays perivascular, perineural, and intraneural invasion.40 The cellular and molecular mechanisms that regulate these processes are not well defined, and further investigation is needed to delineate the possible implication of EMT, protease-dependent invasion, and hypoxia signaling in those alternative routes of tumor dissemination.
Conclusion
Hypoxia is an emerging determinant of pancreatic tumor malignancy. A number of studies have elucidated effector signaling pathways that mediate the pro-invasive and metastatic effects of hypoxia signaling in PDAC, and many new pathways will be identified in the near future. Hypoxia signaling in pancreatic cancer cells induces EMT and the formation of invadopodia, which are phenotypic changes associated with the activation and maintenance of a pro-invasive program, in large part by promoting the ability of cancer cells to cross the epithelial basement membrane and initiate invasion. Hypoxia signaling also affects stromal cells promoting the activation of macrophages, CAFs, and PSCs, and the secretion of specific extracellular matrix components that contribute to generate the extensive desmoplastic stroma characteristic of PDAC. Hypoxia may even promote invasion in a non-cell-autonomous manner through paracrine stimulation of invadopodia formation, suggesting that the effect of hypoxia in cancer cell invasiveness may extend to cells surrounding hypoxic regions of tumors. Furthermore, hypoxia signaling is likely important for completing all the steps of the metastatic cascade, although the effector pathways are not yet well understood. In addition to its effects on cancer malignancy, hypoxia also reduces the efficacy of chemo- and radiotherapy. Furthermore, hypoxia induced by antiangiogenic therapy contributes to therapy failure.142 All these findings point to hypoxia as an important therapeutic target in PDAC, a disease in urgent need of novel therapeutic approaches.
Acknowledgments
Dr Angela Yuen is currently affiliated to the College of Pharmacy, Roseman University of Health Sciences, Henderson, NV, USA. Many important articles were not included in this review due to space restrictions. We are grateful to Christopher Abdullah (Sanford Burnham Medical Research Institute, La Jolla, CA) and Dr Jack Arbiser (Emory University, Atlanta, GA) for critical reading of this manuscript.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer Statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362(17):1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 3.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378(9791):607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pérez-Mancera PA, Guerra C, Barbacid M, Tuveson DA. What we have learned about pancreatic cancer from mouse models. Gastroenterology. 2012;142(5):1079–1092. doi: 10.1053/j.gastro.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Erkan M, Hausmann S, Michalski CW, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol. 2012;9(8):454–467. doi: 10.1038/nrgastro.2012.115. [DOI] [PubMed] [Google Scholar]
- 7.Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63(5):318–348. doi: 10.3322/caac.21190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neesse A, Krug S, Gress TM, Tuveson DA, Michl P. Emerging concepts in pancreatic cancer medicine: targeting the tumor stroma. Onco Targets Ther. 2013;7:33–43. doi: 10.2147/OTT.S38111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8(6):425–437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8(12):967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12(1):9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8(3):180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 13.Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47–71. doi: 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- 14.Rovira M, Scott SG, Liss AS, Jensen J, Thayer SP, Leach SD. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci U S A. 2010;107(1):75–80. doi: 10.1073/pnas.0912589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strobel O, Rosow DE, Rakhlin EY, et al. Pancreatic duct glands are distinct ductal compartments that react to chronic injury and mediate Shh-induced metaplasia. Gastroenterology. 2010;138(3):1166–1177. doi: 10.1053/j.gastro.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apte MV, Haber PS, Applegate TL, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43(1):128–133. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bachem MG, Schneider E, Gross H, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115(2):421–432. doi: 10.1016/s0016-5085(98)70209-4. [DOI] [PubMed] [Google Scholar]
- 18.Crnogorac-Jurcevic T, Efthimiou E, Capelli P, et al. Gene expression profiles of pancreatic cancer and stromal desmoplasia. Oncogene. 2001;20(50):7437–7446. doi: 10.1038/sj.onc.1204935. [DOI] [PubMed] [Google Scholar]
- 19.Tuveson DA, Hingorani SR. Ductal pancreatic cancer in humans and mice. Cold Spring Harb Symp Quant Biol. 2005;70:65–72. doi: 10.1101/sqb.2005.70.040. [DOI] [PubMed] [Google Scholar]
- 20.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cubilla AL, Fitzgerald PJ. Morphological lesions associated with human primary invasive nonendocrine pancreas cancer. Cancer Res. 1976;36(7 PT 2):2690–2698. [PubMed] [Google Scholar]
- 22.Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57(11):2140–2143. [PubMed] [Google Scholar]
- 23.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6(8):2969–2972. [PubMed] [Google Scholar]
- 24.Yanagisawa A, Ohtake K, Ohashi K, et al. Frequent c-Ki-ras oncogene activation in mucous cell hyperplasias of pancreas suffering from chronic inflammation. Cancer Res. 1993;53(5):953–956. [PubMed] [Google Scholar]
- 25.Tada M, Ohashi M, Shiratori Y, et al. Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology. 1996;110(1):227–231. doi: 10.1053/gast.1996.v110.pm8536861. [DOI] [PubMed] [Google Scholar]
- 26.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 27.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol. 2007;171(1):263–273. doi: 10.2353/ajpath.2007.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guerra C, Schuhmacher AJ, Cañamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11(3):291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Esposito I, Seiler C, Bergmann F, Kleeff J, Friess H, Schirmacher P. Hypothetical progression model of pancreatic cancer with origin in the centroacinar-acinar compartment. Pancreas. 2007;35(3):212–217. doi: 10.1097/mpa.0b013e31805d0190. [DOI] [PubMed] [Google Scholar]
- 31.Kopp JL, von Figura G, Mayes E, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(6):737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris JP, 4th, Cano DA, Sekine S, Wang SC, Hebrok M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120(2):508–520. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lowenfels AB, Maisonneuve P, Cavallini G, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328(20):1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 34.Malka D, Hammel P, Maire F, et al. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut. 2002;51(6):849–852. doi: 10.1136/gut.51.6.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hruban RH, Takaori K, Klimstra DS, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28(8):977–987. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 36.Singh M, Maitra A. Precursor lesions of pancreatic cancer: molecular pathology and clinical implications. Pancreatology. 2007;7(1):9–19. doi: 10.1159/000101873. [DOI] [PubMed] [Google Scholar]
- 37.Stanger BZ, Stiles B, Lauwers GY, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8(3):185–195. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 38.Ray KC, Bell KM, Yan J, et al. Epithelial tissues have varying degrees of susceptibility to Kras(G12D)-initiated tumorigenesis in a mouse model. PLoS One. 2011;6(2):e16786. doi: 10.1371/journal.pone.0016786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Bapat AA, Hostetter G, Von Hoff DD, Han H. Perineural invasion and associated pain in pancreatic cancer. Nat Rev Cancer. 2011;11(10):695–707. doi: 10.1038/nrc3131. [DOI] [PubMed] [Google Scholar]
- 41.Johnstone PA, Sindelar WF. Lymph node involvement and pancreatic resection: correlation with prognosis and local disease control in a clinical trial. Pancreas. 1993;8(5):535–539. doi: 10.1097/00006676-199309000-00001. [DOI] [PubMed] [Google Scholar]
- 42.Slidell MB, Chang DC, Cameron JL, et al. Impact of total lymph node count and lymph node ratio on staging and survival after pancreatectomy for pancreatic adenocarcinoma: a large, population-based analysis. Ann Surg Oncol. 2008;15(1):165–174. doi: 10.1245/s10434-007-9587-1. [DOI] [PubMed] [Google Scholar]
- 43.Hruban RH, Fukushima N. Pancreatic adenocarcinoma: update on the surgical pathology of carcinomas of ductal origin and PanINs. Mod Pathol. 2007;20(Suppl 1):S61–S70. doi: 10.1038/modpathol.3800685. [DOI] [PubMed] [Google Scholar]
- 44.Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hüsemann Y, Geigl JB, Schubert F, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernards R, Weinberg RA. A progression puzzle. Nature. 2002;418(6900):823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- 48.Klein CA. Cancer. The metastasis cascade. Science. 2008;321(5897):1785–1787. doi: 10.1126/science.1164853. [DOI] [PubMed] [Google Scholar]
- 49.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 50.Mueller MM, Fusenig NE. Friends or foes – bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4(11):839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 51.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mollenhauer J, Roether I, Kern HF. Distribution of extracellular matrix proteins in pancreatic ductal adenocarcinoma and its influence on tumor cell proliferation in vitro. Pancreas. 1987;2(1):14–24. doi: 10.1097/00006676-198701000-00003. [DOI] [PubMed] [Google Scholar]
- 53.Erkan M, Michalski CW, Rieder S, et al. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2008;6(10):1155–1161. doi: 10.1016/j.cgh.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 54.Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kraman M, Bambrough PJ, Arnold JN, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330(6005):827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 56.Bachem MG, Schünemann M, Ramadani M, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128(4):907–921. doi: 10.1053/j.gastro.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 57.Apte M, Pirola R, Wilson J. The fibrosis of chronic pancreatitis: new insights into the role of pancreatic stellate cells. Antioxid Redox Signal. 2011;15(10):2711–2722. doi: 10.1089/ars.2011.4079. [DOI] [PubMed] [Google Scholar]
- 58.Vonlaufen A, Joshi S, Qu C, et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008;68(7):2085–2093. doi: 10.1158/0008-5472.CAN-07-2477. [DOI] [PubMed] [Google Scholar]
- 59.Hwang RF, Moore T, Arumugam T, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68(3):918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. 2012;11(7):1282–1290. doi: 10.4161/cc.19679. [DOI] [PubMed] [Google Scholar]
- 61.Hamada S, Masamune A, Takikawa T, et al. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem Biophys Res Commun. 2012;421(2):349–354. doi: 10.1016/j.bbrc.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 62.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 63.Kurahara H, Shinchi H, Mataki Y, et al. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res. 2011;167(2):e211–e219. doi: 10.1016/j.jss.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 64.Ino Y, Yamazaki-Itoh R, Shimada K, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108(4):914–923. doi: 10.1038/bjc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu Z, Vonlaufen A, Phillips PA, et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol. 2010;177(5):2585–2596. doi: 10.2353/ajpath.2010.090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schäfer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9(8):628–638. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- 67.Apte MV, Wilson JS. Stellate cell activation in alcoholic pancreatitis. Pancreas. 2003;27(4):316–320. doi: 10.1097/00006676-200311000-00008. [DOI] [PubMed] [Google Scholar]
- 68.Guerra C, Collado M, Navas C, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19(6):728–739. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liou GY, Döppler H, Necela B, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κB and MMPs. J Cell Biol. 2013;202(3):563–577. doi: 10.1083/jcb.201301001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11(6):393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 71.Koong AC, Mehta VK, Le QT, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. 2000;48(4):919–922. doi: 10.1016/s0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- 72.Guillaumond F, Leca J, Olivares O, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2013;110(10):3919–3924. doi: 10.1073/pnas.1219555110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang Q, Jurisica I, Do T, Hedley DW. Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 2011;71(8):3110–3120. doi: 10.1158/0008-5472.CAN-10-4049. [DOI] [PubMed] [Google Scholar]
- 74.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 76.Gustafsson MV, Zheng X, Pereira T, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9(5):617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 77.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1- mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277(41):38205–38211. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- 78.Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 1997;57(23):5328–5335. [PubMed] [Google Scholar]
- 79.Zundel W, Schindler C, Haas-Kogan D, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14(4):391–396. [PMC free article] [PubMed] [Google Scholar]
- 80.Akakura N, Kobayashi M, Horiuchi I, et al. Constitutive expression of hypoxia-inducible factor-1alpha renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 2001;61(17):6548–6554. [PubMed] [Google Scholar]
- 81.Kitada T, Seki S, Sakaguchi H, Sawada T, Hirakawa K, Wakasa K. Clinicopathological significance of hypoxia-inducible factor-1alpha expression in human pancreatic carcinoma. Histopathology. 2003;43(6):550–555. doi: 10.1111/j.1365-2559.2003.01733.x. [DOI] [PubMed] [Google Scholar]
- 82.Shibaji T, Nagao M, Ikeda N, et al. Prognostic significance of HIF-1 alpha overexpression in human pancreatic cancer. Anticancer Res. 2003;23(6C):4721–4727. [PubMed] [Google Scholar]
- 83.Sun HC, Qiu ZJ, Liu J, et al. Expression of hypoxia-inducible factor-1 alpha and associated proteins in pancreatic ductal adenocarcinoma and their impact on prognosis. Int J Oncol. 2007;30(6):1359–1367. [PubMed] [Google Scholar]
- 84.Matsuo Y, Ding Q, Desaki R, et al. Hypoxia inducible factor-1 alpha plays a pivotal role in hepatic metastasis of pancreatic cancer: an immunohistochemical study. J Hepatobiliary Pancreat Sci. 2014;21(2):105–112. doi: 10.1002/jhbp.6. [DOI] [PubMed] [Google Scholar]
- 85.Criscimanna A, Duan LJ, Rhodes JA, et al. PanIN-specific regulation of Wnt signaling by HIF2α during early pancreatic tumorigenesis. Cancer Res. 2013;73(15):4781–4790. doi: 10.1158/0008-5472.CAN-13-0566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339(6219):58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 87.Erkan M, Reiser-Erkan C, Michalski CW, et al. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia. 2009;11(5):497–508. doi: 10.1593/neo.81618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mazur PK, Siveke JT. Genetically engineered mouse models of pancreatic cancer: unravelling tumour biology and progressing translational oncology. Gut. 2012;61(10):1488–1500. doi: 10.1136/gutjnl-2011-300756. [DOI] [PubMed] [Google Scholar]
- 89.Zhou W, Capello M, Fredolini C, et al. Proteomic analysis reveals Warburg effect and anomalous metabolism of glutamine in pancreatic cancer cells. J Proteome Res. 2012;11(2):554–563. doi: 10.1021/pr2009274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cook N, Frese KK, Bapiro TE, et al. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med. 2012;209(3):437–444. doi: 10.1084/jem.20111923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Shimosegawa T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am J Physiol Gastrointest Liver Physiol. 2008;295(4):G709–G717. doi: 10.1152/ajpgi.90356.2008. [DOI] [PubMed] [Google Scholar]
- 92.Brizel DM, Scully SP, Harrelson JM, et al. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996;56(5):941–943. [PubMed] [Google Scholar]
- 93.Kizaka-Kondoh S, Itasaka S, Zeng L, et al. Selective killing of hypoxia-inducible factor-1-active cells improves survival in a mouse model of invasive and metastatic pancreatic cancer. Clin Cancer Res. 2009;15(10):3433–3441. doi: 10.1158/1078-0432.CCR-08-2267. [DOI] [PubMed] [Google Scholar]
- 94.Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342(6159):1234850. doi: 10.1126/science.1234850. [DOI] [PubMed] [Google Scholar]
- 95.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 97.Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor invasion in the absence of epithelial-mesenchymal transition: podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell. 2006;9(4):261–272. doi: 10.1016/j.ccr.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 98.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69(14):5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jiang J, Tang YL, Liang XH. EMT: a new vision of hypoxia promoting cancer progression. Cancer Biol Ther. 2011;11(8):714–723. doi: 10.4161/cbt.11.8.15274. [DOI] [PubMed] [Google Scholar]
- 101.Maier HJ, Wirth T, Beug H. Epithelial-mesenchymal transition in pancreatic carcinoma. Cancers (Basel) 2010;2(4):2058–2083. doi: 10.3390/cancers2042058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG. Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. 2007;13(16):4769–4776. doi: 10.1158/1078-0432.CCR-06-2926. [DOI] [PubMed] [Google Scholar]
- 103.Sabeh F, Ota I, Holmbeck K, et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. 2004;167(4):769–781. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5(8):711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 105.Gaggioli C, Hooper S, Hidalgo-Carcedo C, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9(12):1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 106.Wyckoff JB, Wang Y, Lin EY, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67(6):2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 107.Oliver G, Detmar M. The rediscovery of the lymphatic system: old and new insights into the development and biological function of the lymphatic vasculature. Genes Dev. 2002;16(7):773–783. doi: 10.1101/gad.975002. [DOI] [PubMed] [Google Scholar]
- 108.Erickson AC, Couchman JR. Still more complexity in mammalian basement membranes. J Histochem Cytochem. 2000;48(10):1291–1306. doi: 10.1177/002215540004801001. [DOI] [PubMed] [Google Scholar]
- 109.Rowe RG, Weiss SJ. Breaching the basement membrane: who, when and how? Trends Cell Biol. 2008;18(11):560–574. doi: 10.1016/j.tcb.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 110.Kelley LC, Lohmer LL, Hagedorn EJ, Sherwood DR. Traversing the basement membrane in vivo: a diversity of strategies. J Cell Biol. 2014;204(3):291–302. doi: 10.1083/jcb.201311112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rowe RG, Weiss SJ. Navigating ECM barriers at the invasive front: the cancer cell-stroma interface. Annu Rev Cell Dev Biol. 2009;25:567–595. doi: 10.1146/annurev.cellbio.24.110707.175315. [DOI] [PubMed] [Google Scholar]
- 112.Hotary K, Li XY, Allen E, Stevens SL, Weiss SJ. A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev. 2006;20(19):2673–2686. doi: 10.1101/gad.1451806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schoumacher M, Goldman RD, Louvard D, Vignjevic DM. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J Cell Biol. 2010;189(3):541–556. doi: 10.1083/jcb.200909113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12(7):413–426. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hoshino D, Branch KM, Weaver AM. Signaling inputs to invadopodia and podosomes. J Cell Sci. 2013;126(Pt 14):2979–2989. doi: 10.1242/jcs.079475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Paz H, Pathak N, Yang J. Invading one step at a time: the role of invadopodia in tumor metastasis. Oncogene. 2013 Sep 30; doi: 10.1038/onc.2013.393. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, Condeelis J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci. 2012;125(Pt 3):724–734. doi: 10.1242/jcs.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hagedorn EJ, Ziel JW, Morrissey MA, et al. The netrin receptor DCC focuses invadopodia-driven basement membrane transmigration in vivo. J Cell Biol. 2013;201(6):903–913. doi: 10.1083/jcb.201301091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Clark ES, Brown B, Whigham AS, Kochaishvili A, Yarbrough WG, Weaver AM. Aggressiveness of HNSCC tumors depends on expression levels of cortactin, a gene in the 11q13 amplicon. Oncogene. 2009;28(3):431–444. doi: 10.1038/onc.2008.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Blouw B, Seals DF, Pass I, Diaz B, Courtneidge SA. A role for the podosome/invadopodia scaffold protein Tks5 in tumor growth in vivo. Eur J Cell Biol. 2008;87(8–9):555–567. doi: 10.1016/j.ejcb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Eckert MA, Lwin TM, Chang AT, et al. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19(3):372–386. doi: 10.1016/j.ccr.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Perentes JY, Kirkpatrick ND, Nagano S, et al. Cancer cell-associated MT1-MMP promotes blood vessel invasion and distant metastasis in triple-negative mammary tumors. Cancer Res. 2011;71(13):4527–4538. doi: 10.1158/0008-5472.CAN-10-4376. [DOI] [PubMed] [Google Scholar]
- 123.Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114(1):33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 124.Neel NF, Rossman KL, Martin TD, Hayes TK, Yeh JJ, Der CJ. The RalB small GTPase mediates formation of invadopodia through a GTPase-activating protein-independent function of the RalBP1/RLIP76 effector. Mol Cell Biol. 2012;32(8):1374–1386. doi: 10.1128/MCB.06291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Díaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201(2):279–292. doi: 10.1083/jcb.201209151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Razidlo GL, Schroeder B, Chen J, Billadeau DD, McNiven MA. Vav1 as a central regulator of invadopodia assembly. Curr Biol. 2014;24(1):86–93. doi: 10.1016/j.cub.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lucien F, Brochu-Gaudreau K, Arsenault D, Harper K, Dubois CM. Hypoxia-induced invadopodia formation involves activation of NHE-1 by the p90 ribosomal S6 kinase (p90RSK) PLoS One. 2011;6(12):e28851. doi: 10.1371/journal.pone.0028851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hanna SC, Krishnan B, Bailey ST, et al. HIF1α and HIF2α independently activate SRC to promote melanoma metastases. J Clin Invest. 2013;123(5):2078–2093. doi: 10.1172/JCI66715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Avila JL, Kissil JL. Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol Med. 2013;19(5):320–327. doi: 10.1016/j.molmed.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Navas C, Hernández-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(3):318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ardito CM, Grüner BM, Takeuchi KK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22(3):304–317. doi: 10.1016/j.ccr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ray KC, Moss ME, Franklin JL, et al. Heparin-binding epidermal growth factor-like growth factor eliminates constraints on activated Kras to promote rapid onset of pancreatic neoplasia. Oncogene. 2014;33(7):823–831. doi: 10.1038/onc.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Büchler P, Reber HA, Tomlinson JS, et al. Transcriptional regulation of urokinase-type plasminogen activator receptor by hypoxia-inducible factor 1 is crucial for invasion of pancreatic and liver cancer. Neoplasia. 2009;11(2):196–206. doi: 10.1593/neo.08734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhao X, Gao S, Ren H, et al. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014;74(9):2455–2464. doi: 10.1158/0008-5472.CAN-13-3009. [DOI] [PubMed] [Google Scholar]
- 135.Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, Courtneidge SA. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci Signal. 2009;2(88):ra53. doi: 10.1126/scisignal.2000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Munson JM, Fried L, Rowson SA, et al. Anti-invasive adjuvant therapy with imipramine blue enhances chemotherapeutic efficacy against glioma. Sci Transl Med. 2012;4(127):127ra36. doi: 10.1126/scitranslmed.3003016. [DOI] [PubMed] [Google Scholar]
- 137.Ide T, Kitajima Y, Miyoshi A, et al. The hypoxic environment in tumor-stromal cells accelerates pancreatic cancer progression via the activation of paracrine hepatocyte growth factor/c-Met signaling. Ann Surg Oncol. 2007;14(9):2600–2607. doi: 10.1245/s10434-007-9435-3. [DOI] [PubMed] [Google Scholar]
- 138.Schwartz DL, Bankson JA, Lemos R, Jr, et al. Radiosensitization and stromal imaging response correlates for the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreatic cancer. Mol Cancer Ther. 2010;9(7):2057–2067. doi: 10.1158/1535-7163.MCT-09-0768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Spivak-Kroizman TR, Hostetter G, Posner R, et al. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res. 2013;73(11):3235–3247. doi: 10.1158/0008-5472.CAN-11-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lohse I, Lourenco C, Ibrahimov E, Pintilie M, Tsao MS, Hedley DW. Assessment of hypoxia in the stroma of patient-derived pancreatic tumor xenografts. Cancers (Basel) 2014;6(1):459–471. doi: 10.3390/cancers6010459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Goicoechea SM, García-Mata R, Staub J, et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene. 2014;33(10):1265–1273. doi: 10.1038/onc.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.De Bock K, Mazzone M, Carmeliet P. Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or not? Nat Rev Clin Oncol. 2011;8(7):393–404. doi: 10.1038/nrclinonc.2011.83. [DOI] [PubMed] [Google Scholar]
- 143.Semenza GL. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene. 2013;32(35):4057–4063. doi: 10.1038/onc.2012.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ji RC. Hypoxia and lymphangiogenesis in tumor microenvironment and metastasis. Cancer Lett. 2014;346(1):6–16. doi: 10.1016/j.canlet.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 145.Sipos B, Kojima M, Tiemann K, et al. Lymphatic spread of ductal pancreatic adenocarcinoma is independent of lymphangiogenesis. J Pathol. 2005;207(3):301–312. doi: 10.1002/path.1840. [DOI] [PubMed] [Google Scholar]
- 146.Schneider M, Büchler P, Giese N, et al. Role of lymphangiogenesis and lymphangiogenic factors during pancreatic cancer progression and lymphatic spread. Int J Oncol. 2006;28(4):883–890. [PubMed] [Google Scholar]
- 147.Lei J, Ma J, Ma Q, et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol Cancer. 2013;12:66. doi: 10.1186/1476-4598-12-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shimojo Y, Akimoto M, Hisanaga T, et al. Attenuation of reactive oxygen species by antioxidants suppresses hypoxia-induced epithelial-mesenchymal transition and metastasis of pancreatic cancer cells. Clin Exp Metastasis. 2013;30(2):143–154. doi: 10.1007/s10585-012-9519-8. [DOI] [PubMed] [Google Scholar]
- 149.Salnikov AV, Liu L, Platen M, et al. Hypoxia induces EMT in low and highly aggressive pancreatic tumor cells but only cells with cancer stem cell characteristics acquire pronounced migratory potential. PLoS One. 2012;7(9):e46391. doi: 10.1371/journal.pone.0046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cannito S, Novo E, Compagnone A, et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis. 2008;29(12):2267–2278. doi: 10.1093/carcin/bgn216. [DOI] [PubMed] [Google Scholar]